

20-Hydroxyeicosatetraeonic acid (20-HETE) plays a critical role in the pathogenesis of hypertension. We demonstrate here, to the best to our knowledge for the first time, that it stimulated inositol 1,4,5-triphoshate stores and increased the production of mitochondrial superoxide in the pulmonary arteries. Activation of PKG by glucose-6-phosphate dehydrogenase (G6PD) inhibition blocked the 20-HETE-mediated signaling.
Keywords: 20-HETE, TNF-α, elk-1, mitogen-activated protein kinase 1 and 3, extracellular signal regulated kinase 2 and 1, protein kinase G, miRs
Abstract
20-Hydroxyeicosatetraeonic acid (20-HETE) produced by cytochrome P-450 monooxygenases in NADPH-dependent manner is proinflammatory, and it contributes to the pathogenesis of systemic and pulmonary hypertension. In this study, we tested the hypothesis that inhibition of glucose-6-phosphate dehydrogenase (G6PD), a major source of NADPH in the cell, prevents 20-HETE synthesis and 20-HETE-induced proinflammatory signaling that promotes secretory phenotype of vascular smooth muscle cells. Lipidomic analysis indicated that G6PD inhibition and knockdown decreased 20-HETE levels in pulmonary arteries as well as 20-HETE-induced 1) mitochondrial superoxide production, 2) activation of mitogen-activated protein kinase 1 and 3, 3) phosphorylation of ETS domain-containing protein Elk-1 that activate transcription of tumor necrosis factor-α gene (Tnfa), and 4) expression of tumor necrosis factor-α (TNF-α). Moreover, inhibition of G6PD increased protein kinase G1α activity, which, at least partially, mitigated superoxide production and Elk-1 and TNF-α expression. Additionally, we report here for the first time that 20-HETE repressed miR-143, which suppresses Elk-1 expression, and miR-133a, which is known to suppress synthetic/secretory phenotype of vascular smooth muscle cells. In summary, our findings indicate that 20-HETE elicited mitochondrial superoxide production and promoted secretory phenotype of vascular smooth muscle cells by activating MAPK1-Elk-1, all of which are blocked by inhibition of G6PD.
NEWS & NOTEWORTHY
20-Hydroxyeicosatetraeonic acid (20-HETE) plays a critical role in the pathogenesis of hypertension. We demonstrate here, to the best to our knowledge for the first time, that it stimulated inositol 1,4,5-triphoshate stores and increased the production of mitochondrial superoxide in the pulmonary arteries. Activation of PKG by glucose-6-phosphate dehydrogenase (G6PD) inhibition blocked the 20-HETE-mediated signaling.
20-hydroxyeicosatetraeonic acid (20-HETE), the ω-hydroxylation metabolite of arachidonic acid (AA), is produced by cytochrome P-450 monooxygenases of the (CYP4A and CYP4F gene) families in an NADPH-dependent manner (49). 20-HETE is proinflammatory and it regulates vascular and renal function (49). It also plays a critical role in the pathogenesis of systemic hypertension, renal stenosis, and atherosclerosis (27, 63, 65). 20-HETE increased by hypoxia inhibits hypoxia-induced pulmonary artery constriction (69). However, its role in the hypoxia-induced inflammation of pulmonary arteries or lungs is yet unclear.
Recently, our laboratory also found that 20-HETE is involved in promoting prolonged hypoxia-induced pulmonary vasoconstriction and that glucose-6-phosphate dehydrogenase (G6PD), which is a major producer of NADPH in the cell, and cytochrome P-450 monooxygenase enzymes are functionally coupled in vascular smooth muscle tissue (unpublished observations). G6PD inhibition relaxes pulmonary arteries by decreasing intracellular calcium (20), prevents switching of vascular smooth muscle cells to a synthetic phenotype (9, 10), and reduces pulmonary hypertension (9, 47). However, whether prolonged hypoxia-induced 20-HETE-synthesis is G6PD dependent or independent and whether G6PD modulates 20-HETE signaling in the vascular smooth muscle remain unknown. Various cell types in the lungs produce 20-HETE. Notably, airway and peripheral lung tissues produce the most 20-HETE (70). Since 20-HETE is an autacoid, we studied the effects of G6PD inhibition on extracellular 20-HETE-elicited cell signaling. Proving the existence of such an interaction will be of clinical importance.
Increased production of 20-HETE in vascular tissue or application of 20-HETE to blood vessels stimulates vascular smooth muscle cell contraction (52) as well as reactive oxygen species production (37, 53). 20-HETE activates mitogen-activated protein kinase (MAPK) 1 and 3 (also known as extracellular signal regulated kinase 2 and 1, respectively) and promotes secretion of cytokines, including TNF-α, IL-8, and IL-6 (6, 30, 40, 59). However, the sources of reactive oxygen species stimulated by 20-HETE in vascular smooth muscle cells remain unknown and the mechanisms through which 20-HETE stimulate the secretory or synthetic phenotype of vascular smooth muscle cells are not clearly understood. Therefore, this study was undertaken to test the hypothesis whether the inhibition of G6PD prevents 20-HETE production and 20-HETE-induced proinflammatory signaling that promotes secretory phenotype of vascular smooth muscle cells.
MATERIALS AND METHODS
All physiological buffers [Krebs, 120 mM KCl (HiK), 30 mM KCl (30K), and HEPES] were prepared by using salts (analyzed reagent grade) from Baker Chemical. All gas tanks were purchased from Air Gas (Allentown, PA). Dehydroepiandosterone (DHEA), 6-aminonicotinamide (6-AN), and maleimide were obtained from Sigma Chemical (St. Louis, MO). 20-HETE was purchased from Cayman Chemical (Ann Arbor, MI).
Animal studies.
All experiments were performed following the New York Medical College Animal Care and Use Committee-approved protocol in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Adult male G6PD mutant mice and appropriate age-matched wild-type (WT) controls were used. The mutation results in decreased translation of G6PD and leads to ∼20–40% residual G6PD activity G6PD mutant mice compared with the WT littermate control mice (26, 48).
Tissue preparations.
Bovine lungs were obtained from the local slaughterhouse in ice-cold physiological-buffered saline (PBS). The third order branches of pulmonary artery were used for experiments as published previously (25). Pulmonary arteries were cleaned of their connective tissue and cut into rings of 2- to 3-mm diameter and width. Thoracic aorta was isolated from male Sprague-Dawley rats (340 g) and 2- to 3-mm length rings were used as described in previous studies (23). Endothelium was removed by rubbing the lumen with wooden stick. Freshly prepared blood vessel rings were used in studies for vascular reactivity and Western blot protein analysis.
Measurement of vascular reactivity in bovine pulmonary arteries.
Endothelium denuded artery rings were mounted on Radnoti Instruments' force displacement transducers for recording isometric force development through Powerlab data acquisition system from AD Instruments, as previously described (20). Arterial rings were incubated in Krebs-bicarbonate buffer containing 118 mM NaCl, 4.7 mM KCl, 1.5 mM CaCl2, 25 mM NaHCO3, 1.1 mM MgSO4, 1.2 mM KH2PO4, and 5.6 mM glucose for 1 h under resting tension of 5 g. In all studies, arterial rings were depolarized with KCl (120 mM) containing Krebs bicarbonate buffer and then the rings were equilibrated with Krebs-bicarbonate buffer for 30 min. As mentioned in results, interventions were conducted under prolonged hypoxia (40 Torr) or under aerobic conditions. For acute experiments the blood vessels were exposed to hypoxia or aerobic conditions for 1 h while for prolonged experiments they were exposed for 12 h.
Western blot analysis.
Freshly prepared arterial rings were flash frozen by using liquid nitrogen. These frozen arterial rings were pulverized and homogenized in NP-40 lysis buffer (50 mmol/l Tris·HCl pH 7.4, 150 mmol/l NaCl, and 0.5% NP-40) containing protease and phosphatase inhibitors. The expression level of all the proteins except protien kinase G1α (PKG1α) was obtained under reducing condition as described previously (36). Thiol reducing conditions were avoided in the samples analyzed for PKG1α dimer (by not using β-mercaptoethanol). Maleimide (100 mM) was added in the lysis buffer in samples where PKG was measured to alkylate the thiols to avoid any artifactual disulfide bond formation during homogenization as published previously (42). Protein quantification analysis was conducted by Bradford assay and samples were prepared for gel electrophoresis. Proteins were separated using 10% SDS-polyacrylamide gel (SDS-PAGE). Gels were transferred to PVDF membranes and the membranes were blocked with 5% milk in Tris-buffered saline with Tween-20 for 1 h. Subsequently, membranes were incubated in primary and secondary antibodies as per manufacturer's protocol. Protein bands were visualized with an enhanced chemiluminescence kit (Pierce 32106) on X-OMAT autoradiography film (Kodak). Protein levels were measured using densitometry analysis by the ImageJ software. The following antibodies were used for immunoblotting: total Elk (A-303-530 A; Bethyl Laboratories), PKG1α (SC10338; Santa Cruz Biotechnology), p-ELK-1 (SC8406; Santa Cruz Biotechnology), TNF-α (SC-292640; Santa Cruz Biotechnology), pNFATc3 (SC-8405; Santa Cruz Biotechnology), tNFATc3 (SC-365786; Santa Cruz Biotechnology), β-actin (A5441; Sigma), pErk1/2 (p42/44; 9101; Cell Signaling), tErk1/2 MAP kinase (p42/44; 4695; Cell Signaling), total VASP (3132; Cell Signaling), and phospho VASP (3114; Cell Signaling).
cGMP assay.
Rings made from third order bovine pulmonary arteries were incubated in tissue baths in Krebs solution under aerobic conditions for 12 h and were treated with either 6-AN (1 μM) or DHEA (100 μM) or were left untreated and then frozen in liquid nitrogen after 12 h. Rings were then weighed and crushed in liquid nitrogen and cGMP levels were measured using the cGMP EIA Kit (item no. 581021) purchased from Cayman Chemical Laboratory.
Superoxide measurement using chemiluminescence.
As published previously by our laboratory (24), changes in superoxide were measured from quantifying the chemiluminescence of 5 μM lucigenin in a liquid scintillation counter (LS6000IC; Beckman Instruments, San Diego, CA) with a single active photomultiplier tube in a dark room. Initial background chemiluminescence (blank readings) was measured in plastic scintillation minivials containing only 5 μM lucigenin in 1 ml of Krebs solution buffered with 10 mM HEPES-NaOH (pH 7.4) in the absence of tissue. Right after obtaining the blank readings, arterial rings were added into each vial to measure the chemiluminescence in presence of the tissue (tissue readings). Blank measurement was subtracted from subsequent measurements made in the presence of arterial rings. The rings were weighed at the end of the experiment. The counts (tissue-blank) were divided by weight to gain the final data in counts per minute per gram of tissue.
Superoxide measurement using HPLC.
Measurement of the superoxide-specific hydroxylated products of MitoSox and dihydroethidium were employed for quantifying changes in mitochondrial and extra-mitochondrial superoxide, using previously described methods (71). Before the start of the experiment increasing concentrations of Mito-2-hydroxyethidium or 2-hydroxyethidium were loaded into the column to generate a standard curve. Third order bovine pulmonary arteries were incubated with 20-HETE (1 μM) in the presence and absence of G6PD inhibitors, DHEA (300 μM), and 6-AN (3 mM) for 12 h and were then treated with either 5 μM MitoSox or dihydroethidium (DHE) for 1 h in the dark to measure mitochondrial and extra-mitochondrial superoxide, respectively. They were washed several times with Krebs solution buffered with 10 mM HEPES-NaOH (pH 7.4) and then flash frozen with liquid nitrogen. Tissues were first weighed and then pulverized in the presence of liquid nitrogen, dissolved in a solution of 100% acetonitrile (HPLC grade). These samples were incubated at −20°C for 1 h. After 1 h, samples were centrifuged and the supernatant was used for HPLC analysis of the superoxide-specific hydroxylated product of MitoSox (Mito-2-hydroxyethidium) or of dihydroethidine (2-hydroxyethidium) using an HPLC system with a Jasco FP-1520 fluorescence detector and a Beckman ultrasphere reverse column (C18; 5 μ, 250 × 4.6 mm).
Liquid chromatography-tandem mass spectrometry: analysis of 20-HETE and PGE2.
For the mass spectrometry analysis we used endothelium denuded third order bovine pulmonary arteries and lungs from G6PD mutant and WT mice. The arterial rings were incubated in tissue baths for 12 h under hypoxia or aerobic conditions after giving them passive tension of 5 g and were either treated with DHEA (300 μM) or 6-AN (3 mM) or were left untreated. After 12 h the arterial rings were flash frozen in liquid nitrogen. The arterial rings and lungs were then weighed and pulverized in liquid nitrogen and the lipids were extracted as previously mentioned (15). To test if microsomal G6PD contributed to production of 20-HETE, we treated SD rat liver microsomes with either DHEA (300 μM) or 6-AN (3 mM) and 1 μg AA in the presence and absence of 1 mM NADPH (61).
A liquid chromatography tandem mass spectrometry (LC-MS/MS) method for identification and quantification of eicosanoids was developed on a Shimadzu Triple Quadrupole Mass Spectrometer LCMS-8050 equipped with a Nexera UHPLC using multiple reaction monitoring mode. MS conditions were as follows: ionization mode: negative heated electrospray (HESI); applied voltage: −4.5 to approximately −3 kV; nebulizer gas: 3.0 l/min N2; drying gas: 5.0 l/min N2; heating gas: 12.0 l/min air; interface temperature: 400°C; DL temperature: 100°C; heat block temperature: 500°C; and internal standards: d6 20-HETE, d8 5-HETE, and d4 PGE2. UHPLC conditions were as follows: analytical column: Zorbax Eclipse Plus C18 RRHD (50-mm long × 2.1-mm inner diameter, 1.8 μm); mobile phase A: 95% water 5% acetonitrile 0.05% acetic acid; mobile phase B: acetonitrile 0.05%; time program: 40% B (0 min)→75% B (3 min)→85% B (7.5 min); flow rate: 0.4 ml/min; injection volume: 5 μl; column oven temperature: 40°C. MRM transitions were as follows: 20-HETE, CE 19.5 m/z: 319.2→289.2; 20-HETE, CE 19.5 m/z: 319.2→289.2; 20-HETE, CE 19.5 m/z: 319.2→289.2; d6 20-HETE, CE 19.0 m/z: 325.2→295.2; 15-HETE, CE 13.0 m/z: 319.2→219.2; 12-HETE, CE 13.5 m/z: 319.2→179.1; 5-HETE, CE 15.0 m/z: 319.2→115.0; d8 5-HETE, CE 14.5 m/z: 327.5→116.0; PGE2, CE 16.0 m/z: 351.1→271.2; and d4 PGE2, CE 18.0 m/z: 355.3→275.2.
MicroRNA expression by quantitative PCR.
MicroRNA (miR) analysis was performed by quantitative (Q)PCR as previously described (8). Briefly, total RNA was extracted from aorta using Qiagen miRNEasy kit (No. 217004). Input RNA quality and concentration were measured on NanoDrop and cDNA was prepared using miR-specific TaqMan miR assays (Applied Biosystems, Foster City, CA). Quantitative PCR was performed in triplicates using TaqMan Universal PCR Master mix. Ct values from standard curves were then used to quantify relative expression of specific miR. Standard curves were made for each miR using synthetic miR oligonucleotides (IDT, Coralville, IA) with the following sequence: rno-miR-145: GUCCAGUUUUCCCAGGAAUCCCU; rno-miR-1: UGGAAUGUAAAGAAGUGUGUAU; rno-miR-143: UGAGAUGAAGCACUGUAGCUC; and rno-miR-133a: UUUGGUCCCCUUCAACCAGCUG.
Cell culture.
A7r5 smooth muscle cells were purchased from the American Type Culture Collection (Manassas, VA) and maintained under 5% CO2 at 37°C in Dulbecco's modified Eagle's medium supplemented with l-glutamine, 4.5 g/l glucose, and 10% fetal bovine serum (Invitrogen, Carlsbad, CA). Cells at ∼70% confluence were subcultured weekly using 0.05% trypsin-EDTA (GIBCO cat no. 25300-054, Thermo Fischer Scientific, Grand Island, NY) for up to seven passages.
Statistical analysis.
Values are means ± SE of the number of samples (n) from different animals. Statistical analyses were performed and unpaired Student's t-test was used to compare two groups and one-way ANOVA with Bonferroni correction was used for comparing multiple groups. P < 0.05 was used to establish statistical significance.
RESULTS
G6PD inhibition or knockdown decreased endogenous production of 20-HETE.
Synthesis of 20-HETE by CYP4A and 4F is dependent on NADPH (5). In the vascular smooth muscle cells a majority of NADPH is produced by G6PD and 6PGD. Therefore, we measured 20-HETE levels in the aerobic and prolonged hypoxic pulmonary arteries treated without and with G6PD inhibitors. 20-HETE was undetectable in pulmonary arteries under aerobic conditions, and its levels increased (3.1 ± 0.6 pg/mg tissue) within 30 min after the vessels were exposed to hypoxia, and it remained elevated for 12 h (Fig. 1A). G6PD inhibition reduced (P < 0.05; Fig. 1A) 20-HETE and PGE2 levels were not affected by G6PD inhibition (control: 16.75, DHEA: 17.88, and 6-AN: 17.89 pg/mg). We also found that 20-HETE levels were significantly less in the lungs of G6PD mutant compared with WT mice (Fig. 2B). CYP activity was not modified by 6-AN (3 mM; 61 ± 3 pg 20-HETE/h/25 μg microsomes; n = 5) vs. control (76 ± 3 pg 20-HETE/h/25 μg microsomes; n = 5).
Fig. 1.
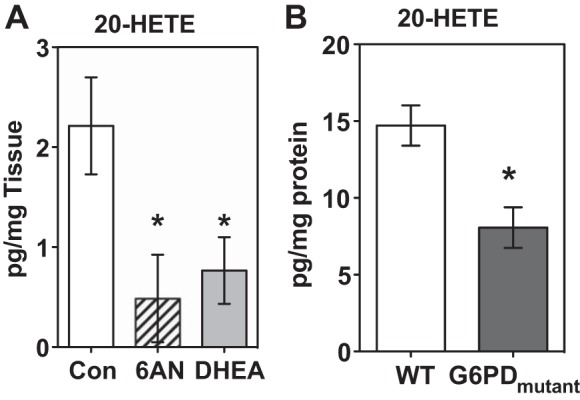
Glucose-6-phosphate dehydrogenase (G6PD) inhibition or knockdown decreased endogenous levels of 20-HETE. A: inhibition of G6PD by dehydroepiandosterone (DHEA) and 6-aminonicotinamide (6-AN) significantly decreased endogenous levels of 20-HETE in the bovine pulmonary arteries after they were exposed to 12-h hypoxia. B: 20-HETE levels were also reduced in the lungs of G6PD-deficient (G6PDmutant) mice. *P < 0.05 vs. wild type (WT); n = 5 in each group.
Fig. 2.

Protein kinase G1α (PKG1α), phosphorylated-VASP, and cGMP levels in bovine pulmonary arteries untreated and treated with G6PD inhibitors. A and B: expression of PKG1α measured as dimer-to-monomer ratios and phosphorylated-VASP increased by inhibiting G6PD with DHEA (100 μM) or 6-AN (1 mM). C: cGMP levels in the samples are not changed. n = 5 in each group. *P < 0.05 vs. control; #P < 0.05 vs. treatments.
G6PD inhibition increased PKG activity without increasing intracellular cGMP and decreased 20-HETE production in a PKG-dependent manner.
Previous work done in our laboratory has demonstrated that G6PD activation contributes to the development of hypoxic pulmonary vasoconstriction (20) and inhibition of G6PD with 6-AN (1 mM) blocked it in a PKG-independent and -dependent manner (10, 21). To determine whether G6PD inhibition activated PKG, we treated the arterial rings with DHEA and 6-AN for 12 h. G6PD inhibition by DHEA (100 μM) and 6-AN (1 mM) did not increase cGMP (Fig. 2A) but concurrently increased PKG1α dimer (Fig. 2C), the physiologically active form of the kinase (17, 34, 35), and augmented phosphorylation of VASP at Ser239 (Fig. 2B), which is an indicator of PKG activity, in the same samples. It is noteworthy that a competitive and irreversible inhibitor of PKG, 2-bromo-3,4-dihydro-3-[3,5-O-[(R)-mercaptophosphinylidene]-β-d-ribofuranosyl]-6-phenyl-9H-imidazo[1,2-a]purin-9-one sodium salt (Rp-cGMPs; 100 nM), decreased DHEA-mediated PKG1α dimer expression (Fig. 2C).
Next, we determined whether PKG signaling-mediated G6PD inhibition induces reduction of 20-HETE levels in pulmonary arteries. We incubated the arteries in the absence or presence of the PKG inhibitor Rp-cGMPs (100 nM) and the G6PD inhibitor DHEA. Interestingly, inhibition of PKG with Rp-cGMPs partially reversed the suppression of 20-HETE by G6PD inhibitors (DHEA + Rp-cGMPs: 1.3 ± 0.3 pg/mg tissue).
G6PD inhibition prevented 20-HETE-mediated downregulation of PKG1α dimer expression and upregulation of pErk1/2.
To determine the signaling pathways through which 20-HETE evokes inflammation, we measured the PKG1α dimer-to-monomer ratio and pErk1/2. Application of 20-HETE (1 μM) to pulmonary arteries decreased PKG1α dimer-to-monomer ratio and increased pErk1/2 (Fig. 3, A and B). G6PD inhibition blocked the 20-HETE-induced decrease in the PKG1α dimer-to-monomer ratio and the increase in pErk1/2.
Fig. 3.

The 20-HETE induced decrease of PKG1α dimer-to-monomer ratios and increased ERK phosphorylation were prevented by G6PD inhibition in bovine pulmonary arteries. A: the PKG1α dimer-to-monomer ratio was decreased by application of 20-HETE (1 μM) to bovine pulmonary arteries for 12 h and this decrease was prevented by G6PD inhibition with DHEA and 6-AN. B: there was increased ERK 1/2 phosphorylation by application of 20-HETE (1 μM) to the bovine pulmonary arteries. This was prevented when the vessels were pretreated with DHEA; n = 5 in each group. *P < 0.05 vs. control; #P < 0.05 vs. 20-HETE.
G6PD inhibitors via activation of PKG blocked 20-HETE-induced production of mitochondrial superoxide in the pulmonary arteries.
20-HETE stimulates reactive oxygen species that are proinflammatory, -migratory, and -proliferative (49, 64, 66). Previous studies reported that 20-HETE increases superoxide production (detected by DHE fluorescence by microscopy) in aortic and pulmonary artery endothelial cells (7, 37). Since this approach to detect superoxide is semiquantitative and is somewhat nonspecific, we employed HPLC methods to determine extra-mitochondrial and mitochondrial superoxide levels in pulmonary arteries in response to 20-HETE. Mitochondrial but not extra-mitochondrial derived superoxide production was stimulated by 20-HETE (1 μM) under aerobic conditions (Fig. 4, A and B) and hypoxia (6585.385 nmol/g of tissue). In contrast, dibromo-dodecenyl-methylsulfimide (DDMS), a potent inhibitor of CYP4A (1 μM), blocked mitochondrial superoxide production in the pulmonary arteries under chronic hypoxia (Fig. 4C).
Fig. 4.

20-HETE increased mitochondrial superoxide in the bovine pulmonary arteries, which was blocked by G6PD inhibitors by activation of PKG. A: incubation of bovine pulmonary arteries with 20-HETE for 12 h did not increase extra-mitochondrial superoxide. B: it significantly increased mitochondrial superoxide. *P < 0.05 vs. control. C: inhibition of endogenous 20-HETE levels by dibromo-dodecenyl-methylsulfimide (DDMS; 1 μM) significantly decreased mitochondrial superoxide. *P < 0.05 vs. control. D: increase in superoxide by 20-HETE was blocked by DHEA and 6-AN. The actions of DHEA and 6-AN were reversed when they were given with Rp-cGMPs (100 nM), a PKG blocker. *P < 0.05 vs. control; $P < 0.05 vs. 20-HETE; @P < 0.05 vs. 20-HETE + DHEA; n = 5 in each group.
Since G6PD-derived NADPH regulates superoxide production from NADPH oxidases (22), we examined whether 20-HETE-elicites generation of superoxide in a G6PD-dependent manner. DHEA and 6-AN blocked the increase in 20-HETE-elicited superoxide production. Next, we investigated whether 20-HETE-induced superoxide production was reduced by G6PD inhibitor(s) in a PKG-dependent manner. Therefore, we treated pulmonary arteries with 20-HETE for 12 h in tissue baths after pretreating them with either DHEA or 6-AN alone or in the presence of Rp-cGMPs and then measured superoxide production by lucigenin chemiluminesence method. Inhibition of 20-HETE-induced superoxide productions by DHEA and 6-AN was partly reversed by Rp-cGMPs treatment (Fig. 4D). Additionally, we found that 2-aminoethoxydiphenyl borate (2APB; 60 μM), an antagonist of the inositol 1,4,5-triphosphate (IP3) receptor, blocked the increase in mitochondrial superoxide elicited by 20-HETE [in arbitrary units/g tissue: control (221,998 ± 32,463); 20-HETE (565,749 ± 123,822); and 2APB + 20-HETE (114,705 ± 42,092)].
20-HETE-induced expression of TNF-α by pulmonary arteries is blocked by G6PD inhibitor(s).
Inflammation is an important contributor to the pathogenesis of systemic and pulmonary hypertension (39). Since proinflammatory cytokine TNF-α is involved in the acute phase reaction and IL-6 evokes late phase reaction, we determined TNF-α and IL-6 expression in the arteries/pulmonary artery smooth muscle cells exposed to 20-HETE. TNF-α has a soluble form and a transmembrane form (28), both of which are involved in the inflammatory response. We were able to detect the transmembrane form in the pulmonary artery smooth muscle. TNF-α expression increased (P < 0.05) in arteries (Fig. 5A) and smooth muscle cells (data not shown) exposed to 20-HETE. 20-HETE increased IL-6 level (in pg/ml; control: 266 ± 44 and 20-HETE: 443 ± 97; P < 0.05).
Fig. 5.

20-HETE increased TNF-α and Elk-1 expression and this was blocked when G6PD was inhibited. A: incubation of bovine pulmonary arteries with 20-HETE (1 μM) significantly increased the expression of TNF-α. The increase in expression of TNF-α was blocked when the vessels were pretreated with the G6PD inhibitors DHEA and 6-AN. *P < 0.05 vs. control; $P < 0.05 vs. 20-HETE. B: inhibition of endogenous 20-HETE levels by DDMS reduced the expression of ELK-1, which is a transcription factor for TNF-α. *P < 0.05 vs. control. C: 20-HETE increased Elk-1 expression in the endothelium denuded bovine pulmonary arteries. Increase in Elk-1 expression by 20-HETE was inhibited by DHEA and MitoTempol (1 μM). *P < 0.05 vs. control; n = 5 in each group.
Next, we investigated if inhibition of G6PD decreased 20-HETE-induced expression of TNF-α and if this is mediated via PKG. We treated pulmonary arteries with 20-HETE for 12 h after pretreating them with either DHEA or 6-AN alone or in the presence of Rp-cGMPs. DHEA and 6-AN decreased 20-HETE-induced expression of TNF-α in a PKG-dependent manner (Fig. 5A).
20-HETE increased Elk-1 expression and pretreatment of arteries with G6PD inhibitor(s) decreased Elk-1 expression.
Elk-1 drives transcription of Tnfa gene (19). Transcriptional activation activity of Elk-1 is increased by Erk1/2 (MAPK)-dependent phosphorylation at Ser383 and conversely is decreased by PKG1α-dependent sumoylation (11, 31). Since 20-HETE decreased PKG1α and increased pErk1/2, we estimated Elk-1 expression status in arteries treated with the CYP4A inhibitor DDMS and in arteries treated with 20-HETE in absence and presence of the G6PD inhibitors DHEA or MitoTempol. DDMS decreased Elk-1 expression in pulmonary arteries under prolonged hypoxia (Fig. 5B). 20-HETE upregulated Elk-1 expression (Fig. 5C), which was decreased by the pretreatment with DHEA and by MitoTempol (1 μM).
20-HETE decreased miR-143 and miR-133a in rat aorta and increased Elk-1 expression in aortic smooth muscle cells. It is established that miR-145/143 and miR-1/133a are involved in the regulation of smooth muscle cell phenotype (32). miR-143 suppresses the expression of Elk-1 (12, 46). Activation of Erk1/2 (MAPK1/3) downregulates miR-133a expression, and this promotes a synthetic phenotype in vascular smooth muscle cells in vitro and in vivo leading to remodeling of blood vessels (57). Therefore, we asked the question whether 20-HETE affects the expression of miR-133a and miR-143? We used rat aorta and aortic smooth muscle cells to study the effect of 20-HETE on miR expression and concomitantly on the miR-143 target, Elk-1. We applied 20-HETE (0.1 and 1 μM) to isolated rat aorta for 12 h and then measured miR-1/133a and miR-145/143 by QPCR. 20-HETE did not significantly affect the expression of miR-1 or miR-145 (Fig. 6, A and C). Expression of miR-133a (Fig. 6B) and miR-143 (Fig. 6D) in aorta was decreased by application of 20-HETE in a dose-dependent manner. Since 20-HETE elevated reactive oxygen species and G6PD inhibitors decreased it, we also examined if H2O2 and G6PD inhibitors regulated miR-143 levels. We found that H2O2 decreased and G6PD inhibitors increased miR-143 levels in aortic smooth muscle cell line (A7r5; Fig. 6E). Furthermore, we observed that G6PD inhibition by DHEA and mitochondrial ROS by MitoTempol decreased the 20-HETE-induced increase in Elk-1 in A7r5 cells (Fig. 6, F and G).
Fig. 6.

20-HETE decreased miR-143 and miR-133a in rat aorta and increased Elk-1 expression in aortic smooth muscle cells. A–C: miR-1 and miR-145 expression was not affected by 20-HETE B–D: miR-133a and miR-143 expression was significantly decreased by 20-HETE (0.1 and 1 μM). E: miR-143 expression levels were significantly decreased by H2O2 (100 μM; n = 5) and increased significantly when G6PD was inhibited by DHEA (100 μM; n = 6–8) or 6-AN (1 mM; n = 5). F: 20-HETE-induced increase of Elk-1 was decreased by G6PD inhibition. G: MitoTempol (1 μM) also decreased 20-HETE induced ELK-1 expression.
DISCUSSION
The detection of G6PD and CYPs complex in the vascular smooth muscle identified by proteomic analysis (Table 1), along with the observation that G6PD inhibition or knockdown decreased 20-HETE levels, suggests G6PD and CYP4-ω-hydroxylases are functionally coupled in vivo These findings complemented earlier studies, which showed that CYP4A and 4F family monooxygenases generated 20-HETE in a NADPH-dependent manner (4, 44). Furthermore, 20-HETE is increased by hypoxia in the pulmonary artery and G6PD inhibition blocked the synthesis of 20-HETE. Since G6PD inhibition blocked 20-HETE signaling, it is reasonable to propose that inhibition of 20-HETE-dependent signaling pathways that elicit constriction and inflammation of the arteries are potential mechanisms through which G6PD inhibition decreases hypoxia-induced pulmonary vasoconstriction and pulmonary hypertension (9). Therefore, our findings indicated that G6PD-derived NADPH is a driver for 20-HETE-synthesis and that upregulation of G6PD expression or activity not only increases endogenous levels of 20-HETE but also modulates 20-HETE signaling in vascular diseases such as pulmonary hypertension.
Table 1.
Proteomic analysis by LC/MS/MS showing a complex of G6PD with various CYP450 enzymes in G6PD pull down from bovine arteries
| Top Ranked Protein Name | Accession No. | Protein MW | Protein PI | Peptide Count | Total Ion Score | Total Ion CI, % |
|---|---|---|---|---|---|---|
| CYP450, family 2, subfamily C, Polypeptide 87 precursor | gi 115497566 | 55.895 | 7.2 | 2 | 54 | 100 |
| (Bos taurus) | ||||||
| CYP450 2C21 | gi 297464426 | 48.686 | 7.6 | 5 | 84 | 100 |
| (Bos taurus) | ||||||
| CYP450 2C19 | gi 297464424 | 48.475 | 6.3 | 2 | 45 | 99 |
| (Bos taurus) | ||||||
| CYPP450 subfamily 2B | gi 296477790 | 50.607 | 8.2 | 2 | 38 | 95 |
| (Bos taurus) |
LC/MS/MS, liquid chromatography tandem mass spectrometry; G6PD, glucose-6-phosphate dehydrogenase; MW molecular weight; CI, confidence interval; PI, isoelectric point.
20-HETE is pro-proliferative, -inflammatory, and -migratory (49, 66), all of which contribute to the pathogenesis of systemic and pulmonary hypertension. Although the increase in 20-HETE by hypoxia inhibited acute hypoxia-induced pulmonary artery constriction (2, 69), it time and dose dependently increased superoxide production from NADPH oxidases in the cultured pulmonary artery endothelial cells (37). Also, studies have reported that 20-HETE-induced superoxide mediated flow-induced constriction of cerebral arteries (58). Our current results further demonstrated that inhibition of 20-HETE biosynthesis by DDMS decreased mitochondrial superoxide generation and conversely application of 20-HETE to endothelium denuded pulmonary arteries for 12 h elicited superoxide generation from mitochondria but not from extra-mitochondrial sources. This led us to the question of how does 20-HETE increase mitochondrial superoxide generation? One potential explanation was 20-HETE passed through the gap junctions (unpublished data) and stimulated mitochondrial superoxide. Alternatively, since 2APB blocked 20-HETE-induced superoxide (see results) and 20-HETE stimulates Ca2+ release from IP3 receptors potentially through a PLA2 or G protein-coupled receptor-dependent mechanism in airway smooth muscle cells (50), the data suggest that 20-HETE potentially triggered mitochondrial Ca2+ overload through SR-mitochondrial coupling, which is well known to increase mitochondrial respiration/metabolic rate and superoxide levels (13, 33). Although the functional role of mitochondrial vs. extra-mitochondrial superoxide in the development of vascular diseases is not yet well characterized, elevated superoxide in the cell has been shown to inactivate nitric oxide (NO) and soluble guanylate cyclase-PKG or activate Erk1/2 signaling pathways (24, 38) and contribute to the pathogenesis of vascular diseases including pulmonary hypertension (18, 36). Therefore, from our current findings it is reasonable to suggest that elevated mitochondrial superoxide mediated, at least partly, 20-HETE-induced pulmonary artery dysfunction.
Superoxide is mitogenic and proinflammatory in vascular smooth muscle cell (56). Along these lines we found that 20-HETE, which increased mitochondrial superoxide, robustly increased expression of proinflammatory cytokine TNF-α and IL-6 in pulmonary artery and pulmonary artery smooth muscle cell, respectively. Interestingly, G6PD inhibition decreased mitochondrial superoxide production and TNF-α expression elicited by 20-HETE. Transcription of the Tnfa gene is elevated by reactive oxygen species-induced NF-κB activation (51) and is also increased by Elk-1 (19). 20-HETE is a known activator of NF-κB (30). Here, we also found that 20-HETE upregulated and DDMS downregulated Elk-1 expression, respectively, in pulmonary artery. Although we did not investigate whether the smooth muscle cells or other types of cells in the arterial wall produced 20-HETE, our findings suggested that signaling pathways stimulated by both endogenous and exogenous 20-HETE regulated Elk-1 expression. Furthermore, 20-HETE applications to the rat aorta decreased miR143, which is known to inhibit Elk-1 expression (12), and suppressed miR-133a, which prevented expression of synthetic (secretory/proinflammatory) phenotype in vascular smooth muscle cells (57). Therefore, these findings suggested that 20-HETE-induced transformation of the vascular smooth muscle cells from contractile to secretory/proinflammatory phenotype that increased Tnfa is mediated through the miR143-Elk-1 pathway and inhibition of G6PD or mitochondrial superoxide generation by MitoTempol prevented this.
The TNF-α-induced increase in cGMP paradoxically downregulates PKG1α expression in the bovine aortic smooth muscle cells (3). Therefore, it is reasonable to speculate that 20-HETE-induced overexpression of TNF-α downregulated PKG1α. Interestingly, G6PD inhibition decreased 20-HETE and rescued 20-HETE-induced downregulation of PKG1α and increase of pErk1/2. Since inhibition of G6PD-derived NADPH redox oxidized Cys42 residues on PKG and activated it (10), we proposed that application of G6PD inhibitors to pulmonary arteries for 12 h oxidized Cys42 residues that either prevented or overruled the 20-HETE-induced decrease of PKG1α leading to an increase in the PKG1α dimer and VASP phosphorylation without increasing intracellular cGMP levels. Previous studies showed that PKG1α dimer is elevated in vascular tissue by DHEA (43, 45). Furthermore, the increase of PKG activity by G6PD inhibition decreased 20-HETE levels as well as 20-HETE-induced mitochondrial superoxide generation. This suggests that either PKG inhibited CYP4 activity or downregulated hypoxia-induced CYP4 expression. In this regards, NO is known to block CYP4-derived 20-HETE (1, 62) and increased NO-mediated activity of cGMP/PKG has been associated with downregulation of CYP4A enzymes in various organs and renal arteries of a LPS-treated rat model of septic shock (60). The increase of PKG activity sumoylates Elk-1 and inactivates it (11). In contrast, the increase of MAPK1/Erk2 activity phosphorylates Elk-1 at Ser383 and stimulates its transcriptional activity (68). Accordingly, since G6PD inhibition upregulated miR-143 expression and decreased 20-HETE-induced Elk-1 expression, we proposed that increased PKG and decreased Erk1/2 activity by G6PD inhibition antagonized the 20-HETE-induced increase of Elk-1 leading to reduction of TNF-α expression. Moreover, DHEA and 6-AN decreased mitochondrial superoxide and increased miR-143, H2O2 downregulated miR-143, and MitoTempol blocked 20-HETE-induced Elk-1 expression; all of this suggested that H2O2 derived from mitochondrial superoxide mediated the action of 20-HETE and this was antagonized by G6PD inhibition. Therefore, our current findings suggested that 20-HETE-induced mitochondrial superoxide production and synthetic (secretory/proinflammatory) phenotype in vascular smooth muscle cells are decreased by G6PD inhibition via activation of PKG-dependent signaling and a concurrent decrease in Erk2/MAPK1 activity.
In vascular smooth muscle cells, 20-HETE has been shown to stimulate PI3K-MAPK (55) and RAS-MAPK (41) pathways that are activated by reactive oxygen species and that contribute to development of hypertension (67). 20-HETE-dependent hypertension is associated with microvascular remodeling (14, 16), a contributor to the development and maintenance of hypertension. Microvascular remodeling is promoted by a variety of stimuli resulting in structural changes including collagen synthesis and deposition, reorganization of the extracellular matrix, increased proinflammatory signaling, and altered matrix metalloproteinase activity rendering vessels stiffer and thicker and noncompliant, thus further exacerbating hypertension (29). Therefore, it is reasonable to suggest that increased 20-HETE-mediated mitochondrial superoxide generation potentially contributes to the pathogenesis of pulmonary hypertension.
In summary, the salient findings of this study are summarized in Fig. 7 and they are as follows: 1) CYP4-ω-hydroxylases (or 20-HETE biosynthesis is) are redox sensitive and are regulated by G6PD-derived NADPH; 2) inhibition of G6PD decreased endogenous levels of 20-HETE levels; 3) 20-HETE increased mitochondrial superoxide, which was blocked when G6PD was inhibited; 4) G6PD inhibition blocked 20-HETE-induced expression of Elk-1, which promotes the secretory phenotype of vascular smooth muscle cells; and 5) 20-HETE application to rat aorta decreased miR-143, which suppressed Elk-1 expression, and potentially promoted TNF-α secretion from vascular smooth muscle cells.
Fig. 7.

Summary model. 20-HETE is synthesized from arachidonic acid (AA) by CYP4 group of enzymes, which utilize NADPH generated by G6PD. 20-HETE causes an increase in mitochondrial reactive oxygen species [ROS; superoxide (O2·−) and hydrogen peroxide (H2O2)] and a decrease in miR-143, p42/44 MAP kinase phosphorylation, which then leads to increased expression of ELK-1. Increased expression of ELK-1 then leads to an increase in expression of TNF-α, which can then block PKG signaling in the vascular smooth muscle. Inhibition of G6PD depletes the NADPH available to the CYP4 group of enzymes thereby decreasing synthesis of 20-HETE. Inhibition of G6PD also activates PKG without increasing intracellular cGMP, which then inhibits 20-HETE mediated increase in mitochondrial ROS. Decrease in mitochondrial ROS leads to an increased expression of miR-143, which then inhibits ELK-1 expression. Activation of PKG by G6PD inhibition also leads to decreased ELK-1 expression, which in turn leads to a decreased expression of TNF-α. Solid lines indicate known and broken lines indicate unknown pathways.
GRANTS
The study was supported by intramural funds (to S. A. Gupte) and National Heart, Lung, and Blood Institute Grants R01-HL-115124 (to M. S. Wolin) P01-HL-34300 (to M. L. Schwartzman).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.L., V.D., S.R.J., K.H.G., and D.P. performed experiments; A.L., V.D., S.R.J., K.H.G., D.P., and D.S. analyzed data; A.L., S.R.J., D.S., M.S.W., M.L.S., and S.A.G. interpreted results of experiments; A.L. and V.D. prepared figures; A.L., S.R.J., and S.A.G. drafted manuscript; A.L., V.D., S.R.J., K.H.G., D.P., D.S., M.S.W., M.L.S., and S.A.G. edited and revised manuscript; A.L., V.D., S.R.J., K.H.G., D.P., D.S., M.S.W., M.L.S., and S.A.G. approved final version of manuscript; S.A.G. conception and design of research.
ACKNOWLEDGMENTS
A part of these results were presented at the Federation of American Societies for Experimental Biology (FASEB) meeting (Boston, MA, 2012).
REFERENCES
- 1.Alonso-Galicia M, Hudetz AG, Shen H, Harder DR, Roman RJ. Contribution of 20-HETE to vasodilator actions of nitric oxide in the cerebral microcirculation. Stroke 30: 2727–2734; discussion 2734, 1999. [DOI] [PubMed] [Google Scholar]
- 2.Birks EK, Bousamra M, Presberg K, Marsh JA, Effros RM, Jacobs ER. Human pulmonary arteries dilate to 20-HETE, an endogenous eicosanoid of lung tissue. Am J Physiol Lung Cell Mol Physiol 272: L823–L829, 1997. [DOI] [PubMed] [Google Scholar]
- 3.Browner NC, Sellak H, Lincoln TM. Downregulation of cGMP-dependent protein kinase expression by inflammatory cytokines in vascular smooth muscle cells. Am J Physiol Cell Physiol 287: C88–C96, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Bylund J, Finnstrom N, Oliw EH. Gene expression of a novel cytochrome P450 of the CYP4F subfamily in human seminal vesicles. Biochem Biophys Res Commun 261: 169–174, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Capdevila JH, Falck JR, Estabrook RW. Cytochrome P450 and the arachidonate cascade. FASEB J 6: 731–736, 1992. [DOI] [PubMed] [Google Scholar]
- 6.Cheng J, Edin ML, Hoopes SL, Li H, Bradbury JA, Graves JP, DeGraff LM, Lih FB, Garcia V, Shaik JS, Tomer KB, Flake GP, Falck JR, Lee CR, Poloyac SM, Schwartzman ML, Zeldin DC. Vascular characterization of mice with endothelial expression of cytochrome P450 4F2. FASEB J 28: 2915–2931, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng J, Ou JS, Singh H, Falck JR, Narsimhaswamy D, Pritchard KA Jr, Schwartzman ML. 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am J Physiol Heart Circ Physiol 294: H1018–H1026, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Chettimada S, Ata H, Rawat DK, Gulati S, Kahn AG, Edwards JG, Gupte SA. Contractile protein expression is upregulated by reactive oxygen species in aorta of Goto-Kakizaki rat. Am J Physiol Heart Circ Physiol 306: H214–H224, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L287–L300, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chettimada S, Rawat DK, Dey N, Kobelja R, Simms Z, Wolin MS, Lincoln TM, Gupte SA. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am J Physiol Lung Cell Mol Physiol 303: L64–L74, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi C, Sellak H, Brown FM, Lincoln TM. cGMP-dependent protein kinase and the regulation of vascular smooth muscle cell gene expression: possible involvement of Elk-1 sumoylation. Am J Physiol Heart Circ Physiol 299: H1660–H1670, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460: 705–710, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Decuypere JP, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB. The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta 1813: 1003–1013, 2011. [DOI] [PubMed] [Google Scholar]
- 14.Ding Y, Wu CC, Garcia V, Dimitrova I, Weidenhammer A, Joseph G, Zhang F, Manthati VL, Falck JR, Capdevila JH, Schwartzman ML. 20-HETE induces remodeling of renal resistance arteries independent of blood pressure elevation in hypertension. Am J Physiol Renal Physiol 305: F753–F763, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia V, Cheng J, Weidenhammer A, Ding Y, Wu CC, Zhang F, Gotlinger K, Falck JR, Schwartzman ML. Androgen-induced hypertension in angiotensinogen deficient mice: role of 20-HETE and EETS. Prostaglandins Other Lipid Mediat 116–117: 124–130, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia V, Joseph G, Shkolnik B, Ding Y, Zhang FF, Gotlinger K, Falck JR, Dakarapu R, Capdevila JH, Bernstein KE, Schwartzman ML. Angiotensin II receptor blockade or deletion of vascular endothelial ACE does not prevent vascular dysfunction and remodeling in 20-HETE-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 309: R71–R78, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gill GN, Holdy KE, Walton GM, Kanstein CB. Purification and characterization of 3′:5′-cyclic GMP-dependent protein kinase. Proc Natl Acad Sci USA 73: 3918–3922, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am J Respir Cell Mol Biol 47: 718–726, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guha M, O'Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, Stern D, Mackman N. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor alpha expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood 98: 1429–1439, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Gupte RS, Rawat DK, Chettimada S, Cioffi DL, Wolin MS, Gerthoffer WT, McMurtry IF, Gupte SA. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem 285: 19561–19571, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupte SA, Arshad M, Viola S, Kaminski PM, Ungvari Z, Rabbani G, Koller A, Wolin MS. Pentose phosphate pathway coordinates multiple redox-controlled relaxing mechanisms in bovine coronary arteries. Am J Physiol Heart Circ Physiol 285: H2316–H2326, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol 288: H13–H21, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Gupte SA, Li KX, Okada T, Sato K, Oka M. Inhibitors of pentose phosphate pathway cause vasodilation: involvement of voltage-gated potassium channels. J Pharmacol Exp Ther 301: 299–305, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Gupte SA, Rupawalla T, Mohazzab HK, Wolin MS. Regulation of NO-elicited pulmonary artery relaxation and guanylate cyclase activation by NADH oxidase and SOD. Am J Physiol Heart Circ Physiol 276: H1535–H1542, 1999. [DOI] [PubMed] [Google Scholar]
- 25.Gupte SA, Rupawalla T, Phillibert D Jr, Wolin MS. NADPH and heme redox modulate pulmonary artery relaxation and guanylate cyclase activation by NO. Am J Physiol Lung Cell Mol Physiol 277: L1124–L1132, 1999. [DOI] [PubMed] [Google Scholar]
- 26.Hecker PA, Lionetti V, Ribeiro RF Jr, Rastogi S, Brown BH, O'Connell KA, Cox JW, Shekar KC, Gamble DM, Sabbah HN, Leopold JA, Gupte SA, Recchia FA, Stanley WC. Glucose 6-phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circ Heart Fail 6: 118–126, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoopes SL, Garcia V, Edin ML, Schwartzman ML, Zeldin DC. Vascular actions of 20-HETE. Prostaglandins Other Lipid Mediat 120: 9–16, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology 49: 1215–1228, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension 38: 581–587, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Ishizuka T, Cheng J, Singh H, Vitto MD, Manthati VL, Falck JR, Laniado-Schwartzman M. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. J Pharmacol Exp Ther 324: 103–110, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Janknecht R, Ernst WH, Pingoud V, Nordheim A. Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J 12: 5097–5104, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joshi SR, Comer BS, McLendon JM, Gerthoffer WT. MicroRNA regulation of smooth muscle phenotype. Mol Cell Pharmacol 4: 1–16, 2012. [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z, Ji G, Neugebauer V. Mitochondrial reactive oxygen species are activated by mGluR5 through IP3 and activate ERK and PKA to increase excitability of amygdala neurons and pain behavior. J Neurosci 31: 1114–1127, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lincoln TM, Dills WL Jr, Corbin JD. Purification and subunit composition of guanosine 3':5'-monophosphate-dependent protein kinase from bovine lung. J Biol Chem 252: 4269–4275, 1977. [PubMed] [Google Scholar]
- 35.Lincoln TM, Hall CL, Park CR, Corbin JD. Guanosine 3′:5′-cyclic monophosphate binding proteins in rat tissues. Proc Natl Acad Sci USA 73: 2559–2563, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 290: L2–L10, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Medhora M, Chen Y, Gruenloh S, Harland D, Bodiga S, Zielonka J, Gebremedhin D, Gao Y, Falck JR, Anjaiah S, Jacobs ER. 20-HETE increases superoxide production and activates NAPDH oxidase in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 294: L902–L911, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montezano AC, Burger D, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, He Y, Callera GE, He G, Krause KH, Lambeth D, Quinn MT, Touyz RM. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res 106: 1363–1373, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morrell NW, Bloch DB, Ten Dijke P, Goumans MT, Hata A, Smith J, Yu PB, Bloch KD. Targeting BMP signalling in cardiovascular disease and anaemia. Nat Rev Cardiol 13: 106–120, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muthalif MM, Benter IF, Karzoun N, Fatima S, Harper J, Uddin MR, Malik KU. 20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin-dependent protein kinase II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Proc Natl Acad Sci USA 95: 12701–12706, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muthalif MM, Uddin MR, Fatima S, Parmentier JH, Khandekar Z, Malik KU. Small GTP binding protein Ras contributes to norepinephrine-induced mitogenesis of vascular smooth muscle cells. Prostaglandins Other Lipid Mediat 65: 33–43, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Neo BH, Kandhi S, Wolin MS. Roles for soluble guanylate cyclase and a thiol oxidation-elicited subunit dimerization of protein kinase G in pulmonary artery relaxation to hydrogen peroxide. Am J Physiol Heart Circ Physiol 299: H1235–H1241, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neo BH, Patel D, Kandhi S, Wolin MS. Roles for cytosolic NADPH redox in regulating pulmonary artery relaxation by thiol oxidation-elicited subunit dimerization of protein kinase G1α. Am J Physiol Heart Circ Physiol 305: H330–H343, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen X, Wang MH, Reddy KM, Falck JR, Schwartzman ML. Kinetic profile of the rat CYP4A isoforms: arachidonic acid metabolism and isoform-specific inhibitors. Am J Physiol Regul Integr Comp Physiol 276: R1691–R1700, 1999. [DOI] [PubMed] [Google Scholar]
- 45.Patel D, Kandhi S, Kelly M, Neo BH, Wolin MS. Dehydroepiandrosterone promotes pulmonary artery relaxation by NADPH oxidation-elicited subunit dimerization of protein kinase G1α. Am J Physiol Lung Cell Mol Physiol 306: L383–L391, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rangrez AY, Massy ZA, Metzinger-Le Meuth V, Metzinger L. miR-143 and miR-145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet 4: 197–205, 2011. [DOI] [PubMed] [Google Scholar]
- 47.Rawat DK, Alzoubi A, Gupte R, Chettimada S, Watanabe M, Kahn AG, Okada T, McMurtry IF, Gupte SA. Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension 64: 1266–1274, 2014. [DOI] [PubMed] [Google Scholar]
- 48.Rawat DK, Hecker P, Watanabe M, Chettimada S, Levy RJ, Okada T, Edwards JG, Gupte SA. Glucose-6-phosphate dehydrogenase and NADPH redox regulates cardiac myocyte l-type calcium channel activity and myocardial contractile function. PLoS One 7: e45365, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82: 131–185, 2002. [DOI] [PubMed] [Google Scholar]
- 50.Rousseau E, Cloutier M, Morin C, Proteau S. Capsazepine, a vanilloid antagonist, abolishes tonic responses induced by 20-HETE on guinea pig airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 288: L460–L470, 2005. [DOI] [PubMed] [Google Scholar]
- 51.Sanlioglu S, Williams CM, Samavati L, Butler NS, Wang G, McCray PB Jr, Ritchie TC, Hunninghake GW, Zandi E, Engelhardt JF. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-alpha secretion through IKK regulation of NF-kappa B. J Biol Chem 276: 30188–30198, 2001. [DOI] [PubMed] [Google Scholar]
- 52.Schwartzman ML, Falck JR, Yadagiri P, Escalante B. Metabolism of 20-hydroxyeicosatetraenoic acid by cyclooxygenase. Formation and identification of novel endothelium-dependent vasoconstrictor metabolites. J Biol Chem 264: 11658–11662, 1989. [PubMed] [Google Scholar]
- 53.Singh H, Cheng J, Deng H, Kemp R, Ishizuka T, Nasjletti A, Schwartzman ML. Vascular cytochrome P450 4A expression and 20-hydroxyeicosatetraenoic acid synthesis contribute to endothelial dysfunction in androgen-induced hypertension. Hypertension 50: 123–129, 2007. [DOI] [PubMed] [Google Scholar]
- 55.Stec DE, Gannon KP, Beaird JS, Drummond HA. 20-Hydroxyeicosatetraenoic acid (20-HETE) stimulates migration of vascular smooth muscle cells. Cell Physiol Biochem 19: 121–128, 2007. [DOI] [PubMed] [Google Scholar]
- 56.Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension 42: 1075–1081, 2003. [DOI] [PubMed] [Google Scholar]
- 57.Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res 109: 880–893, 2011. [DOI] [PubMed] [Google Scholar]
- 58.Toth P, Rozsa B, Springo Z, Doczi T, Koller A. Isolated human and rat cerebral arteries constrict to increases in flow: role of 20-HETE and TP receptors. J Cereb Blood Flow Metab 31: 2096–2105, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tunctan B, Korkmaz B, Sari AN, Kacan M, Unsal D, Serin MS, Buharalioglu CK, Sahan-Firat S, Cuez T, Schunck WH, Falck JR, Malik KU. 5,14-HEDGE, a 20-HETE mimetic, reverses hypotension and improves survival in a rodent model of septic shock: contribution of soluble epoxide hydrolase, CYP2C23, MEK1/ERK1/2/IKKbeta/IkappaB-alpha/NF-kappaB pathway, and proinflammatory cytokine formation. Prostaglandins Other Lipid Mediat 102–103: 31–41, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tunctan B, Korkmaz B, Sari AN, Kacan M, Unsal D, Serin MS, Buharalioglu CK, Sahan-Firat S, Cuez T, Schunck WH, Manthati VL, Falck JR, Malik KU. Contribution of iNOS/sGC/PKG pathway, COX-2, CYP4A1, and gp91(phox) to the protective effect of 5,14-HEDGE, a 20-HETE mimetic, against vasodilation, hypotension, tachycardia, and inflammation in a rat model of septic shock. Nitric Oxide 33: 18–41, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang MH, Brand-Schieber E, Zand BA, Nguyen X, Falck JR, Balu N, Schwartzman ML. Cytochrome P450-derived arachidonic acid metabolism in the rat kidney: characterization of selective inhibitors. J Pharmacol Exp Ther 284: 966–973, 1998. [PubMed] [Google Scholar]
- 62.Wang MH, Wang J, Chang HH, Zand BA, Jiang M, Nasjletti A, Laniado-Schwartzman M. Regulation of renal CYP4A expression and 20-HETE synthesis by nitric oxide in pregnant rats. Am J Physiol Renal Physiol 285: F295–F302, 2003. [DOI] [PubMed] [Google Scholar]
- 63.Williams JM, Murphy S, Burke M, Roman RJ. 20-hydroxyeicosatetraeonic acid: a new target for the treatment of hypertension. J Cardiovasc Pharmacol 56: 336–344, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu CC, Cheng J, Zhang FF, Gotlinger KH, Kelkar M, Zhang Y, Jat JL, Falck JR, Schwartzman ML. Androgen-dependent hypertension is mediated by 20-hydroxy-5,8,11,14-eicosatetraenoic acid-induced vascular dysfunction: role of inhibitor of kappaB kinase. Hypertension 57: 788–794, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu CC, Gupta T, Garcia V, Ding Y, Schwartzman ML. 20-HETE and blood pressure regulation: clinical implications. Cardiol Rev 22: 1–12, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu CC, Schwartzman ML. The role of 20-HETE in androgen-mediated hypertension. Prostaglandins Other Lipid Mediat 96: 45–53, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yaghini FA, Zhang C, Parmentier JH, Estes AM, Jafari N, Schaefer SA, Malik KU. Contribution of arachidonic acid metabolites derived via cytochrome P4504A to angiotensin II-induced neointimal growth. Hypertension 45: 1182–1187, 2005. [DOI] [PubMed] [Google Scholar]
- 68.Yang SH, Yates PR, Whitmarsh AJ, Davis RJ, Sharrocks AD. The Elk-1 ETS-domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol Cell Biol 18: 710–720, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu D, Birks EK, Dawson CA, Patel M, Falck JR, Presberg K, Roman RJ, Jacobs ER. Hypoxic pulmonary vasoconstriction is modified by P-450 metabolites. Am J Physiol Heart Circ Physiol 279: H1526–H1533, 2000. [DOI] [PubMed] [Google Scholar]
- 70.Zhu D, Effros RM, Harder DR, Roman RJ, Jacobs ER. Tissue sources of cytochrome P450 4A and 20-HETE synthesis in rabbit lungs. Am J Respir Cell Mol Biol 19: 121–128, 1998. [DOI] [PubMed] [Google Scholar]
- 71.Zielonka J, Hardy M, Kalyanaraman B. HPLC study of oxidation products of hydroethidine in chemical and biological systems: ramifications in superoxide measurements. Free Radic Biol Med 46: 329–338, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]