

Abstract
Myofibroblast differentiation is a key process in pathogenesis of fibrotic diseases. Cardiac glycosides (ouabain, digoxin) inhibit Na+-K+-ATPase, resulting in increased intracellular [Na+]-to-[K+] ratio in cells. Microarray analysis suggested that increased intracellular [Na+]/[K+] ratio may promote the expression of cyclooxygenase-2 (COX-2), a critical enzyme in the synthesis of prostaglandins. Given antifibrotic effects of prostaglandins through activation of protein kinase A (PKA), we examined if cardiac glycosides stimulate COX-2 expression in human lung fibroblasts and how they affect myofibroblast differentiation. Ouabain stimulated a profound COX-2 expression and a sustained PKA activation, which was blocked by COX-2 inhibitor or by COX-2 knockdown. Ouabain-induced COX-2 expression and PKA activation were abolished by the inhibitor of the Na+/Ca2+ exchanger, KB-R4943. Ouabain inhibited transforming growth factor-β (TGF-β)-induced Rho activation, stress fiber formation, serum response factor activation, and the expression of smooth muscle α-actin, collagen-1, and fibronectin. These effects were recapitulated by an increase in intracellular [Na+]/[K+] ratio through the treatment of cells with K+-free medium or with digoxin. Although inhibition of COX-2 or of the Na+/Ca2+ exchanger blocked ouabain-induced PKA activation, this failed to reverse the inhibition of TGF-β-induced Rho activation or myofibroblast differentiation by ouabain. Together, these data demonstrate that ouabain, through the increase in intracellular [Na+]/[K+] ratio, drives the induction of COX-2 expression and PKA activation, which is accompanied by a decreased Rho activation and myofibroblast differentiation in response to TGF-β. However, COX-2 expression and PKA activation are not sufficient for inhibition of the fibrotic effects of TGF-β by ouabain, suggesting that additional mechanisms must exist.
Keywords: ouabain, digoxin, intracellular sodium-to-potassium ion ratio, cyclooxygenase-2
idiopathic pulmonary fibrosis (IPF) is a progressive fatal disease characterized by parenchymal fibrosis and structural distortion of the lungs. Age-adjusted mortality due to pulmonary fibrosis is increasing, and it poses a vexing clinical challenge given the lack of efficacious therapy. IPF is thought to be a disorder of abnormal wound healing (54), in which the initial trigger to the fibrotic response is injury to the alveolar epithelial cell, followed by an exuberant nonresolving wound-healing response (50). Injury of alveolar epithelial cells is thought to result in the elaboration of a fibrinous provisional matrix and activation of several proinflammatory, procoagulant, and profibrotic mediators, of which transforming growth factor-β1 (TGF-β1) is the most established (47). Fibroblasts, under stimulation of TGF-β1, respond by altering their gene expression profile with de novo expression of cytoskeletal and contractile proteins normally found within smooth muscle (SM) cells, modified focal adhesion complexes, and components of the extracellular matrix (18, 27). These SM-like fibroblasts are called myofibroblasts and display a phenotype that is in an intermediate state between fibroblasts and SM cells (10, 19). Several cytoskeletal and SM proteins are expressed in myofibroblasts, including SM α-actin, the most established marker for myofibroblast differentiation (19, 52). Additionally, induction of myofibroblast phenotype is also associated with secretion of extracellular matrix proteins (collagen isoforms, cellular fibronectin, etc.) and of profibrotic factors (connective tissue growth factor, insulin-like growth factor-II, etc.), thus perpetuating the ongoing tissue remodeling and fibrosis. Myofibroblasts are invariably found in histological sections of human lung specimens from patients with pulmonary fibrosis and are thought to be a critical pathogenic mechanism responsible for the progressive nature of IPF (27). Therefore, disrupting cellular mechanisms that induce and maintain the myofibroblast phenotype may be a potential strategy to attenuate the ongoing fibrotic response in pulmonary fibrosis.
In vitro studies have demonstrated that differentiation, proliferation, and extracellular matrix deposition by pulmonary myofibroblasts can be diminished by activators of cAMP and protein kinase A (PKA) (21, 32, 44), suggesting that cAMP/PKA signaling may be a potential therapeutic target for the treatment of pulmonary fibrosis. In vivo studies have shown a protective effect of cAMP-raising agonists such as prostaglandin E2 (PGE2), prostacyclin analog, iloprost (8, 59), or adrenomedullin (11, 23) in a bleomycin model of pulmonary fibrosis. However, identification of suitable agonists acting through cAMP/PKA signaling that could be effective in humans remains unaccomplished.
Cyclooxygenases are the rate-limiting enzymes in the production of prostaglandins (PGE2, PGI2, PGD2) from arachidonic acid (48). It is well documented that the expression of inducible cyclooxygenase isoform COX-2 is decreased in lungs from IPF patients (42, 56, 57). COX-2 inhibition or genetic deficiency worsens the degree of pulmonary fibrosis in animal models of pulmonary fibrosis (6, 17, 20). Therefore, upregulation of COX-2 expression in the lung providing production of endogenous prostanoids and resulting in a sustained PKA activation in fibroblasts could be an attractive strategy for treatment of pulmonary fibrosis.
Data on the beneficial effect of extracts from the leaves of Digitalis on the patients with heart failure documented more than 200 years ago led to the isolation of plant-derived cardiac glycosides known also as cardenolides (12). Digoxin isolated from Digitalis lanata has been widely employed in clinics, whereas ouabain isolated from Strophanthus gratus and possessing much higher water solubility is commonly used for in vitro studies. Mg2+-dependent Na+ + K+-stimulated adenosine triphosphatase (Na+-K+-ATPase) is the only target that is inhibited by cardiac glycosides (49). Na+-K+-ATPase is an integral plasma membrane protein consisting of α- and β-subunits and detected in all types of animal cells. It was shown that ATP hydrolysis by the larger α-subunit leads to its phosphorylation, conformational transition, and pumping of 3Na+ out of the cell and 2K+ into the cell against their gradients (51). Recently performed microarray analysis suggested that COX-2 mRNA is upregulated in ouabain-treated vascular SM cells from the rat aorta, human umbilical vein endothelial cells, and the HeLa cancer cell line (26). Therefore, we examined whether cardiac glycosides affect COX-2 expression in human lung fibroblasts (HLF) and whether this translates to PKA activation and regulation of myofibroblast differentiation.
EXPERIMENTAL PROCEDURES
Isolation and culture of primary human lung fibroblasts.
Human lung fibroblasts were isolated as described previously (44). Briefly, tissue samples from explanted lungs from patients undergoing lung transplantation were obtained and placed in Dulbecco's modified Eagle medium (DMEM) with 100 U/ml streptomycin, 250 ng/ml amphotericin B, and 100 U/ml penicillin. Alveolated lung tissue was minced, washed in PBS, and plated on 10-cm plates in growth media containing DMEM supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml streptomycin, 250 ng/ml amphotericin B, and 100 U/ml penicillin. Expanded populations of fibroblasts were subsequently subcultured after 4–5 days, resulting in the development of a homogenous fibroblast population. All primary cultures were used from passage 3 to 10.
Transfection and luciferase assay.
Subconfluent cells were cotransfected with desired firefly luciferase reporter plasmid, and thymidine kinase promoter-driven Renilla luciferase plasmid. Cells were serum-starved, followed by stimulation with desired agonists. Cells were washed and then lysed in protein extraction reagent. Lysates were assayed for firefly and Renilla luciferase activity using the dual luciferase assay kit (Promega). To account for differences in transfection efficiency, firefly luciferase activity of each sample was normalized to Renilla luciferase activity.
Knockdown of COX2.
For COX2 knockdown, the following small-interfering RNA (siRNA) was used: 5′-UAGGGCUUCAGCAUAAAGCGU-3′ (Qiagen, Valencia, CA). COX siRNA or scrambled RNA were transfected using use Lipofectamine RNAiMAX transfection reagent (Life Technologies, Grand Island, NY) following the manufacturer's standard protocol.
Cell lysis and Western blotting.
After stimulation of cells with desired agonists, cells were lysed in the radioimmunoprecipitation buffer containing 25 mM HEPES (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 2 mM EDTA, 2 mM EGTA, 10% glycerol, 1 mM NaF, 200 μM sodium orthovanadate, and protease inhibitor cocktail (Sigma). Cells were scraped and sonicated, a sample was taken for the measurement of protein concentration, and the remainder was boiled in Laemmli buffer for 5 min. The samples were normalized to the protein content, subjected to polyacrylamide gel electrophoresis, analyzed by Western blotting with desired primary antibodies and corresponding horseradish peroxidase-conjugated secondary antibodies, and developed by an enhanced chemilumeniscence reaction (Pierce). The digital chemiluminescent pictures were imaged by the Luminescent Image Analyzer LAS-4000 (Fujifilm).
Reverse transcription-quantitative real-time PCR.
RNA STAT-60 (TEL-Test) was used to isolate total RNA following the manufacturer's protocol. RNA was randomly primed and reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) according to the manufacturer's protocols. Real-time PCR analysis was performed using iTaq SYBR Green supermix with ROX (Bio-Rad) in a MyIQ single-color real-time PCR detection system (Bio-Rad). The COX-2 primers were as follows: AGAAAACTGCTCAACACCGGA (forward) and CAAGGGAGTCGGGCAATCAT (reverse).
Fluorescent microscopy of stress fibers.
For visualization of stress fibers, cells grown on cover slips were fixed in 4% paraformaldehyde in PBS followed by permeabilization with 0.2% Triton X-100 in PBS. Cells were then incubated in 2% BSA in PBS, followed by incubation in rhodamine-conjugated phalloidin (Life Technologies) for 1 h at room temperature. Cells were then washed five times with PBS, and cover slips were mounted using Vectashield mounting medium containing DAPI nuclear stain (Vector Laboratories, Burlingame, CA). Fluorescence images were obtained using an Olympus 1X71 fluorescent microscope.
In vitro isolation of stress fibers.
Stress fibers were isolated as previously described (45). All procedures were performed on ice using the buffers containing protease inhibitor cocktail (Sigma). After stimulation with desired agonists, cells were washed with PBS and then extracted with a buffer containing 2.5 mM triethanolamine (pH 8.2) for 30 min with six buffer changes followed by extraction with 0.05% Nonidet P-40 (pH 7.2) for 5 min and subsequent extraction with 0.5% Triton X-100 (pH 7.2) for an additional 5 min. Cells were then immediately washed with cold PBS, scraped, and suspended in PBS, followed by centrifugation at 100,000 g for 1 h. Supernatant was removed, and the pellet was sonicated in 0.5% Triton X-100, 50 mM NaCl, 20 mM HEPES (pH 7.0), and 1 mM EDTA. Laemmli buffer was added, and samples were boiled for 5 min before further Western blot analysis as described above.
Intracellular content of monovalent ions.
Intracellular K+ and Na+ content was measured as the steady-state distribution of extra- and intracellular 86Rb and 22Na, respectively. To establish isotope equilibrium, cells growing in 12-well plates were preincubated for 3 h in control or K+-free medium (Sp-DMEM + Ca) containing 0.5 μCi/ml 86RbCl or 4 μCi/ml 22NaCl with high concentration ouabain added for the next 3 h. To test the action of K+-free medium, the cells were washed two times with ice-cold Sp-DMEM + Ca. Next, cells were transferred to Sp-DMEM + Ca medium containing 22NaCl. After 3 h, cells were transferred on ice, washed four times with 2 ml of ice-cold medium W containing 100 mM MgCl2 and 10 mM HEPES-Tris buffer (pH 7.4). The washing medium was aspirated, and the cells were lysed with 1% SDS and 4 mM EDTA solution. Radioactivity of the incubation media and cell lysates was quantified, and intracellular cation content was calculated as A/am, where A was the radioactivity of the samples [counts/min (cpm)], “a” was the specific radioactivity of 86Rb (K+) or 22Na (cpm/nmol), and “m” was protein content (mg). For more details, see Ref. 2.
45Ca2+ influx.
Confluent quiescent cultures of HLF seeded in 12-well plates were treated as indicated in the legend for Fig. 10A and washed two times at room temperature with 2-ml aliquots of medium containing 150 mM NaCl and 10 mM HEPES-Tris (pH 7.4),followed by addition of 0.5 ml of medium containing 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 0.1 mM CaCl2, 5 mM glucose, 20 mM HEPES-Tris 20 (pH 7.4), 4 mCi/ml 45Ca with or without 1 μM nicardipine or 3 μM KB-R4943. After 5 min, isotope uptake was terminated by the addition of 2.5 ml ice-cold medium W. The dishes were transferred onto ice, and the cells were washed five times with 2.5 ml of ice-cold medium W. The cells were lysed with 1 ml of 4 mM EDTA/1% SDD, and radioactivity was quantified by liquid scintillation counting. 45Ca influx was calculated as A/am, where “A” is radioactivity in the cell lysate (cpm), a is specific radioactivity of the incubation medium (cpm/pmol), and m is the protein content per well (mg). The activity of L-type Ca2+ channel and Na+/Ca2+ exchanger was quantified as nicardipine- and KB-R7943-sensitive components of the rate of 45Ca influx, respectively. For more details, see Refs. 37 and 40.
Cytotoxicity assay.
Cytotoxicity of drugs was measured by a release of lactate dehydrogenase (LDH) using the colorimetric CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega) and following the manufacturer's protocol.
Reagents.
TGF-β1 and vasodilator-stimulated phosphoprotein (VASP) antibodies were from EMD Millipore (Billerica, MA). COX-2 antibodies were from Cell Signaling Technology (Danvers, MA); collagen-1 antibodies were from Cedarlane (Burlington, NC); and fibronectin antibodies were from BD Biosciences (San Jose, CA). Ouabain, digoxin, nicardipine, and antibodies against SM α-actin and β-tubulin were provided by Sigma-Aldrich (St. Louis, MO). 22NaCl, 86RbCl, and 45CaCl2 were obtained from PerkinElmer (Waltham, MA).
RESULTS
Ouabain increases COX-2 expression and activates PKA in human lung fibroblasts.
As shown in Fig. 1, quiescent HLFs do not express detectable levels of COX-2. Treatment of HLF with 100 nM ouabain resulted in a profound and sustained expression of COX-2 at both the mRNA and protein levels (Fig. 1A). Given the established role of COX-2 in the production of prostanoids that act through receptor-mediated activation of cAMP/PKA signaling, we assessed PKA activation in HLFs by examining phosphorylation of the PKA substrate VASP using electrophoretic mobility shift assay as a reporter for PKA activity (9). As shown in Fig. 1B, 100 nM ouabain stimulated a sustained VASP phosphorylation at 24–48 h that paralleled COX-2 expression in HLF cells. Furthermore, the specific inhibitor of COX-2, NS-398, completely abolished the ouabain-induced VASP shift while increasing COX-2 expression (Fig. 1B). To confirm the specificity of NS-398, we performed a knockdown of COX-2 mRNA. Figure 1C demonstrates a highly efficient knockdown of COX-2 expression in HLF resulting in suppression of VASP shift triggered by ouabain. Together, these data demonstrate that ouabain induces the expression of COX-2 in HLF, which functionally translates to activation of PKA.
Fig. 1.
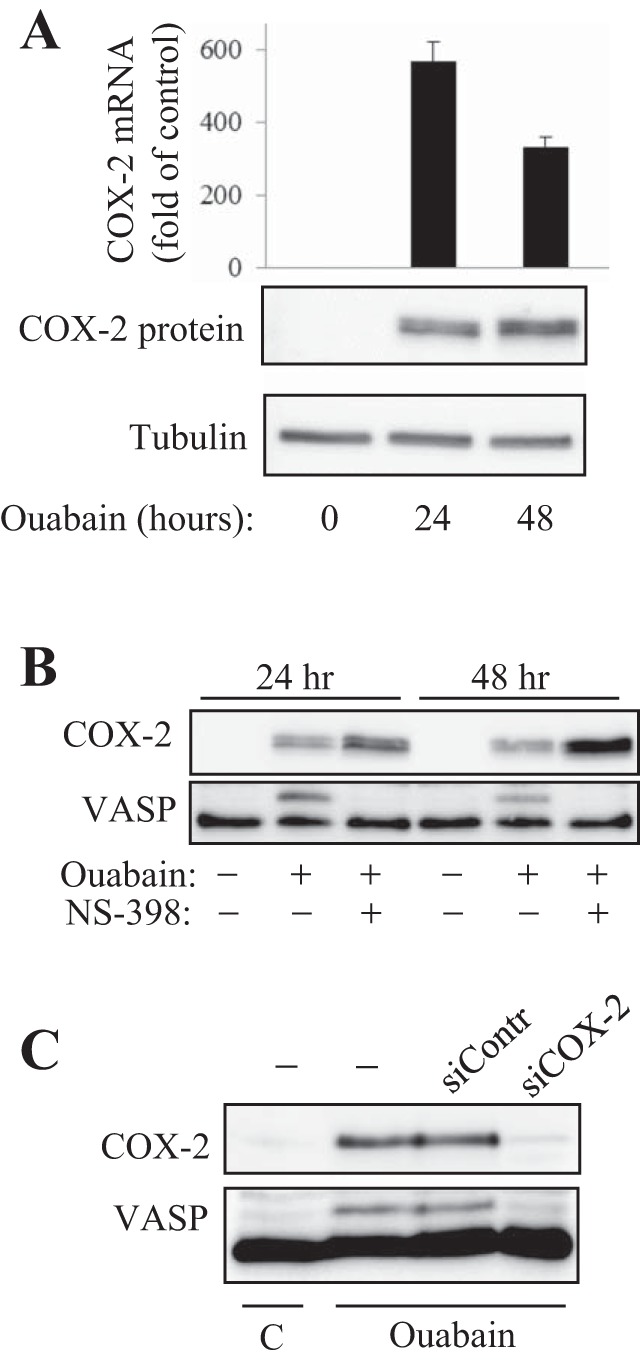
Ouabain-induced cyclooxygenase (COX)-2 expression and protein kinase A (PKA) activation in human lung fibroblasts (HLF). Serum-starved HLFs were treated with 100 nM ouabain with or without 1 μM NS-398 for the times indicated. Cells were analyzed by real-time qPCR for COX-2 mRNA levels (A) or by Western blotting for COX-2 expression and vasodilator-stimulated phosphoprotein (VASP) shift (A–C). C: HLFs were transfected with a scramble or COX-2 small-interfering RNAs (siRNAs), serum-starved for 24 h, and treated with 100 nM ouabain for 24 h. Shown are the Western blots for COX-2 and VASP.
Activation of Na+/Ca exchanger mediates COX-2 expression and PKA activation in HLF.
It is well documented that elevation of the [Na+]i/[K+]i ratio as a result of inhibition of Na+-K+-ATPase typically leads to increases in [Ca2+]i via activation of the reverse mode of the Na+/Ca2+ exchanger (5) and/or of voltage-gated Ca2+ channels (35), a phenomenon termed excitation-transcription coupling (39). It has been shown also that agonist-induced COX-2 expression is mediated by intracellular Ca2+ (36, 58). To explore the role of Ca2+ in COX-2 expression triggered by ouabain, we used inhibitors of voltage-gated L-type Ca2+ channels, nicardipine, and of Na+/Ca2+ exchanger compound, KB-R4943. We found that exposure of HLF to 100 nM ouabain increased the rate of 45Ca influx by ∼50% (Fig. 2A). These differences were preserved in the presence of nicardipine but were abolished by KB-R4943. Figure 2B shows that KB-R4943 sharply decreases COX-2 expression and VASP shift in response to ouabain. Neither COX-2 expression nor VASP phosphorylation was affected by nicardipine.
Fig. 2.
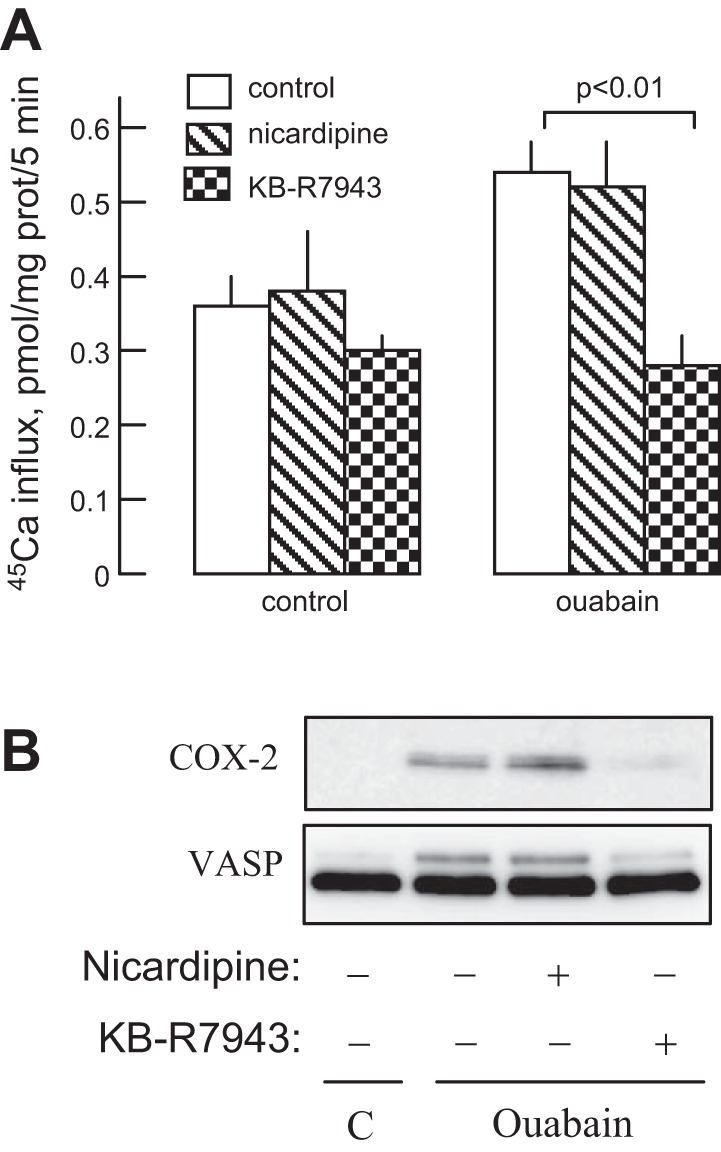
Effect of ouabain, nicardipine, and KB-R7943 on 45Ca2+ influx and on COX-2 expression and VASP shift. A: HLFs were preincubated for 24 h in the presence of 100 nM ouabain, 1 mM nicardipine, or 3 mM KB-R7943, and the rate of 45Ca2+ influx was measured as described in experimental procedures. Means ± SE from experiments performed in triplicate are shown. B: HLFs were treated with 100 nM ouabain, 1 mM nicardipine, or 3 mM KB-R7943 for 24 h, and cell extracts were examined by Western blotting for COX-2 expression and VASP shift.
Disruption of actin stress fibers by ouabain in HLF.
Microscopy analysis showed that ouabain treatment resulted in cell shape transition of human lung fibroblasts (Fig. 3A), the effect that was observed at long-term treatment (24–48 h) but not at short-term treatment (up to 6 h). Because such cell shape changes may associate with cell death, and because ouabain is known to induce apoptosis of various but not all cell types (38, 39), we examined a potential cytotoxic effect of ouabain on HLF by measuring the release of LDH. As shown in Fig. 3B, 100 nM ouabain had no significant cytotoxic effect for up to 48 h, whereas the positive control H2O2 drove a drastic increase in LDH release. We also did not observe membrane blebbing or accumulation of apoptotic bodies (that commonly accompany apoptosis) in ouabain-treated HLF (Fig. 3A). On the other hand, a similar shape change in oubain-treated HLF was previously described as “arborization” or “stellation” in vascular SM cells treated with agents that disrupt actin filaments or activate PKA (7, 41). Given that, we examined a potential effect of ouabain on stress fibers in HLF. As shown in Fig. 3C, ouabain treatment halved the total content of polymerized actin and clearly disrupted the structure of stress fibers, as assessed by phalloidin staining.
Fig. 3.

Ouabain distorts actin stress fibers without affecting HLF viability. A: serum-starved HLFs were treated with or without 100 nM ouabain for times indicated. Shown are the phase-contrast microscopy images of the cells. B: effect of 100 nM ouabain or 1 mM H2O2 on HLF viability as assessed by lactate dehydrogenase (LDH) release. C: phalloidin staining of HLFs treated with or without 100 nM ouabain for 48 h. Percentile intensity of phalloidin is indicated.
Ouabain and digoxin inhibit TGF-β1-induced myofibroblast differentiation.
We and others have demonstrated that myofibroblast differentiation is highly sensitive to regulation by activators of PKA (21, 44). Given the activation of PKA by ouabain in HLF, we examined its effect on myofibroblast differentiation in response to TGF-β. As shown in Fig. 4A, treatment of HLF with TGF-β resulted in a profound induction of the expression of myofibroblast differentiation markers, such as SM-specific α-actin, fibronectin, and collagen-1. Ouabain inhibited the expression of myofibroblast markers in response to TGF-β1, which paralleled its effect on COX-2 expression. We also examined the effect of clinically relevant cardiac glycoside, digoxin, on myofibroblast differentiation. As shown in Fig. 4B, digoxin similarly stimulated expression of COX-2 and phosphorylation of VASP and completely blocked myofibroblast differentiation in response to TGF-β.
Fig. 4.
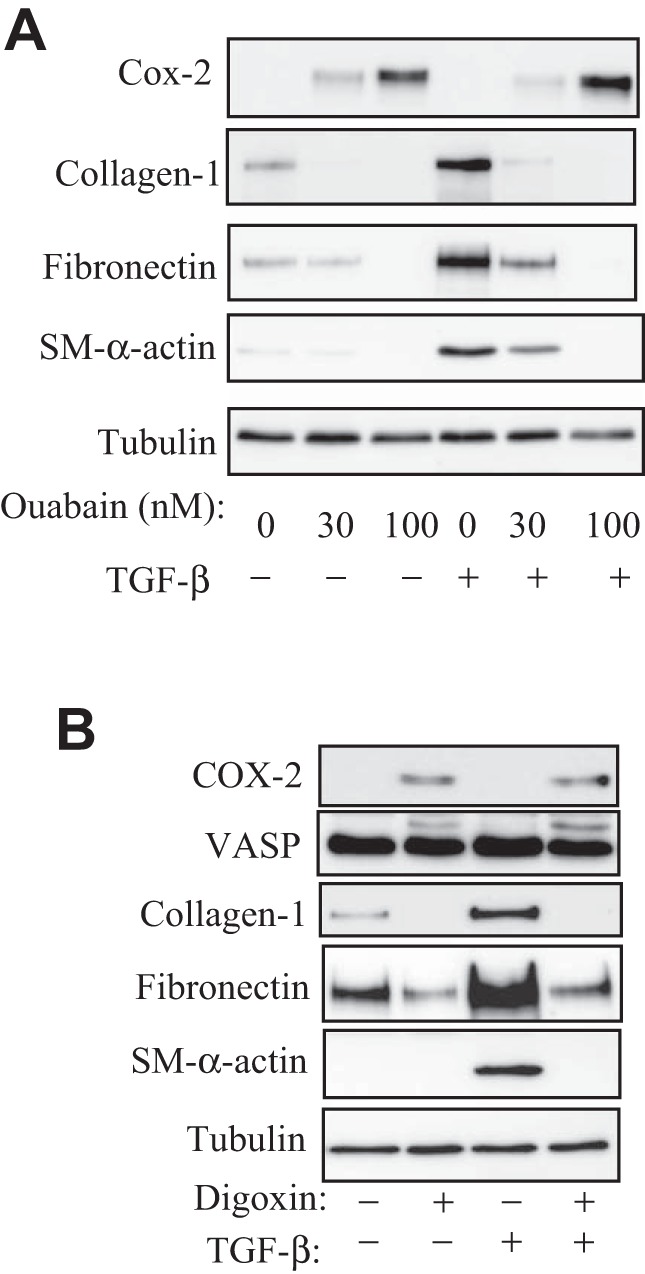
Ouabain and digoxin inhibit myofibroblast differentiation. Serum-starved HLFs were treated with or without 1 ng/ml transforming growth factor-β (TGF-β) in the presence or absence of 30 or 100 nM ouabain (A) or 100 nM digoxin (B) for 48 h. Cell extracts were assessed by Western blotting for COX-2 expression, VASP shift, and myofibroblast differentiation markers as indicated.
Upon stimulation with TGF-β1, fibroblasts respond by altering their ultrastructure by formation of prominent actin stress fibers and modified focal adhesion complexes, which provide mechanical coupling to the surrounding matrix and increased contractility of myofibroblasts (18, 53). In addition, actin stress fiber formation plays a signaling role in driving the activation of serum response factor (SRF) and SRF-dependent expression of several myofibroblast markers, including SM α-actin (44, 45). Therefore, we examined the effect of ouabain on this pathway. Consistent with previous data (45), a 48-h exposure to TGF-β1 resulted in stress fiber formation, as quantitatively examined by biochemical isolation of stress fibers (Fig. 5A). Importantly, ouabain decreased TGF-β-induced stress fiber formation. Furthermore, ouabain attenuated TGF-β-induced SRF-luciferase reporter activity (Fig. 5B). Together, these data suggest that ouabain attenuates TGF-β-induced myofibroblast differentiation through a disruption of actin stress fibers and inhibition of SRF activity.
Fig. 5.
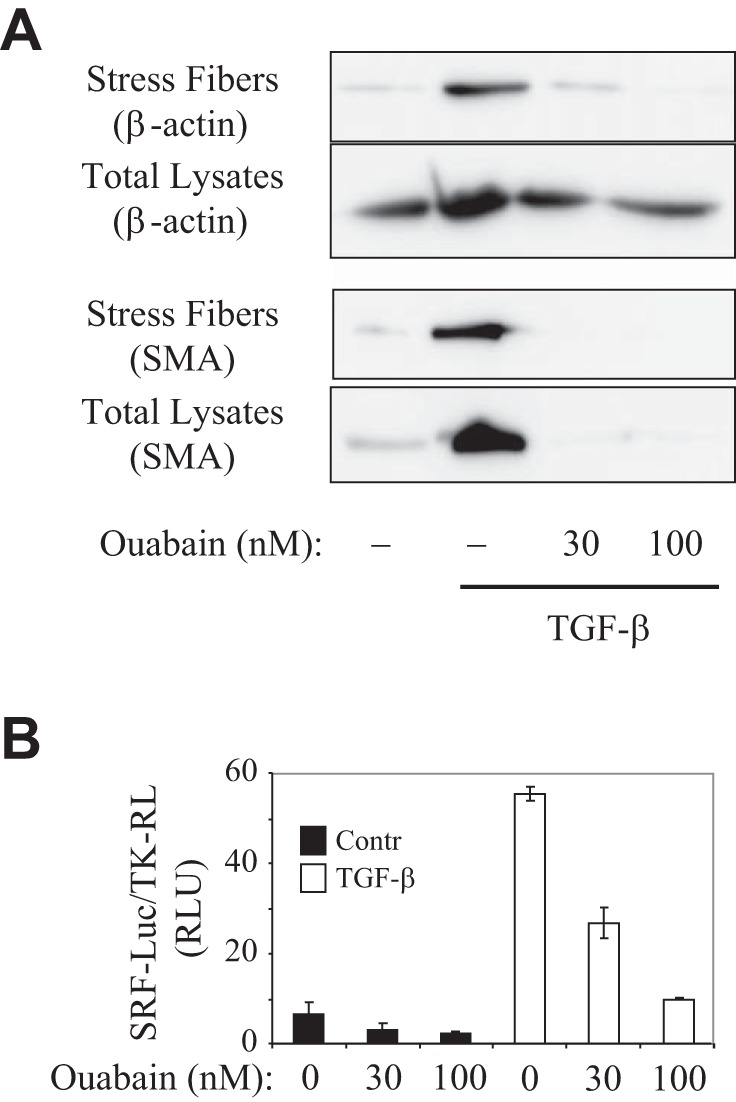
Ouabain inhibits TGF-β-induced stress fiber formation and serum response factor (SRF) activation. A: serum-starved HLF cells were treated with 1 ng/ml TGF-β in the presence or absence of ouabain for 48 h. Cell extracts were either lysed (total) or were processed for isolation of stress fibers, followed by Western blotting with desired antibodies. B: effect of increasing concentrations of ouabain (100 nM, 48 h) on SRF-luciferase reporter activity.
Role of intracellular monovalent cations in regulation of COX-2 expression and myofibroblast differentiation.
The roles of Nai+/Ki+-mediated and Nai+/Ki+-independent signaling mechanisms in cellular responses triggered by cardiac glycosides are widely disputed (39). Keeping this in mind, we 1) employed K+-free medium as an alternative approach for Na+-K+-ATPase inhibition and 2) compared dose-dependent action of ouabain on the [Na+]i/[K+]i ratio, COX-2 content, and expression of myofibroblast markers. As expected, inhibition of the Na+-K+-ATPase in K+-free media resulted in the same elevation of intracellular Na+ content as it was detected in HLFs subjected to Na+-K+-ATPase inhibition by 100 nM ouabain (Fig. 6A). Importantly, similarly to ouabain-treated cells, inhibition of the Na+-K+-ATPase by K+-free medium was accompanied by the expression of COX-2, phosphorylation of VASP, and downregulation of TGF-β1-induced expression of myofibroblast markers (Fig. 6B).
Fig. 6.
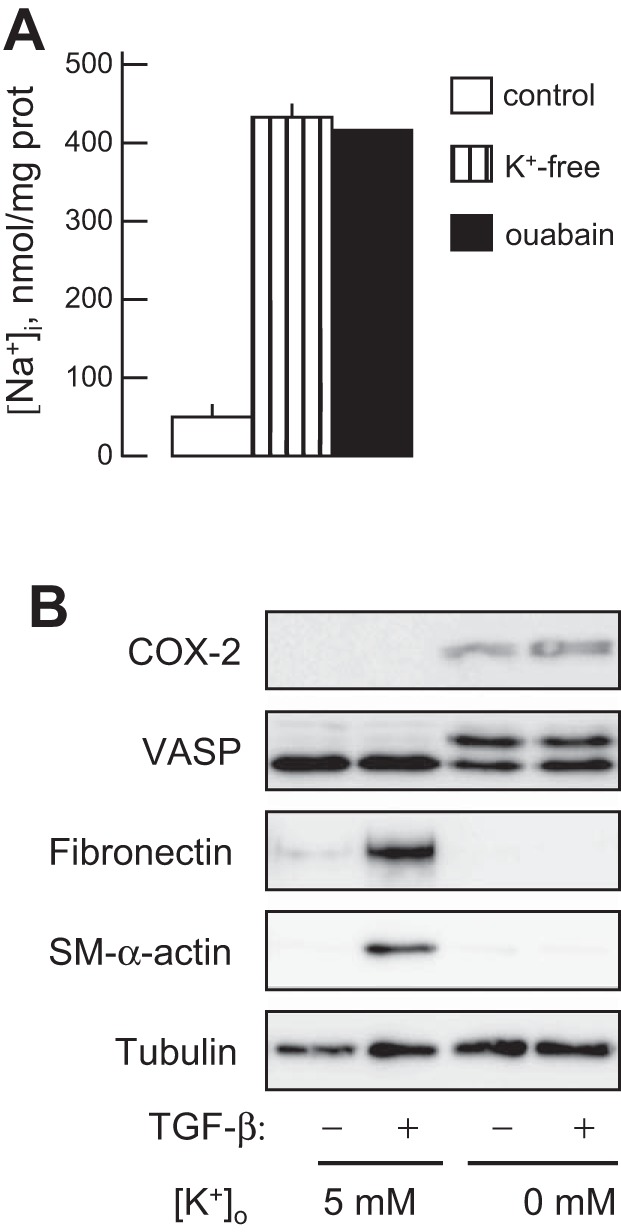
Effect of K+-free medium on intracellular Na+ content and on COX-2 expression, VASP phosphorylation, and myofibroblast differentiation. HLFs were treated with a control medium ([K+]o = 5 mM), K+-free medium, or with 100 nM ouabain in a control medium for 48 h, with or without 1 ng/ml TGF-β for 48 h as indicated. A: intracellular Na+ concentration was measured as described in experimental procedures. B: cell extracts were analyzed by Western blotting with desired antibodies as indicated.
Figure 7A shows that, at 3 nM, ouabain did not change [K+]i and slightly but not significantly increased [Na+]i by 74 ± 28% (P = 0.6). At higher concentrations (10–100 nM), ouabain dose-dependently increased [Na+]i and decreased [K+]i by eight- and sevenfold, respectively. The similar dose-response analysis of the effect of ouabain on TGF-β-induced myofibroblast differentiation revealed the following (Fig. 7, B and C). 1) Ouabain, at doses of 30–100 nM, induced COX-2 expression, which was parallel to inhibition of SM α-actin expression; and 2) 10 nM ouabain, resulting in a threefold gain of [Na+]i and twofold loss of [K+]i (Fig. 7A), had no detectable effect on COX-2 expression, but significantly inhibited the expression of SM α-actin (P = 0.047), collagen-1 (P = 0.025), or fibronectin (P = 0.006) (Fig. 7, B and C).
Fig. 7.

Dose-dependent actions of ouabain on intracellular Na+ and K+ content, COX-2 expression, and expression of myofibroblast differentiation markers. A: HLFs were treated with ouabain for 48 h and assessed for intracellular Na+ and K+ content. Means ± SE from experiments performed in triplicate are shown. B: HLFs were treated with 1 ng/ml TGF-β in the presence of increasing doses of ouabain for 48 h. Cell extracts were examined by Western blotting for COX-2 expression and myofibroblast differentiation markers. Shown are representative images from three independent experiments. C: densitometry of ECL for collagen-1, SM α-actin, and fibronectin Western blots (mean ± SD from 3 independent experiments). *P < 0.05 and **P < 0.01.
COX-2 expression is not sufficient for inhibition of HLF differentiation by ouabain.
Last, we examined the role of COX-2-dependent PKA activation in the inhibitory effect of ouabain on myofibroblast differentiation, first using COX-2 inhibitor NS-398. As shown above, this compound at a concentration of 1 μM completely blocked ouabain-induced VASP phosphorylation as an indicator of PKA activity in HLF (Fig. 1B). However, NS-398 failed to abolish the inhibitory effect of ouabain on myofibroblast differentiation (Fig. 8A). We and others have demonstrated that Rho/Rho kinase (ROCK) signaling is critical for TGF-β-induced stress fiber formation and SRF-dependent expression of myofibroblast marker proteins. Therefore, we examined the contribution of COX-2 expression on the regulation of this signaling pathway by ouabain by assessing phosphorylation of cofilin as a downstream target of ROCK and a reporter of its activity, as we performed previously (45). As shown in Fig. 8A, ouabain inhibited TGF-β-induced phosphorylation of cofilin; however, NS-398 failed to reverse this effect.
Fig. 8.

Inhibition of COX-2 or of Na+/Ca2+ exchanger does not abolish the inhibitory action of ouabain on TGF-β-induced myofibroblast differentiation. HLFs were treated with 1 μM NS-398 (A), pretransfected with a control or COX-2 siRNA (B), or treated with 3 μM KB-R7943 (C) followed by incubation with 100 nM ouabain or 1 ng/ml TGF-β for 48 h as indicated. Cells were lysed, and the cell extracts were analyzed by Western blotting with the desired antibodies as indicated.
Considering the possibility of prostaglandin-independent effects of COX-2, we then used the knockdown approach. As shown in Fig. 8B, transfection of HLF with COX-2 siRNA resulted in a complete attenuation of ouabain-induced COX-2 expression. However, this did not affect the ability of ouabain to inhibit TGF-β-induced myofibroblast differentiation or cofilin phosphorylation as a reporter of Rho signaling. Finally, we considered the role of [Ca2+]i increase via activation of the reverse mode of the Na+/Ca2+ exchanger in the action of ouabain (Fig. 2). As show in Fig. 8C, the inhibitor of Na+/Ca2+ exchanger, KB-R4943, while inhibiting the effect of ouabain on COX-2 expression, failed to reverse the inhibition of TGF-β-induced Rho activation (cofilin phosphorylation) and myofibroblast differentiation by ouabain.
DISCUSSION
The present study describes two major findings. 1) We demonstrate a profound induction of COX-2 expression and PKA activation under various conditions that inhibit Na+-K+-ATPase and elevate the [Na+]i/[K+]i ratio (treatment with ouabain, digoxin, or K+-free media) in human lung fibroblasts, and this effect is largely mediated by an increase in intracellular Ca2+ concentration through activation of a reverse mode of Na+/Ca2+ exchanger. 2) Inhibition of Na+-K+-ATPase by ouabain results in attenuation of TGF-β-induced fibrotic signaling (Rho activation, stress fiber formation, and SRF activation) and expression of myofibroblast differentiation markers, demonstrating a novel function of Na+-K+-ATPase in control of fibroblast phenotype. However, inhibition of COX-2 or of Na+/Ca2+ exchanger failed to reverse the inhibitory effect of ouabain on TGF-β-induced fibrotic signaling and myofibroblast differentiation. This may suggest that either the antifibrotic effects of ouabain are independent of COX-2, or COX-2 expression and PKA activation are not sufficient for inhibition of the fibrotic effects of TGF-β by ouabain, but may act together with other mechanisms yet to be identified. Given the previously established antifibrotic role of COX-2 and PKA (8, 22, 23, 33, 59), the latter possibility is quite plausible.
Our results show for the first time that cardiac glycosides suppress TGF-β-induced fibrotic signaling (Rho activation, stress fiber formation, and SRF activation; Figs. 5 and 8) and myofibroblast differentiation of cultured HLFs (expression of collagen-1, fibronectin, and SM α-actin) (Fig. 4) without any impact on their survival (Fig. 3B). During the last two decades, a number of studies reported that, along with Na+-K+ pump inhibition and modulation of canonical Nai+/Ki+-dependent cell functions, cardiac glycosides may have Nai+/Ki+-independent effects, i.e., promoting the interaction of Na+-K+-ATPase with the membrane-associated nonreceptor tyrosine kinase Src, activation of Ras/Raf/ERK1,2, phosphatidylinositol 3-kinase [PI(3)K], PI(3)K-dependent protein kinase B, phospholipase C, [Ca2+]i oscillations, and augmented production of reactive oxygen species (for review, see Refs. 3 and 31). We show that TGF-β1-induced myofibroblast differentiation is also blocked by sustained inhibition of the Na+-K+-ATPase in K+-free medium (Fig. 6). We also demonstrate that inhibition of myofibroblast differentiation by ouabain dose-dependently associates with increased [Na+]i/[K+]i ratio (Fig. 7). These data strongly suggest that cardiac glycosides inhibit myofibroblast differentiation via an Nai+/Ki+-dependent mechanism.
It is noteworthy that actions of cardiac glycosides on fibrotic response may be cell specific. It was shown that certain inhibitors of Na+-K+-ATPase increase collagen synthesis by cultured rat cardiac fibroblasts, human dermal fibroblasts, or rat vascular SM cells (13–16). However, these studies have not rigorously examined the effect of these compounds on the expression of myofibroblast differentiation markers, on the magnitude of their effect relative to that of TGF-β, or on their effect on TGF-β-induced myofibroblast differentiation. Furthermore, the association of the effects of these compounds on collagen synthesis with their actions on the [Na+]i/[K+]i ratio was not performed. Finally, these studies have largely employed marinobufagenin, which causes distinct from ouabain structural changes in the α1-subunit of Na+-K+-ATPase (25) and may act through Nai+/Ki+-independent mechanisms (15).
It was shown that the increase in the [Na+]i/[K+]i ratio affects the expression of genes via Cai2+-mediated and Cai2+-independent mechanisms of excitation-transcription coupling (39). We found that COX-2 expression in ouabain-treated HLF is abolished by KB-R7943, a potent inhibitor of Na+/Ca2+ exchange, suggesting that elevation of the [Na+]i/[K+]i ratio triggers COX-2 accumulation via Cai2+-mediated signaling pathways. It should be noted, however, that, along with inhibition of three isoforms of Na+/Ca2+ exchanger (NCX1–NCX3) (4), KB-R7943 affects other molecules/processes, including ATP-dependent K+ current (1), nonselective cation channels (43), and the mitochondrial permeability transition pore (55). Thus, additional experiments should be performed to examine the role of intracellular Ca2+ in the expression of COX-2 and to identify other transcriptomic changes contributing to the suppression of myofibroblast differentiation by cardiac glycosides.
The antifibrotic action of COX-2, its products, prostaglandins, as well as of PKA is well established (8, 22, 23, 59). It was shown that genetic disruption or pharmacological inhibition of COX-2 induces an exaggerated accumulation of myofibroblasts in mouse models of pulmonary fibrosis (6, 17, 20, 24). Our data suggest that antifibrotic action of elevated [Na+]i/[K+]i ratio parallels the increased expression of COX-2, which, in turn, leads to activation of PKA. Thus, treatment of HLF with cardiac glycosides (ouabain or digoxin) or with K+-free medium sharply augmented the content to COX-2 mRNA and its immunoreactive protein as well as the activity of PKA as estimated by VASP mobility shift (Figs. 1, 4, and 6). Furthermore, ouabain blocked TGF-β-induced Rho activation (Fig. 7), stress fiber formation, and SRF activation (Fig. 5), the processes critical for myofibroblast differentiation and known to be regulated by PKA (44, 45). We also showed that both COX-2 inhibitor NS-398 and siRNA-induced COX-2 knockdown completely abolished the VASP shift seen in ouabain-treated HLF (Fig. 1). However, neither COX-2 inhibitor NS-398 nor the knockdown of this enzyme abolished the inhibitory action of ouabain on Rho activation and myofibroblast differentiation of HLF in response to TGF-β1. This may suggest that the antifibrotic effects of ouabain are independent of COX-2. However, given the established antifibrotic role of COX-2 (8, 22, 23, 59), we propose that augmented COX-2 expression may be an important but not sufficient mechanism in the inhibition of myofibroblast differentiation by elevated [Na+]i/[K+]i ratio. Furthermore, our data demonstrate that inhibition of myofibroblast differentiation by ouabain is not mediated by the increase in [Ca2+]i, since it was not reversed by inhibition of Na+/Ca2+ exchanger (Fig. 8C), which largely contributed to the increase in [Ca2+]i in response to ouabain (Fig. 2A). Thus, yet unknown potential actions of elevated [Na+]i/[K+]i ratio may be responsible for the regulation of myofibroblast differentiation by cardiac glycosides, which is currently under investigation in our laboratory.
While this study focused on TGF-β-induced myofibroblast differentiation, myofibroblast phenotype and function depend on many factors, including matrix stiffness (30, 34). Thus, it would be important to determine if cardiac glycosides affect myofibroblast differentiation driven by other factors, especially given that myofibroblast differentiation induced by stiff matrix is associated with a decreased COX-2 expression and PGE2 synthesis (30). Furthermore, while this study proposes the inhibition of the Rho/stress fiber/SRF pathway as a potential antifibrotic mechanism of cardiac glycosides, their effect on other mechanosensitive signaling pathways that also contribute to myofibroblast differentiation should be examined, including the YAP/TAZ pathway (29).
Finally, it would be fundamentally and practically important to demonstrate the antifibrotic effect of cardiac glycosides in vivo. However, numerous studies have demonstrated that, in rodents, cardiac glycosides inhibit α1-Na+-K+-ATPase at concentrations of up to four orders of magnitude higher than in other mammals, whereas the affinities of rodent α2- and α3-subunits for cardiac glycosides are similar to those in other mammals (46). Given that α1-subunit is a predominant isoform expressed in mouse lung fibroblasts (data not shown), wild-type rodents could not be used for assessing the antifibrotic effect of cardiac glycoside models of pulmonary fibrosis. The resistance to cardiac glycosides is caused by a substitution of Gln111 and Asn122 of human α1-subunit for Arg and Asp, respectively, in rodents. Based on this, Lingrel and coworkers have generated a mouse with knockin of ouabain-sensitive α1-isoform (α1s/s) of Na+-K+-ATPase and have used it to delineate the role of α1 in blood pressure regulation, cardiac and skeletal muscle contraction, and renal salt handling (28). These mice can be also used for elucidation of the antifibrotic effect of cardiac glycosides in vivo in the models of pulmonary fibrosis, which is our future goal.
GRANTS
Research reported in this publication was supported by the National Institutes of Health Award 1R56-HL-127395 (N. O. Dulin) and National Center For Advancing Translational Sciences of the National Institutes of Health under Award UL1-TR-000430 (N. O. Dulin). This study was also supported by American Heart Association Predoctoral Fellowship Award 14PRE18360017 (J. La), the Russian Foundation for Fundamental Research (14-04-31705 and 15-04-00101), and the Russian Scientific Foundation (No. 14–15-0006; O. Akimova, S. Koltsova, and S. N. Orlov).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
AUTHOR CONTRIBUTIONS
J.L., E.B.R., S.V.K., O.A., R.B.H., and N.O.D. performed experiments; J.L., E.B.R., S.V.K., O.A., R.B.H., G.M.M., S.N.O., and N.O.D. analyzed data; J.L., E.B.R., S.V.K., O.A., R.B.H., G.M.M., S.N.O., and N.O.D. interpreted results of experiments; J.L., S.V.K., O.A., R.B.H., S.N.O., and N.O.D. prepared figures; J.L., S.N.O., and N.O.D. drafted manuscript; J.L., G.M.M., S.N.O., and N.O.D. edited and revised manuscript; J.L., E.B.R., S.V.K., O.A., R.B.H., G.M.M., S.N.O., and N.O.D. approved final version of manuscript; G.M.M., S.N.O., and N.O.D. conception and design of research.
REFERENCES
- 1.Abramochkin DV, Vornanen M. Inhibition of the cardiac ATP-dependent potassium current by KB-R7943. Comp Biochem Physiol A Mol Integr Physiol 175: 38–45, 2014. [DOI] [PubMed] [Google Scholar]
- 2.Akimova OA, Bagrov AY, Lopina OD, Kamernitsky AV, Tremblay J, Hamet P, Orlov SN. Cardiotonic steroids differentially affect intracellular Na+ and [Na+]i/[K+]i-independent signaling in C7-MDCK cells. J Biol Chem 280: 832–839, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Aperia A. New roles for an old enzyme: Na,K-ATPase emerges as an interesting drug target. J Intern Med 261: 44–52, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Billman GE. KB-R7943. Kanebo. Curr Opin Investig Drugs 2: 1740–1745, 2001. [PubMed] [Google Scholar]
- 5.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev 79: 763–854, 1999. [DOI] [PubMed] [Google Scholar]
- 6.Bonner JC, Rice AB, Ingram JL, Moomaw CR, Nyska A, Bradbury A, Sessoms AR, Chulada PC, Morgan DL, Zeldin DC, Langenbach R. Susceptibility of cyclooxygenase-2-deficient mice to pulmonary fibrogenesis. Am J Pathol 161: 459–470, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaldakov GN, Nabika T, Nara Y, Yamori Y. Cyclic AMP- and cytochalasin B-induced arborization in cultured aortic smooth muscle cells: its cytopharmacological characterization. Cell Tissue Res 255: 435–442, 1989. [DOI] [PubMed] [Google Scholar]
- 8.Dackor RT, Cheng J, Voltz JW, Card JW, Ferguson CD, Garrett RC, Bradbury JA, DeGraff LM, Lih FB, Tomer KB, Flake GP, Travlos GS, Ramsey RW Jr, Edin ML, Morgan DL, Zeldin DC. Prostaglandin E(2) protects murine lungs from bleomycin-induced pulmonary fibrosis and lung dysfunction. Am J Physiol Lung Cell Mol Physiol 301: L645–L655, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis A, Hogarth K, Fernandes D, Solway J, Niu J, Kolenko V, Browning D, Miano JM, Orlov SN, Dulin NO. Functional significance of protein kinase A activation by endothelin-1 and ATP: negative regulation of SRF-dependent gene expression by PKA. Cell Signal 15: 597–604, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen 13: 7–12, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Di Paola R, Talero E, Galuppo M, Mazzon E, Bramanti P, Motilva V, Cuzzocrea S. Adrenomedullin in inflammatory process associated with experimental pulmonary fibrosis. Respir Res 12: 41, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dmitrieva RI, Doris PA. Cardiotonic steroids: potential endogenous sodium pump ligands with diverse function. Exp Biol Med (Maywood) 227: 561–569, 2002. [DOI] [PubMed] [Google Scholar]
- 13.El-Okdi N, Smaili S, Raju V, Shidyak A, Gupta S, Fedorova L, Elkareh J, Periyasamy S, Shapiro AP, Kahaleh MB, Malhotra D, Xie Z, Chin KV, Shapiro JI. Effects of cardiotonic steroids on dermal collagen synthesis and wound healing. J Appl Physiol 105: 30–36, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, Smaili S, Periyasamy SM, Hariri IM, Fedorova L, Liu J, Wu L, Kahaleh MB, Xie Z, Malhotra D, Fedorova OV, Kashkin VA, Bagrov AY, Shapiro JI. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 49: 215–224, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Elkareh J, Periyasamy SM, Shidyak A, Vetteth S, Schroeder J, Raju V, Hariri IM, El-Okdi N, Gupta S, Fedorova L, Liu J, Fedorova OV, Kahaleh MB, Xie Z, Malhotra D, Watson DK, Bagrov AY, Shapiro JI. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli-1: implications for uremic cardiomyopathy. Am J Physiol Renal Physiol 296: F1219–F1226, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fedorova OV, Emelianov IV, Bagrov KA, Grigorova YN, Wei W, Juhasz O, Frolova EV, Marshall CA, Lakatta EG, Konradi AO, Bagrov AY. Marinobufagenin-induced vascular fibrosis is a likely target for mineralocorticoid antagonists. J Hypertens 33: 1602–1610, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giri SN, Hyde DM. Increases in severity of lung damage and mortality by treatment with cyclo and lipoxygenase inhibitors in bleomycin and hyperoxia model of lung injury in hamsters. Pathology 19: 150–158, 1987. [DOI] [PubMed] [Google Scholar]
- 18.Hinz B. Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur J Cell Biol 85: 175–181, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple rrigins. Am J Pathol 170: 1807–1816, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hodges RJ, Jenkins RG, Wheeler-Jones CP, Copeman DM, Bottoms SE, Bellingan GJ, Nanthakumar CB, Laurent GJ, Hart SL, Foster ML, McAnulty RJ. Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E(2) production. Am J Pathol 165: 1663–1676, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang SK, Wettlaufer SH, Chung J, Peters-Golden M. Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. Am J Respir Cell Mol Biol 39: 482–489, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kach J, Sandbo N, La J, Denner D, Reed EB, Akimova OA, Koltsova S, Orlov SN, Dulin NO. Antifibrotic effects of noscapine through activation of prostaglandin E2 receptors and protein kinase A. J Biol Chem 289: 7505–7513, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kach J, Sandbo N, Sethakorn N, Williams J, Reed EB, La J, Tian X, Brain SD, Rajendran K, Krishnan R, Sperling AI, Birukov K, Dulin NO. Regulation of myofibroblast differentiation and bleomycin-induced pulmonary fibrosis by adrenomedullin. Am J Physiol Lung Cell Mol Physiol 304: L757–L764, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keerthisingam CB, Jenkins RG, Harrison NK, Hernandez-Rodriguez NA, Booth H, Laurent GJ, Hart SL, Foster ML, McAnulty RJ. Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-beta in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am J Pathol 158: 1411–1422, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klimanova EA, Petrushanko IY, Mitkevich VA, Anashkina AA, Orlov SN, Makarov AA, Lopina OD. Binding of ouabain and marinobufagenin leads to different structural changes in Na,K-ATPase and depends on the enzyme conformation. FEBS Lett 589: 2668–2674, 2015. [DOI] [PubMed] [Google Scholar]
- 26.Koltsova SV, Trushina Y, Haloui M, Akimova OA, Tremblay J, Hamet P, Orlov SN. Ubiquitous [Na+]i/[K+]i-sensitive transcriptome in mammalian cells: evidence for Ca(2+)i-independent excitation-transcription coupling. PLoS One 7: e38032, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J 18: 816–827, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Lingrel JB. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu Rev Physiol 72: 395–412, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu F, Lagares D, Choi KM, Stopfer L, Marinkovic A, Vrbanac V, Probst CK, Hiemer SE, Sisson TH, Horowitz JC, Rosas IO, Fredenburgh LE, Feghali-Bostwick C, Varelas X, Tager AM, Tschumperlin DJ. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 308: L344–L357, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol 190: 693–706, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Xie ZJ. The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim Biophys Acta 1802: 1237–1245, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Ostrom RS, Insel PA. cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Physiol Cell Physiol 286: C1089–C1099, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Lovgren AK, Jania LA, Hartney JM, Parsons KK, Audoly LP, Fitzgerald GA, Tilley SL, Koller BH. COX-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 291: L144–L156, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Marinkovic A, Mih JD, Park JA, Liu F, Tschumperlin DJ. Improved throughput traction microscopy reveals pivotal role for matrix stiffness in fibroblast contractility and TGF-β responsiveness. Am J Physiol Lung Cell Mol Physiol 303: L169–L180, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev 74: 365–507, 1994. [DOI] [PubMed] [Google Scholar]
- 36.Nakao S, Ogata Y, Modeer T, Segawa M, Furuyama S, Sugiya H. Bradykinin induces a rapid cyclooxygenase-2 mRNA expression via Ca2+ mobilization in human gingival fibroblasts primed with interleukin-1 beta. Cell Calcium 29: 446–452, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Orlov S, Resink TJ, Bernhardt J, Ferracin F, Buhler FR. Vascular smooth muscle cell calcium fluxes. Regulation by angiotensin II and lipoproteins. Hypertension 21: 195–203, 1993. [DOI] [PubMed] [Google Scholar]
- 38.Orlov SN, Hamet P. The death of cardiotonic steroid-treated cells: evidence of Na+i,K+i-independent H+i-sensitive signalling. Acta Physiol (Oxf) 187: 231–240, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Orlov SN, Hamet P. Salt and gene expression: evidence for [Na+]i/[K+]i-mediated signaling pathways. Pflugers Arch 467: 489–498, 2015. [DOI] [PubMed] [Google Scholar]
- 40.Orlov SN, Tremblay J, Hamet P. cAMP signaling inhibits dihydropyridine-sensitive Ca2+ influx in vascular smooth muscle cells. Hypertension 27: 774–780, 1996. [DOI] [PubMed] [Google Scholar]
- 41.Orlov SN, Tremblay J, Hamet P. Cell volume in vascular smooth muscle is regulated by bumetanide-sensitive ion transport. Am J Physiol Cell Physiol 270: C1388–C1397, 1996. [DOI] [PubMed] [Google Scholar]
- 42.Petkova DK, Clelland CA, Ronan JE, Lewis S, Knox AJ. Reduced expression of cyclooxygenase (COX) in idiopathic pulmonary fibrosis and sarcoidosis. Histopathology 43: 381–386, 2003. [DOI] [PubMed] [Google Scholar]
- 43.Pezier A, Bobkov YV, Ache BW. The Na+/Ca2+ exchanger inhibitor, KB-R7943, blocks a nonselective cation channel implicated in chemosensory transduction. J Neurophysiol 101: 1151–1159, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandbo N, Kregel S, Taurin S, Bhorade S, Dulin NO. Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-beta. Am J Respir Cell Mol Biol 41: 332–338, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandbo N, Lau A, Kach J, Ngam C, Yau D, Dulin NO. Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-β. Am J Physiol Lung Cell Mol Physiol 301: L656–L666, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol 293: C509–C536, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest 100: 768–776, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 56: 387–437, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Skou JC. Further investigation on a Mg2+ + Na+-activated adenosinetriphosphatase possibly related to the active transport of Na+ and K+ across the nerve cell membrane. Biochim Biophys Acta 42: 6–23, 1960. [Google Scholar]
- 50.Thannickal VJ, Toews GB, White ES, Lynch JP 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med 55: 395–417, 2004. [DOI] [PubMed] [Google Scholar]
- 51.Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol 279: C541–C566, 2000. [DOI] [PubMed] [Google Scholar]
- 52.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3: 349–363, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res 257: 180–189, 2000. [DOI] [PubMed] [Google Scholar]
- 54.White ES, Lazar MH, Thannickal VJ. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J Pathol 201: 343–354, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiczer BM, Marcu R, Hawkins BJ. KB-R7943, a plasma membrane Na(+)/Ca(2+) exchanger inhibitor, blocks opening of the mitochondrial permeability transition pore. Biochem Biophys Res Commun 444: 44–49, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest 95: 1861–1868, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xaubet A, Roca-Ferrer J, Pujols L, Ramirez J, Mullol J, Marin-Arguedas A, Torrego A, Gimferrer JM, Picado C. Cyclooxygenase-2 is up-regulated in lung parenchyma of chronic obstructive pulmonary disease and down-regulated in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 21: 35–42, 2004. [PubMed] [Google Scholar]
- 58.Zhu Y, Hua P, Rafiq S, Waffner EJ, Duffey ME, Lance P. Ca2+- and PKC-dependent stimulation of PGE2 synthesis by deoxycholic acid in human colonic fibroblasts. Am J Physiol Gastrointest Liver Physiol 283: G503–G510, 2002. [DOI] [PubMed] [Google Scholar]
- 59.Zhu Y, Liu Y, Zhou W, Xiang R, Jiang L, Huang K, Xiao Y, Guo Z, Gao J. A prostacyclin analogue, iloprost, protects from bleomycin-induced pulmonary fibrosis in mice. Respir Res 11: 34, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]