

Abstract
AIM: To study whether the inflammatory bowel disease (IBD) colon which exhibits varying severity and cytokine levels across its mucosa create varying types of transepithelial leak.
METHODS: We examined the effects of tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), interleukin-1-β (IL1β) and hydrogen peroxide (H2O2) - singly and in combinations - on barrier function of CACO-2 cell layers. Our focus was on the type (not simply the magnitude) of transepithelial leak generated by these agents as measured by transepithelial electrical resistance (TER) and transepithelial flux of 14C-D-mannitol, 3H-Lactulose and 14C-Polyethylene glycol as radiolabeled probe molecules. The isoquinoline alkaloid, berberine, was then examined for its ability to reduce specific types of transepithelial leak.
RESULTS: Exposure to TNF-α alone (200 ng/mL; 48 h) induced a 50% decrease in TER, i.e., increased leak of Na+ and Cl- - with only a marginal but statistically significant increase in transepithelial leak of 14C-mannitol (Jm). Exposure to TNF-α + IFN-γ (200 ng/mL; 48 h) + IL1β (50 ng/mL; 48 h) did not increase the TER change (from TNF-α alone), but there was now a 100% increase in Jm. There however was no increase in transepithelial leak of two larger probe molecules, 3H-lactulose and 14C-polyethylene glycol (PEG). However, exposure to TNF-α + IFN-γ + IL1β followed by a 5 h exposure to 2 mmol/L H2O2 resulted in a 500% increase in 14C-PEG leak as well as leak to the luminal mitogen, epidermal growth factor.
CONCLUSION: This model of graded transepithelial leak is useful in evaluating therapeutic agents reducing IBD morbidity by reducing barrier leak to various luminal substances.
Keywords: Intestine, Crohn’s disease, Tight junction, Ulcerative colitis, CACO-2, Berberine, Micronutrient, Cytokine
Core tip: A cell culture model of graded transepithelial leak can be very valuable in evaluating the various types and magnitudes of leak that can exhibit across the inflammatory bowel disease mucosa. This graded leak can be achieved through various combinations of proinflammatory cytokines and peroxide. Berberine provides an example of a micronutrient that can be more effective against one type of induced leak than another.
INTRODUCTION
The idiopathic inflammatory bowel diseases (IBD), the major types being Crohn’s disease (CD) and ulcerative colitis (UC), are autoimmune diseases affecting the gastrointestinal tract and causing chronic intestinal inflammation. CD and UC have similar key characteristics, perhaps the most important being an observed compromise of epithelial barrier function. The crucial role of the mucosal layer of the gastrointestinal tract is to actively separate gut luminal contents from the underlying interstitium. The epithelial cell layer that lines the gastrointestinal (GI) tract functions as a selectively permeable barrier. A major component of this barrier is the tight junctional (TJ) protein complex, which prevents free diffusion along the paracellular pathway. In the case of IBD, the integrity of the TJ barrier is compromised in part as a result of the inflammatory response increasing local and systemic pro-inflammatory cytokine [tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), interleukin-1-β (IL1β)] production. Luminal antigens now able to traverse the “leaky” barrier exacerbate the inflammatory response in the mucosa and submucosa, which in turn further worsens the integrity of the TJ complex and the cell layer overall[1-6]. It is debated whether intestinal leak is causal or simply a result of the disease. A study by Hollander et al[7] found a two-fold increase in permeability to 14C-polyethylene glycol (PEG)-400 of CD patients and their healthy relatives as compared to normal controls, suggesting intestinal barrier leak may precede clinical intestinal inflammation and be an etiologic factor in IBD. However the overall involvement of compromised barrier function in IBD is not debated.
TNF-α is a key proinflammatory cytokine involved in intestinal inflammation in IBD[6]. Experimentally it has been shown to increase epithelial TJ permeability in several cell types including the human intestinal epithelial monolayers: CACO-2, T84, and HT29/B6[1,8-11]. In CACO-2, TNF-α decreased transepithelial electrical resistance in both a dose- and time- dependent manner[1,9,10]. A decrease in transepithelial electrical resistance (TER) was shown after 24 h incubation with 100 ng/mL TNF-α which reached a maximum at 48 h, and was sustained for up to 8 d after TNF-α removal[10]. TNF-α reduction of TER was associated with an increase in mannitol (182 MW) as well as inulin (5000 MW) permeability[9]. The exact characteristics of the TNF-α effect on barrier function vary across different cell lines. In the renal epithelium model, LLC-PK1, TNF-α produces a rapidly reversible reduction of TER within 2 h and is accompanied by increased permeability to molecules as large as PEG (4000 MW)[11]. TNF-α was also capable of producing a pronounced effect on HT29/B6 cell layers, reducing TER by 81% of the control[12]. The mechanism by which TNF-α generates leak in CACO-2 cell layers has been shown not to be simply a direct result of apoptosis, but rather attributed to TNF-α’s ability to activate NF-κB, induce myosin light chain kinase (MLCK) protein expression and activity, and engender TJ leak[9].
The proinflammatory cytokine IFN-γ has also been reported to increase TJ permeability across T84, HT29/B6, and CACO-2 cell layers[1,13]. T84 cells treated basal-laterally with IFN-γ for 24 h showed a decrease in TER that continued for five days after exposure[13]. Watson et al[14] (2005) demonstrated that in T84 cell layers, IFN-γ caused a greater increase in permeability to larger-sized than to smaller-sized molecules, proposing that IFN-γ selectively activates specific permeation pathways within the TJ. In several studies, using both HT29/B6 and CACO-2 cell layers, TNF-α has been used in combination with IFN-γ to produce a synergistic effect on TER[11,15]. In the CACO-2 model, IFN-γ (10 ng/mL) and TNF-α (2.5 ng/mL) individually did not have an effect on TER or paracellular flux of 3 kD dextran. However, when the cell layers were first primed with IFN-γ for 24 h followed by treatment with TNF-α for 8 h, the cells exhibited both a significant decrease in TER and an increase in 3 kD dextran flux[15].
Increased levels of IL1β in IBD patients have also been associated with increased intestinal inflammation[16]. In CACO-2 cell layers, IL1β caused a drop in TER that was maximal after 48 h treatment. This decrease in TER was accompanied by approximately a 20-fold increase in paracellular permeability to inulin. IL1β was also shown to affect TJ proteins, inducing a decrease in occludin protein expression and an increase in claudin-1 expression. The CACO-2 TJ permeability increase as a result of IL1β exposure involved NF-κB activation and MLCK gene regulation, not induction of apoptosis. Studies by Al-Sadi et al[17,18] suggest a role for p38-kinase dependent activation of the nuclear transcription factor, activating transcription factor-2.
Increased production of mucosal-damaging oxygen radicals by white blood cells has been shown in IBD[19]. Decreased nutritional intake of a variety of antioxidants in IBD patients can lead to an imbalance that causes an additional increase in reactive oxygen species levels resulting in exacerbated oxidative stress of the inflamed intestinal tissue[20,21]. Furthermore, Strus et al[22] (2009) demonstrated that hydrogen peroxide-producing bacteria, present in samples from IBD patients, may be another contributing factor behind increased hydrogen peroxide in the IBD mucosa. In the literature, oxidative stress of CACO-2 cells induced by treatment with H2O2 causes increased paracellular permeability as evidenced by a decrease in TER, as well as increases in both mannitol and inulin flux. Hydrogen peroxide is capable of disrupting the TJ through a specific mechanism that involves protein tyrosine phosphorylation[23-25].
The objectives of the following study were to: (1) Observe the effects of the proinflammatory cytokines, TNF-α, IFN-γ, IL1β, and H2O2, alone and in combination, on CACO-2 barrier function in order to create an in vitro model of graded leak that can reflect the clinical situation in IBD at different sites along the intestinal mucosa; (2) determine if this leak allows for barrier breakdown to biologically active proteins such as epidermal growth factor (EGF); and (3) determine if a previously described nutraceutical capable of barrier protection can in fact reduce barrier compromise under these extreme conditions.
MATERIALS AND METHODS
Cell culture
The CACO-2 cell culture, an epithelial cell line derived from human colon adenocarcinoma[26], was used between passages 52 and 64. Upon confluence, cells were passaged on a weekly basis by trypsinization [0.25% trypsin, 2.2 mmol/L EDTA (Corning Cellgro, Manassas, VA)] and were seeded at 7.5 × 105 cells/Falcon 75-cm2 culture flask with 25 mL of Dulbecco’s Modified Minimum Essential Medium (Corning Cellgro) supplemented with 2 mmol/L L-Glutamine, 1% non essential amino acids, 1 mmol/L Sodium Pyruvate (all culture medium additives, Corning Cellgro) and 10% defined fetal bovine serum (HyClone, Logan, UT). Cultures were incubated at 37 °C in 95% air-5% CO2 atmosphere.
Treatment with cytokines and/or hydrogen peroxide
Human recombinant proteins TNF-α, IL1β and IFN-γ were obtained from Life Technologies (Frederick, MD). For individual exposures and combinations of cytokine treatment, 200 ng/mL TNF-α, 50 ng/mL IL1β, and between 100 and 200 ng/mL IFN-γ were applied (in complete medium) to both the apical and basal-lateral compartments for 48 h. This combination of three cytokines is referred to in this manuscript as “cytomix” for purposes of brevity. Media was first filter sterilized with a 0.2 μm disc filter unit (Corning). Seven- and twenty-one day post-confluent CACO-2 cell layers were used in our studies, as barrier function at these days is highly similar in CACO-2 monolayers, and we did not observe a difference between their responses to cytokines. For hydrogen peroxide (Sigma Life Science, St. Louis, MO) exposure, with or without a prior 48 h incubation with cytokines, the CACO-2 cells were treated both apically and basal-laterally with 2 mmol/L hydrogen peroxide in Dulbecco’s Phosphate Buffered Saline containing calcium and magnesium (Corning Cellgro) supplemented with 5 mmol/L glucose for 5 h. The dosages of cytokines and hydrogen peroxide were fixed in a given experiment; however, over the course of experiments, concentration and exposure time were reduced, yet conditions remained capable of achieving the maximum effect, as we had been using saturating levels of cytokines in a receptor-mediated response.
Transepithelial electrophysiology and permeability
Cells were seeded into sterile Millipore Millicell polycarbonate (PCF) permeable supports (30 mm diameter with 0.4 μm pore size) on day 0 at a seeding density of 5 × 105 cells/insert. Four sterile Millicell PCF inserts were placed into a 100 mm petri dish. On day 1, all cell layers were refed (2 mL apical/15 mL basal-lateral) with control medium containing penicillin (50 U/mL) and streptomycin (50 mcg/mL), followed by refeedings every 2-3 d until exposure. Depending on the specific exposure combination, cells were fed medium supplemented with the appropriate cytokines for 48 h treatment, followed in certain experiments with 5 h of peroxide exposure, then followed by transepithelial electrophysiological measurements and radiotracer flux studies with 0.1 mmol/L, 0.1 μCi/mL 14C-D-mannitol (PerkinElmer, Boston, MA), 0.1 mmol/L, 0.25 μCi/mL 3H-Lactulose (American Radiolabeled Chemicals, Inc., St Louis, MO) and/or 0.1 mmol/L, 0.3 μCi/mL 14C-Polyethylene glycol (PerkinElmer, Waltham, MA).
On the day of transepithelial experiments (for cells treated with cytokines only), the cell layers were re-fed with fresh control medium and allowed to incubate at 37 °C for 1 to 1.5 h prior to electrophysiological readings. Potential difference, TER, and short-circuit current (Isc) were measured using 1 s, 40 μA direct current pulses, with TER calculated using Ohm’s law. As soon as electrical measurements were completed, the basal-lateral medium was aspirated and replaced with 15 mL of medium containing the appropriate radioisotope and incubated at 37 °C. Triplicate basal-lateral medium samples were taken for liquid scintillation counting (LSC) for specific activity (cpm/micromole) determination. Duplicate samples were taken from the apical medium at 60 and 120 min for LSC to determine radioisotope flux rates. The media lost due to sampling from the apical compartment were replaced with fresh medium of the same sample volume. The flux rate (in cpm/min per square centimeter and pmol/min per square centimetre) was calculated for the radioisotope diffusing across the cell layer. In experiments that included exposure to hydrogen peroxide, the cell layers were rinsed in saline before being refed with saline, with or without hydrogen peroxide. After five hours of incubation, the basal-lateral saline was aspirated and replaced with 15 mL of saline containing 0.1 mmol/L, 0.3 μCi/mL 14C-polyethylene glycol. Triplicate basal-lateral samples and duplicate apical samples were taken at 75 min, and the flux rate was calculated as before.
Paracellular flux of 125I-EGF
CACO-2 cell layers, treated as described above prior to exposure, were refed in control medium or medium containing 50 ng/mL TNF-α, 100 ng/mL IFN-γ, and 50 ng/mL IL1β in the apical and basal-lateral compartments for 48 h. On the day of experimental measurements, the cell layers were exposed to control saline or saline containing 1 mmol/L hydrogen peroxide for 3 h. The apical saline was then replaced with medium containing 0.5 μCi/mL, 10 mmol/L 125I-EGF and the basal-lateral saline was replaced with control medium. After a 2 h incubation period, apical and basal-lateral samples were taken for LSC to determine EGF flux rates. Basal-lateral medium was also sampled for column (G-25) chromatography analysis. Total 125I-EGF flux rates (as cpm/min per square centimetre) were adjusted based upon the percent of intact 125I-EGF in the basal-lateral compartment.
Pretreatment with berberine chloride prior to cytokine exposure
Seven-day post-confluent CACO-2 cell layers were refed in control medium or medium containing 100 μmol/L berberine chloride in the apical and basal-lateral compartments. A berberine chloride (Sigma-Aldrich) stock solution (2.7 mmol/L) was prepared in deionized distilled water, but was made each day at the time of use. Following a 24 h berberine pretreatment, the appropriate cell layers were continued in control medium or berberine medium and additionally exposed to either no cytokines, TNF-α, or cytomix for 48 h prior to transepithelial electrophysiology and permeability measurements. For studies that included hydrogen peroxide exposure, on the day of the experiment the cell layers were treated for 5 h with control saline or saline containing 1 mmol/L hydrogen peroxide ± berberine.
Statistical analysis
For electrophysiology and radiotracer flux studies, cytokine- and/or hydrogen peroxide-exposed cell samples were compared against appropriate matched controls within the same experiment. All data are expressed as the mean ± standard error of the mean with the number of replicates provided for each set of studies. Differences between means are evaluated by two-sided Student’s t tests for two groups or by one-way ANOVA followed by Tukey’s post hoc testing where multiple conditions existed.
RESULTS
Exposure to TNF-α
Treatment for 48 h of 7-d post-confluent CACO-2 monolayers with apical and basal-lateral 200 ng/mL TNF-α resulted in a 50% decrease in TER (Figure 1A). The reduction in TER was associated with only a marginal statistically significant increase in transepithelial leak of 14C-D-mannitol (Figure 1B). Unlike the consistent decrease in TER, this increase in mannitol flux did not always achieve statistical significance within each individual experiment, as exemplified by the lack of statistical significance in Figure 2A.
Figure 1.

The effect of tumor necrosis factor-α on CACO-2 transepithelial electrical resistance and transepithelial flux of 14C-D-mannitol. A: Seven-day post-confluent CACO-2 cell layers on Millipore polycarbonate filters were refed in control medium or medium containing 200 ng/mL tumor necrosis factor-α (TNF-α) (apical and basal-lateral compartments) 48 h prior to electrical measurements. Data shown represent the mean ± SE of 16 cell layers per condition. Data represent the percent of control resistance normalized across 3 experiments; B: After electrical measurements, radiotracer flux studies with 0.1 mmol/L, 0.1 μCi/mL 14C-D-mannitol were performed on CACO-2 cell layers, as described in Materials and Methods. Data represent the percent of control flux rate normalized across 4 experiments, and is expressed as the mean ± SE of 20 cell layers per condition. bP < 0.001 vs control (Student’s t test, one-tailed).
Figure 2.
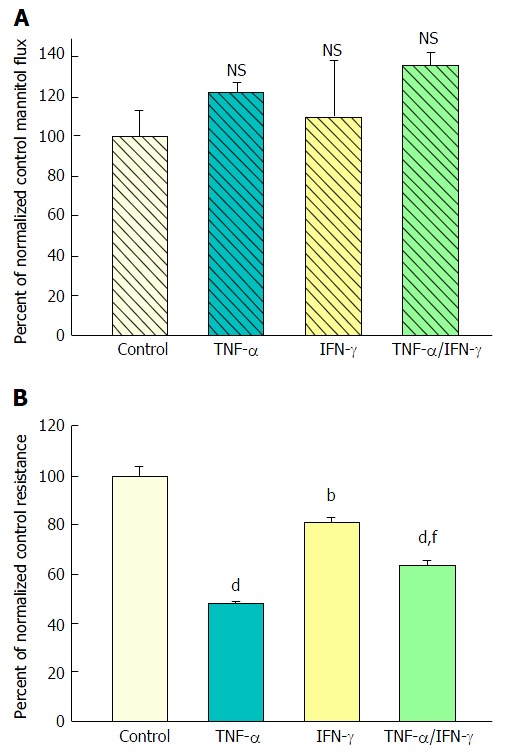
The effect of tumor necrosis factor-α and interferon-γ on CACO-2 transepithelial electrical resistance and transepithelial flux of 14C-D-mannitol. A: Radiotracer flux studies were conducted as described in Figure 1 with the treatment conditions listed above. Data represent the percent of control flux rate, and is expressed as the mean ± SE of 4 cell layers per condition. NS indicates non significance vs control; B: CACO-2 cell layers were cultured and treated as described in Figure 1, using the following conditions: Control medium; medium containing 200 ng/mL tumor necrosis factor-α (TNF-α); medium containing 200 ng/mL Interferon-γ (IFN-γ); or medium containing a combination of 200 ng/mL TNF-α and 200 ng/mL IFN-γ. Data shown represent the mean ± SE of 4 cell layers per condition, with data expressed as the percent of control resistance. bP < 0.01 vs control; dP < 0.001 vs control; fP < 0.01 vs TNF-α alone (one-way ANOVA followed by Tukey’s post hoc testing).
Exposure to TNF-α and IFN-γ
Forty-eight hours combined exposure to 200 ng/mL TNF-α and 100-200 ng/mL IFN-γ (apical and basal-lateral) also caused a significant decrease in TER. Interestingly, this 35% reduction of TER was consistently less than that produced by TNF-α alone (50%) (Figure 2B). A simultaneous slight increase in mannitol flux (Jm) was again observed (Figure 2A), although not quite achieving statistical significance. Exposure to IFN-γ alone reduced TER by only 20% and did not have a significant effect on Jm.
Exposure to TNF-α, IFN-γ, and IL1β
Exposure of 21-d post-confluent CACO-2 cell layers to TNF-α, IFN-γ and 50 ng/mL IL1β (cytomix) on both cell surfaces led to a similar decrease of TER as seen with the combination of TNF-α and IFN-γ (approximately 30%-35% decrease) (Figure 3A). IL1β alone did not generate leak any greater than TNF-α or IFN-γ achieved individually (data not shown). Upon adding IL1β to the mixture of TNF-α and IFN-γ, there was now however a dramatic and consistent 100% increase in Jm (Figure 3B). Further investigation into the impact of cytomix on paracellular permeability showed that the leak pathway produced did not however allow for an increase in flux of the larger probe molecules, 3H-lactulose (MW 342) or 14C-PEG (MW 4000) (Figure 4).
Figure 3.

The effect of tumor necrosis factor-α + interferon-γ + interleukin-1β on CACO-2 transepithelial electrical resistance and transepithelial flux of 14C-D-mannitol. A: Twenty-one day post-confluent CACO-2 cell layers cultured and treated as described in Figure 1, were refed in control medium or medium containing the combination of 200 ng/mL tumor necrosis factor-α, 150 ng/mL interferon-γ, and 50 ng/mL interleukin-1β. Data shown represent the mean ± SE of 16 cell layers per condition. Data represent the percent of control resistance normalized across 4 experiments; B: Radiotracer flux studies were conducted as described in Figure 1, with the same conditions listed above for panel A. Data represent the percent of control flux rate normalized across 2 experiments, and is expressed as the mean ± SE of 8 cell layers per condition. bP < 0.001 vs control (Student’s t test, one-tailed).
Figure 4.

The effect of tumor necrosis factor-α + interferon-γ + interleukin-1β on transepithelial flux of 14C-D-mannitol, 3H-lactulose, and 14C-polyethylene glycol across CACO-2 cell layers. Twenty-one day post-confluent CACO-2 cell layers on Millipore PCF filters were refed in control medium or medium containing the combination of 200 ng/mL tumor necrosis factor-α, 150 ng/mL interferon-γ, and 50 ng/mL interleukin-1β (apical and basal-lateral compartments) 48 h prior to radiotracer flux studies. These studies were performed using 0.1 mmol/L, 0.1 μCi/mL 14C-D-mannitol; 0.1 mmol/L, 0.25 μCi/mL 3H-lactulose; and 0.1 mmol/L, 0.3 μCi/mL 14C-polyethylene glycol as described in Materials and Methods. Data represent the percent of control flux rate normalized across 2 experiments, and is expressed as the mean ± SE of 8 cell layers per condition for the mannitol flux and 4 cell layers per condition for both the lactulose and polyethylene glycol fluxes. NS indicates non significance. bP < 0.001 vs control (Student’s t test, one-tailed).
Exposure to cytomix and hydrogen peroxide
Treatment of both 7- and 21-d post-confluent CACO-2 cell layers with cytomix for 48 h followed by 5 h exposure to 2 mmol/L H2O2 (apical and basal-lateral) induced on average a 500% increase in PEG transepithelial leak. H2O2 alone caused only a 35% increase in PEG leak (Figure 5). In the PEG flux studies, column (G-25) chromatography was used to verify leak of a 4000 MW species of PEG (data not shown). After the 5 h H2O2 exposure, the combination of cytomix and H2O2 resulted in an 80%-90% decrease of TER (data not shown).
Figure 5.
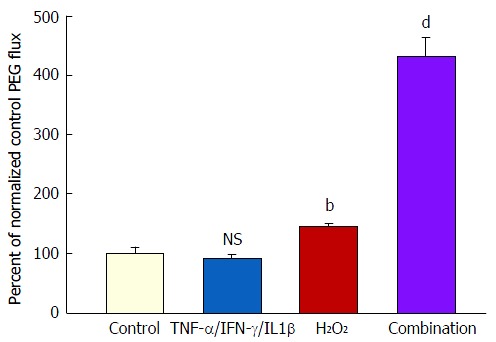
The effect of tumor necrosis factor-α + interferon-γ + interleukin-1β plus hydrogen peroxide on transepithelial flux of 14C-polyethylene glycol. Seven-day and 21-d post-confluent CACO-2 cell layers on Millipore PCF filters were refed in control medium or medium containing the combination of 200 ng/mL TNF-α, 200 ng/mL IFN-γ, and 50 ng/mL IL1β (apical and basal-lateral compartments) 48 h prior to radiotracer flux studies. On the day of the experiment, the cell layers were treated for 5 h with control saline or saline containing 2 mmol/L H2O2. Paracellular permeability was assessed using 0.1 mmol/L, 0.3 μCi/mL 14C-polyethylene glycol as described in materials and methods. Data represent the percent of control flux rate normalized across 2 experiments, and is expressed as the mean ± SE for 8 cell layers per condition. NS indicates non significance vs control. bP < 0.01 vs control; dP < 0.001 vs control (one-way ANOVA followed by Tukey’s post hoc testing). IFN-γ: Interferon-γ; IL1β: Interleukin-1β; TNF-α: Tumor necrosis factor-α.
Hematoxylin and eosin-stained cross sections of CACO-2 cell layers were used to evaluate the histological effects of the various cytokine/H2O2 exposure regimens. As shown in Figure 6, cytomix alone produced no observable morphological changes in cross sections of the epithelial cell layer. Exposure to 2 mmol/L H2O2 resulted in increased blebbing of membranes from the apical surface of occasional cells. Exposure to both cytomix and peroxide induced not only blebbing of apical membranes in occasional cells but also frequent apoptotic nuclei and rare, though occasional, sites of cell detachment.
Figure 6.
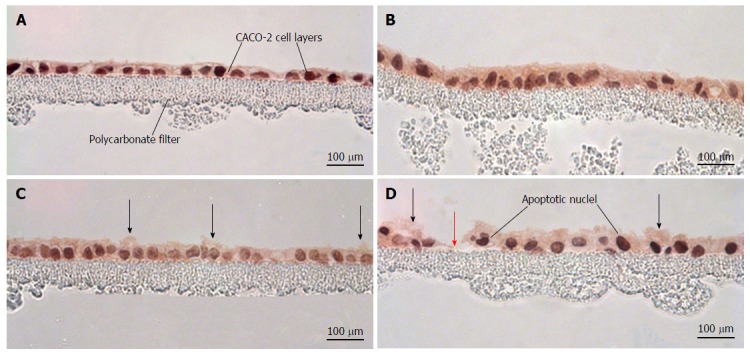
Morphological effects of cytokines and hydrogen peroxide on CACO-2 cell layers. CACO-2 cell layers were cultured at confluent density onto Millipore polycarbonate filters. Seven day post-seeding, the cell layers were refed with control medium or medium containing the combination of 200 ng/mL tumor necrosis factor-α, 200 ng/mL interferon-γ, and 50 ng/mL interleukin-1β (“cytomix”) (apical and basal-lateral compartments) for 48 h, followed by exposure to saline or saline containing 2 mmol/L H2O2 for 5 h. Cell layers were then fixed in formalin and stained with hematoxylin and eosin. A: CACO-2 cell layers exposed to control medium and control saline; B: CACO-2 cell layers exposed to cytomix medium and control saline; C: CACO-2 cell layers exposed to control medium and saline containing hydrogen peroxide; D: CACO-2 cell layers exposed to cytomix medium and saline containing hydrogen peroxide. In C and D, black arrows indicate instances of cytoplasmic blebbing. The red arrow points at a gap in the epithelial barrier arising from cell death and detachment.
Transepithelial leak of EGF
CACO-2 cell layers exposed for 48 h to 50 ng/mL TNF-α, 100 ng/mL IFN-γ, and 50 ng/mL IL1β followed by a 3 h 1 mmol/L H2O2 treatment manifested a transepithelial leak pathway that allowed for not only a leak to 4000 MW PEG, but also an over 30-fold increase in 125I-EGF permeation. This EGF flux was performed in an apical to basal-lateral direction to mimic the diffusion gradient for EGF that would exist in vivo (Table 1). In these EGF studies, the leak of 125I isotope across the cell layer was analyzed by gel filtration chromatography to determine the amount of transepithelial isotope diffusion that corresponded solely with the compound of interest, 6100 MW EGF. Transepithelial leak of actual 6100 MW EGF - and not simply 125I-EGF degradation products - was thus verified.
Table 1.
The effect of tumor necrosis factor-α + interferon-γ + interleukin-1β and hydrogen peroxide on transepithelial flux of 14C-polyethylene glycol and 125I-epidermal growth factor
|
14C-PEG flux
|
125I-EGF flux
|
|||
| cpm/min per square centimeter | pmol/min per square centimeter | cpm/min per square centimeter | fmol/min per square centimeter | |
| Control | 3.74 ± 0.07 | 7.54 ± 0.20 | 43.7 ± 2.0 | 0.034 ± 0.005 |
| Cytomix | 3.40 ± 0.12 | 6.87 ± 0.27 | 30.3 ± 1.4 | 0.015 ± 0.001 |
| Cytomix/H2O2 | 9.80 ± 0.28a | 20.97 ± 0.64a | 192.0 ± 4.0c | 1.21 ± 0.029c |
P < 0.05 vs control;
P < 0.05 vs Cytomix-only condition (one-way ANOVA followed by Tukey’s post hoc testing). CACO-2 cell layers on Millipore polycarbonate filters were refed in control medium or medium containing the combination of 50 ng/mL tumor necrosis factor-α, 100 ng/mL interferon-γ, and 50 ng/mL interleukin-1β (apical and basal-lateral compartments) for 48 h. On the day of radiotracer flux studies, the cell layers were exposed to control saline or saline containing 1 mmol/L hydrogen peroxide for 3 h. These studies were performed using 0.1 mmol/L, 0.025 μCi/mL
C-polyethylene glycol (MW 4000) and 10 nmol/L, 0.5 μCi/mL
I-EGF (MW 6100), as described in Materials and Methods. Total 125I flux rates (as cpm/min per square centimeter) were adjusted, based upon the percent of intact 125I-EGF in the basal-lateral compartment (using column chromatography), and expressed finally as fmol/min per square centimeter. Similar gel chromatography analyses were performed for 14C-PEG experiments, but here all isotopes that diffused across the epithelial cell layer was found to be 4000 MW PEG. Data shown represent the mean standard error for an n = 8 in all cases. PEG: Polyethylene glycol; EGF: Epidermal growth factor.
Evaluation of berberine as a potential therapeutic agent using the graded transepithelial leak model
A major benefit of this in vitro model of graded epithelial barrier leak is the capability of evaluating a great number of potentially efficacious micronutrients - or combinations thereof - for reducing each type of leak that may occur across the surface of the IBD mucosa. In this study, 100 μmol/L berberine chloride pretreatment and simultaneous exposure was evaluated for its effects on the ability of TNF-α and cytomix to impair CACO-2 barrier function. Berberine treatment not only increased basal TER, but also reduced both the TNF-α- and cytomix-induced decrease in TER (Figure 7A). Additionally, berberine reduced the TNF-α- and cytomix-induced increase in Jm (Figure 7B). Berberine also effectively - and significantly - reduced the macromolecule leak resulting from cell layer exposure to cytomix and H2O2 (Figure 8A). Berberine likewise reduced both the H2O2- and cytomix + H2O2-induced decrease in resistance (Figure 8B).
Figure 7.
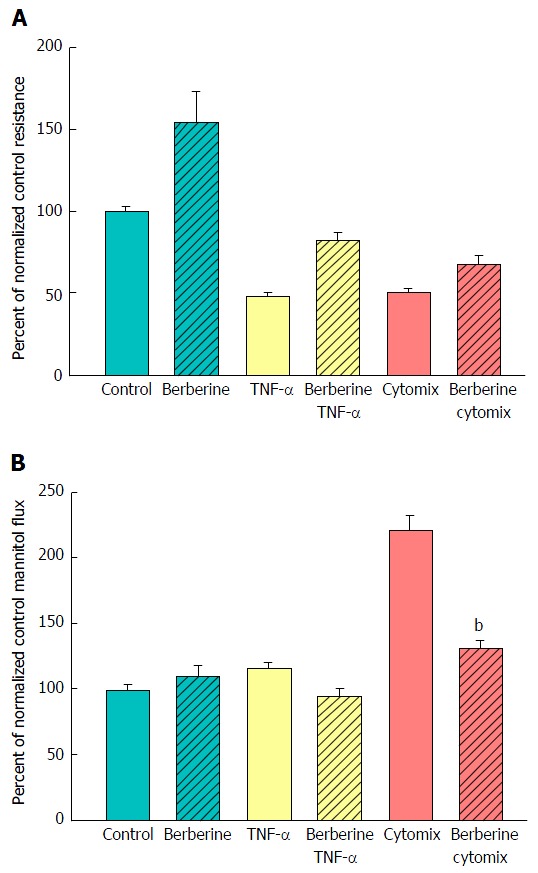
The effect of berberine on cytokine-induced leak of CACO-2 cell layers. A: Seven-day post-confluent CACO-2 cell layers on Millipore polycarbonate filters were refed in control medium or medium containing 100 μmol/L berberine. After 24 h treatment with berberine alone, the cell layers were given either control or berberine medium in addition to being exposed to no cytokines, TNF-α alone, or cytomix (apical and basal-lateral compartments) for 48 h prior to electrical measurements. Data shown represent the mean ± SE of 8 cell layers per condition. Data represent the percent of control resistance normalized across experiments; B: After electrical measurements, the same CACO-2 cell layers represented in A were used to perform radiotracer flux studies with 0.1 mmol/L, 0.1 μCi/mL 14C-D-mannitol, as described in Materials and Methods. Data represent the percent of control flux rate normalized across experiments and is expressed as the mean ± SE of 8 cell layers per condition. bP < 0.001 vs cytomix alone (one-way ANOVA followed by Tukey’s post hoc testing). TNF-α: Tumor necrosis factor-α.
Figure 8.
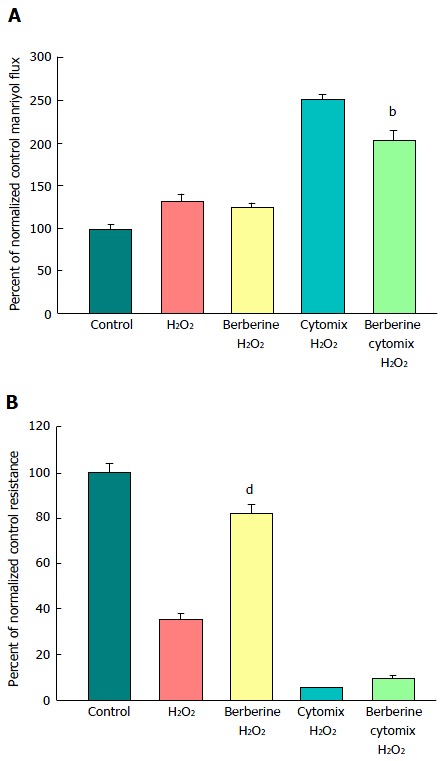
The effect of berberine on cytokine- and peroxide-induced leak of CACO-2 cell layers. A: After electrical measurements, the same CACO-2 cell layers represented in B were used to perform radiotracer flux studies with 0.1 mmol/L, 0.025 μCi/mL 14C-PEG. Data represent the percent of control flux rate, and is expressed as the mean ± SE of 4 cell layers per condition (3 cell layers for the condition of cytomix + peroxide, due to removal of one outlier data point, as determined by a 90% confidence level in the Dixon’s Q test); B: Seven-day post-confluent CACO-2 cell layers on Millipore polycarbonate filters were refed in control medium or medium containing 100 μmol/L berberine. After 24 h treatment with berberine alone, the cell layers were given either control or berberine medium, in addition to being exposed to either cytomix (50 ng/mL tumor necrosis factor-α, 100 ng/mL interferon-γ, 50 ng/mL interleukin-1β) or no cytokines (apical and basal-lateral compartments) for 48 h. On the day of the experiment, the cell layers were treated for 5 h with control saline or saline containing 1 mmol/L hydrogen peroxide ± berberine. Data shown represent the mean ± SE of 4 cell layers per condition, with data expressed as the percent of control resistance. bP < 0.01 vs cytomix + H2O2; dP < 0.001 vs H2O2 alone (one-way ANOVA followed by Tukey’s post hoc testing). Experiment was repeated with similar results.
DISCUSSION
Colon mucosa in active IBD is rarely homogeneously inflamed; rather, there is typically much heterogeneity across the apical surface. This can range from grossly normal, non-inflamed tissue, to normal-appearing tissue but with histological evidence of inflammation (seen, e.g., in white blood cell infiltration in stained tissue sections), to tissue that appears obviously inflamed grossly. In addition, there can be granulomas, pseudopolyps and even micro-ulceration areas that are denuded of epithelium[27-29]. It is well known that inflammation can lead to increased epithelial barrier leak through elevated cytokine levels and other mechanisms[30]. Such grossly observed heterogeneity reflects itself in different degrees of barrier compromise. This variability in barrier compromise can manifest itself not only in the quantitative magnitude of leak, but also in the types of solutes able to leak across the epithelial barrier. As described in Mullin et al[31] (1997) and Watson et al[14] (2005), induction of transepithelial paracellular leak by different agents can result in leak to only small molecules or it can extend to paracellular permeation of macromolecules.
A decrease in TER typically signifies (in “low resistance” epithelial tissues like ileum or colon or CACO-2 cell layers) increased paracellular conductance or diffusion of Na+ and Cl- ions. It implies nothing about potential leak to larger (or uncharged) molecules. And as shown in Figure 1, such was the situation that we observed when CACO-2 cell layers were treated with TNF-α - a significant decrease in TER with only a marginal (and variable) increase in leak to D-mannitol [and no significant leak to the disaccharide lactulose (MW 342) (data not shown)]. This implies that paracellular pathways to Na+ and Cl- ions were increased by TNF-α treatment of the cell layers, but the paracellular pathways that would allow D-mannitol (MW 182) (or larger molecules) to pass, were hardly affected.
However, when cell layers were also treated with IL1β and IFN-γ as well as TNF-α, a sizable increase (100%) in D-mannitol leak was combined with the TER decrease. Obviously, paracellular leak pathways that would admit D-mannitol, were now being induced. This signifies a different type of paracellular pathway that would likely allow for transepithelial paracellular leak of monosaccharides and perhaps neutral amino acids as well as inorganic salts. This could have an effect on the efficiency of gastrointestinal nutrient absorption, as well as ATP consumption by gastrointestinal mucosa. But it is difficult to see how this type of leak would induce inflammation in GI mucosa, since luminal molecules (antigens) capable of eliciting an inflammatory response are typically much larger. It is noteworthy that in our study, even with the increased leak of D-mannitol, leak to the larger probe molecules, lactulose and PEG (4000 MW), was unaffected (Table 2). This not only implies distinct paracellular leak pathways for these different molecules, but also shows that induction of paracellular leak can be a staged, graded phenomenon. Only when exposure of cell layers to cytokines was combined with subsequent treatment with hydrogen peroxide (a situation that reflects certain inflamed tissue in IBD) was a paracellular leak to large molecules observed.
Table 2.
Summary of the magnitude of effect of combinations of cytokines and hydrogen peroxide on transepithelial electrical resistance, mannitol leak, lactulose leak and polyethylene glycol leak
| Transepithelial electrical resistance decrease | Mannitol leak increase | Lactulose leak increase | Polyethylene glycol leak increase | |
| TNF | ++ | 1 | 0 | ND |
| TNF/IFN | + | 1 | 0 | ND |
| TNF/IFN/IL1β | + | ++ | 0 | 0 |
| TNF/IFN/IL1β + hydrogen peroxide | ++++ | ++++ | ++++ | ++++ |
CACO-2 cell layers having been exposed to the above combinations of cytokines and hydrogen peroxide were evaluated for type (salts, mannitol, lactulose, PEG) and magnitude of leak produced. All treatments increased leakage of salts as seen by a decrease in TER, but large molecule leak was seen only in the presence of peroxide alone or both cytokines and hydrogen peroxide, never in the presence of cytokines alone.
Indicates that the observed effect did not routinely achieve statistical significance;
Indicates that an effect was measured but no significant change was observed. ND: Not determined; IFN-γ: Interferon-γ; IL1β: Interleukin-1β; TNF-α: Tumor necrosis factor-α; PEG: Polyethylene glycol.
Transepithelial leak can be both a manifestation of morbidity as well as a driver of morbidity, depending upon the nature of the molecules that are leaking. If leak is induced only to Na+ and Cl- ions (as we observed with TNF-α treatment of CACO-2 cell layers), a situation exists that may not generate serious morbidity. Induced leak to D-mannitol along with decreased TER, however, has implications for the physiological efficiency of nutrient absorption that may then have metabolic/bioenergetic implications for the organism. However, induced leak to molecules larger than D-mannitol, as we observed only for combined treatment with cytokines and peroxide, is the situation most problematic in IBD, because now there can be leak of peptides/proteins present in the GI lumen, into the interstitial fluid compartment, where activation of inflammatory cascades is possible. Substances normally sequestered in the GI lumen such as bacterial toxins and antigens, could - at least on a basis of size - now leak across the epithelial barrier into the interstitial compartment under the epithelium. Bacterial toxins such as Clostridium perfringens enterotoxin (CPE) - which is active from only the abluminal compartment[32] - as well as simple lipopolysaccharide endotoxin, might begin to permeate and either simply raise an immune response (and more cytokine production) in the interstitium or further damage the epithelial barrier directly (in the case of CPE) and compound the barrier compromise even further.
A protein not often considered in the paracellular leak scenarios out of the GI lumen and across a compromised GI barrier is the potent mitogenic growth factor, EGF. EGF (MW 6100) exists in the GI lumen at concentrations over a thousand fold greater than that in the bloodstream, as a result of EGF synthesis and vectorial secretion by salivary glands and Brunner’s glands[33,34]. This EGF is typically biologically inactive however because its receptors are found on the abluminal side of GI epithelia and on interstitial fibroblasts - not on the apical surface of epithelia[35]. However, we show (Table 1) that combined treatment with cytokines and peroxide allows for EGF leak out of the luminal compartment - a situation predicted from the increased leak of PEG (Figure 5). This leakage of luminally situated EGF (down a very steep concentration gradient) into the interstitium may be a major contributing cause for the increased risk of neoplasia in UC, because one could now be putting colonic epithelial cells (whose DNA may be compromised by increased free radical generation) under a near constant replication stimulus.
The greatest utility of the in vitro model of graded transepithelial leak being presented here may derive however from the recent research surrounding improvement/recovery of epithelial barrier function by a diverse - and growing - array of natural (e.g., micronutrients) and synthetic enhancers of TJ barrier function. There have been numerous recent reviews and publications focused on the ability of certain micronutrients like zinc, berberine, quercetin, butyrate, indole, etc., to modify TJ protein composition and in the process yield a TJ - and epithelial barrier - that is less leaky[36-39]. The list of naturally occurring compounds with this capability being reported in the biomedical literature is expanding year by year. One must consider not only the action of these agents in isolation, but also contend with the possibility that certain combinations of these agents can display even greater efficacy than single agents alone in remodeling and enhancing TJs[40]. In short, it is a situation that demands the testing of a large number of agents and permutations of agents. Moreover, one needs to test not only for improvement of basal barrier function, but also for ability to offset the action of proinflammatory proteins and molecules that compromise barrier function. And as we described above, those agents can induce different states of leak, further complicating the testing situation. An in vitro system like what we describe here is ideal for the testing of a large number of different enhancers of barrier function in a range of distinct leak states. This is particularly needed because agents that successfully enhance barrier function in one permeability state of a barrier may or may not be effective in yet another permeability state. As an example, the effectiveness of berberine in reducing leak was tested in this in vitro system and found to not only enhance basal CACO-2 barrier integrity but also to reduce the proinflammtory cytokine - induced compromise in epithelial barrier function (Figure 7). It is the first demonstrated effectiveness of berberine in the context of selective leak induction by treating epithelial cell layers with combinations of cytokines + peroxide. Berberine’s effectiveness in attenuating not only cytokine-induced leak to small molecules but also the transepithelial leak to larger molecules that is seen with cytokines + peroxide, portends potential clinical therapeutic value for IBD.
In our studies, berberine was presented to both cell surfaces simultaneously, as has also been done in earlier studies with berberine and CACO-2 cell layers, studies also showing berberine effectiveness in reducing cytokine-induced barrier disruption[41]. Future studies by our group will evaluate potential sidedness aspects to berberine’s effectiveness in this model. Basal-lateral effectiveness of berberine has been shown pointedly by Taylor et al[42,43] in colon tissue studies. In vivo studies where berberine has been proven effective when given orally[44] may suggest an action from the apical surface, but it is equally possible that berberine is diffusing across damaged epithelial mucosa and engaging the cell from the basal-lateral surface. This is in fact suggested in studies where mucosal barrier damage is modeled through the use of cytochalasin-D treatment[43], and studies showing poor intestinal absorption of berberine into the bloodstream[45].
In conclusion, the expanding array of compounds that are effective in reducing leak across epithelial barriers, both basal as well as induced leak, may be the source of an entirely new class of therapeutics in IBD, therapeutics that could work in complementarity to agents that reduce inflammatory response directly, such as the anti-TNF-α drugs or the salicylates. A need exists for a cell culture model system that could allow large numbers of such compounds (and their combinations) to be tested in vitro before animal model and human studies are undertaken.
COMMENTS
Background
In inflammation and inflammatory diseases impacting epithelial cell layers, there are many molecular agents assaulting the epithelial barrier. Pro-inflammatory cytokines such as tumor necrosis factor-α, interferon-γ and interleukin-1β, and the chemical hydrogen peroxide, are four very common entities in the inflammatory microenvironment that impact an epithelium. In this study, the authors considered that each agent and combinations of agents may have unique effects in terms of the barrier leak that they produce. The authors asked the question of whether these varying effects would confer leakiness to different sizes of molecules. The authors then asked if an agent capable of improving barrier integrity and resisting leak would confer its benefits on specific types of leak.
Research frontiers
It has been well established by numerous research groups that the above three cytokines and hydrogen peroxide all possess ability to induce transepithelial leak, but the nature of the leak produced by the different agents-and especially their combinations-has not been clearly delineated.
Innovations and breakthroughs
In a barrier-related disease such as inflammatory bowel disease (IBD), transepithelial leakage of small molecules (salts, water, sugars, amino acids) will have different medical implications than leak of macromolecules such as protein growth factors, food antigens, and bacterial toxins and antigens. In the testing of various agents capable of barrier protection it is important to note what types of leak are reduced by the agent under study. Berberine, for instance, is shown here to be able to redress the most severe form of leak, namely the leak to macromolecules produced by the combination of cytokines and peroxide.
Applications
Certain aspects of IBD could be alleviated specifically by shutting down or reducing the unregulated leak of molecules across the inflammation-damaged mucosal lining. This manuscript highlights the issue that specific therapeutic agents will be uniquely able to target certain types of leak.
Terminology
Transepithelial leak is not a monolithic entity. It can result from altered tight junctions giving rise to greater leak to small molecules. Or it can result from full disappearance of tight junctions, giving rise to unrestricted paracellular leak, as can happen in epithelial-to-mesenchymanl transition. Or it can result from the disappearance of whole cells (by death and/or detachment), which gives rise to similarly unrestricted leak, as in tight junction disappearance, but here requiring very different mechanisms (including cell replication and motility) to close the leak. All forms are likely in play in IBD, and each-by being regulated differently-can be affected by unique agents capable of restoring aspects of barrier function.
Peer-review
This paper indicated hyper permeability of colon cancer cell layer by cytokine and inhibition by berberine. The results are interesting and the manuscript is well written.
Footnotes
Institutional review board statement: No human subjects were used in this study.
Institutional animal care and use committee statement: No animals were used in this study.
Conflict-of-interest statement: None of the authors of this manuscript have any conflicts of interest, financial or non-financial to declare.
Data sharing statement: Additional data will be shared upon request concerning the action of other micronutrients on cytokine and peroxide-induced leak across gastrointestinal cell layers.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: January 2, 2016
First decision: February 2, 2016
Article in press: March 18, 2016
P- Reviewer: Can G, Suzuki H, Touil-Boukoffa C S- Editor: Ji FF L- Editor: A E- Editor: Li D
References
- 1.Al-Sadi R, Boivin M, Ma T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front Biosci (Landmark Ed) 2009;14:2765–2778. doi: 10.2741/3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruewer M, Samarin S, Nusrat A. Inflammatory bowel disease and the apical junctional complex. Ann N Y Acad Sci. 2006;1072:242–252. doi: 10.1196/annals.1326.017. [DOI] [PubMed] [Google Scholar]
- 3.Nusrat A, Turner JR, Madara JL. Molecular physiology and pathophysiology of tight junctions. IV. Regulation of tight junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:G851–G857. doi: 10.1152/ajpgi.2000.279.5.G851. [DOI] [PubMed] [Google Scholar]
- 4.Shen L, Turner JR. Role of epithelial cells in initiation and propagation of intestinal inflammation. Eliminating the static: tight junction dynamics exposed. Am J Physiol Gastrointest Liver Physiol. 2006;290:G577–G582. doi: 10.1152/ajpgi.00439.2005. [DOI] [PubMed] [Google Scholar]
- 5.Thoreson R, Cullen JJ. Pathophysiology of inflammatory bowel disease: an overview. Surg Clin North Am. 2007;87:575–585. doi: 10.1016/j.suc.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Fakhoury M, Negrulj R, Mooranian A, Al-Salami H. Inflammatory bowel disease: clinical aspects and treatments. J Inflamm Res. 2014;7:113–120. doi: 10.2147/JIR.S65979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. Increased intestinal permeability in patients with Crohn’s disease and their relatives. A possible etiologic factor. Ann Intern Med. 1986;105:883–885. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]
- 8.Fish SM, Proujansky R, Reenstra WW. Synergistic effects of interferon gamma and tumour necrosis factor alpha on T84 cell function. Gut. 1999;45:191–198. doi: 10.1136/gut.45.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 10.Marano CW, Lewis SA, Garulacan LA, Soler AP, Mullin JM. Tumor necrosis factor-alpha increases sodium and chloride conductance across the tight junction of CACO-2 BBE, a human intestinal epithelial cell line. J Membr Biol. 1998;161:263–274. doi: 10.1007/s002329900333. [DOI] [PubMed] [Google Scholar]
- 11.Mullin JM, Laughlin KV, Marano CW, Russo LM, Soler AP. Modulation of tumor necrosis factor-induced increase in renal (LLC-PK1) transepithelial permeability. Am J Physiol. 1992;263:F915–F924. doi: 10.1152/ajprenal.1992.263.5.F915. [DOI] [PubMed] [Google Scholar]
- 12.Schmitz H, Fromm M, Bentzel CJ, Scholz P, Detjen K, Mankertz J, Bode H, Epple HJ, Riecken EO, Schulzke JD. Tumor necrosis factor-alpha (TNFalpha) regulates the epithelial barrier in the human intestinal cell line HT-29/B6. J Cell Sci. 1999;112(Pt 1):137–146. doi: 10.1242/jcs.112.1.137. [DOI] [PubMed] [Google Scholar]
- 13.Adams RB, Planchon SM, Roche JK. IFN-gamma modulation of epithelial barrier function. Time course, reversibility, and site of cytokine binding. J Immunol. 1993;150:2356–2363. [PubMed] [Google Scholar]
- 14.Watson CJ, Hoare CJ, Garrod DR, Carlson GL, Warhurst G. Interferon-gamma selectively increases epithelial permeability to large molecules by activating different populations of paracellular pores. J Cell Sci. 2005;118:5221–5230. doi: 10.1242/jcs.02630. [DOI] [PubMed] [Google Scholar]
- 15.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reinecker HC, Steffen M, Doehn C, Petersen J, Pflüger I, Voss A, Raedler A. Proinflammatory cytokines in intestinal mucosa. Immunol Res. 1991;10:247–248. doi: 10.1007/BF02919700. [DOI] [PubMed] [Google Scholar]
- 17.Al-Sadi R, Guo S, Ye D, Dokladny K, Alhmoud T, Ereifej L, Said HM, Ma TY. Mechanism of IL-1β modulation of intestinal epithelial barrier involves p38 kinase and activating transcription factor-2 activation. J Immunol. 2013;190:6596–6606. doi: 10.4049/jimmunol.1201876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Sadi RM, Ma TY. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol. 2007;178:4641–4649. doi: 10.4049/jimmunol.178.7.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rugtveit J, Haraldsen G, Høgåsen AK, Bakka A, Brandtzaeg P, Scott H. Respiratory burst of intestinal macrophages in inflammatory bowel disease is mainly caused by CD14+L1+ monocyte derived cells. Gut. 1995;37:367–373. doi: 10.1136/gut.37.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J Pathol. 2003;201:28–36. doi: 10.1002/path.1409. [DOI] [PubMed] [Google Scholar]
- 21.Alzoghaibi MA. Concepts of oxidative stress and antioxidant defense in Crohn’s disease. World J Gastroenterol. 2013;19:6540–6547. doi: 10.3748/wjg.v19.i39.6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strus M, Gosiewski T, Fyderek K, Wedrychowicz A, Kowalska-Duplaga K, Kochan P, Adamski P, Heczko PB. A role of hydrogen peroxide producing commensal bacteria present in colon of adolescents with inflammatory bowel disease in perpetuation of the inflammatory process. J Physiol Pharmacol. 2009;60 Suppl 6:49–54. [PubMed] [Google Scholar]
- 23.Basuroy S, Seth A, Elias B, Naren AP, Rao R. MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem J. 2006;393:69–77. doi: 10.1042/BJ20050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao RK, Baker RD, Baker SS, Gupta A, Holycross M. Oxidant-induced disruption of intestinal epithelial barrier function: role of protein tyrosine phosphorylation. Am J Physiol. 1997;273:G812–G823. doi: 10.1152/ajpgi.1997.273.4.G812. [DOI] [PubMed] [Google Scholar]
- 25.Rao RK, Li L, Baker RD, Baker SS, Gupta A. Glutathione oxidation and PTPase inhibition by hydrogen peroxide in Caco-2 cell monolayer. Am J Physiol Gastrointest Liver Physiol. 2000;279:G332–G340. doi: 10.1152/ajpgi.2000.279.2.G332. [DOI] [PubMed] [Google Scholar]
- 26.Grasset E, Pinto M, Dussaulx E, Zweibaum A, Desjeux JF. Epithelial properties of human colonic carcinoma cell line Caco-2: electrical parameters. Am J Physiol. 1984;247:C260–C267. doi: 10.1152/ajpcell.1984.247.3.C260. [DOI] [PubMed] [Google Scholar]
- 27.Feldman M, Sleisenger MH, Friedman LS, Brandt LJ. Sleisenger & Fordtran’s Gastrointestinal and Liver Disease. Vol. 2. Philadelphia, PA: Saunders/Elsevier; 2010. pp. 1948–1986. [Google Scholar]
- 28.Fenoglio-Preiser CM. Gastrointestinal Pathology: An Atlas and Text. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2008. pp. 610–647. [Google Scholar]
- 29.Geboes K. Satsangi J, Sutherland LR, ed. Inflammatory Bowel Diseases, London, England: Churchill Livingstone; 2003. Histopathology of Crohn’s Disease and Ulcerative Colitis; pp. 257–261. [Google Scholar]
- 30.Fries W, Belvedere A, Vetrano S. Sealing the broken barrier in IBD: intestinal permeability, epithelial cells and junctions. Curr Drug Targets. 2013;14:1460–1470. doi: 10.2174/1389450111314120011. [DOI] [PubMed] [Google Scholar]
- 31.Mullin JM, Marano CW, Laughlin KV, Nuciglio M, Stevenson BR, Soler P. Different size limitations for increased transepithelial paracellular solute flux across phorbol ester and tumor necrosis factor-treated epithelial cell sheets. J Cell Physiol. 1997;171:226–233. doi: 10.1002/(SICI)1097-4652(199705)171:2<226::AID-JCP14>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 32.Sonoda N, Furuse M, Sasaki H, Yonemura S, Katahira J, Horiguchi Y, Tsukita S. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: Evidence for direct involvement of claudins in tight junction barrier. J Cell Biol. 1999;147:195–204. doi: 10.1083/jcb.147.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heitz PU, Kasper M, van Noorden S, Polak JM, Gregory H, Pearse AG. Immunohistochemical localisation of urogastrone to human duodenal and submandibular glands. Gut. 1978;19:408–413. doi: 10.1136/gut.19.5.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Playford RJ, Wright NA. Why is epidermal growth factor present in the gut lumen? Gut. 1996;38:303–305. doi: 10.1136/gut.38.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheving LA, Shiurba RA, Nguyen TD, Gray GM. Epidermal growth factor receptor of the intestinal enterocyte. Localization to laterobasal but not brush border membrane. J Biol Chem. 1989;264:1735–1741. [PubMed] [Google Scholar]
- 36.Amasheh M, Andres S, Amasheh S, Fromm M, Schulzke JD. Barrier effects of nutritional factors. Ann N Y Acad Sci. 2009;1165:267–273. doi: 10.1111/j.1749-6632.2009.04063.x. [DOI] [PubMed] [Google Scholar]
- 37.Gu L, Li N, Li Q, Zhang Q, Wang C, Zhu W, Li J. The effect of berberine in vitro on tight junctions in human Caco-2 intestinal epithelial cells. Fitoterapia. 2009;80:241–248. doi: 10.1016/j.fitote.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Bansal T, Alaniz RC, Wood TK, Jayaraman A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc Natl Acad Sci USA. 2010;107:228–233. doi: 10.1073/pnas.0906112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Valenzano MC, Mercado JM, Zurbach EP, Mullin JM. Zinc supplementation modifies tight junctions and alters barrier function of CACO-2 human intestinal epithelial layers. Dig Dis Sci. 2013;58:77–87. doi: 10.1007/s10620-012-2328-8. [DOI] [PubMed] [Google Scholar]
- 40.Mercado J, Valenzano MC, Jeffers C, Sedlak J, Cugliari MK, Papanikolaou E, Clouse J, Miao J, Wertan NE, Mullin JM. Enhancement of tight junctional barrier function by micronutrients: compound-specific effects on permeability and claudin composition. PLoS One. 2013;8:e78775. doi: 10.1371/journal.pone.0078775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li N, Gu L, Qu L, Gong J, Li Q, Zhu W, Li J. Berberine attenuates pro-inflammatory cytokine-induced tight junction disruption in an in vitro model of intestinal epithelial cells. Eur J Pharm Sci. 2010;40:1–8. doi: 10.1016/j.ejps.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Taylor CT, Baird AW. Berberine inhibition of electrogenic ion transport in rat colon. Br J Pharmacol. 1995;116:2667–2672. doi: 10.1111/j.1476-5381.1995.tb17224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor CT, Winter DC, Skelly MM, O’Donoghue DP, O’Sullivan GC, Harvey BJ, Baird AW. Berberine inhibits ion transport in human colonic epithelia. Eur J Pharmacol. 1999;368:111–118. doi: 10.1016/s0014-2999(99)00023-0. [DOI] [PubMed] [Google Scholar]
- 44.Chen C, Yu Z, Li Y, Fichna J, Storr M. Effects of berberine in the gastrointestinal tract - a review of actions and therapeutic implications. Am J Chin Med. 2014;42:1053–1070. doi: 10.1142/S0192415X14500669. [DOI] [PubMed] [Google Scholar]
- 45.Pan GY, Wang GJ, Liu XD, Fawcett JP, Xie YY. The involvement of P-glycoprotein in berberine absorption. Pharmacol Toxicol. 2002;91:193–197. doi: 10.1034/j.1600-0773.2002.t01-1-910403.x. [DOI] [PubMed] [Google Scholar]