

Abstract
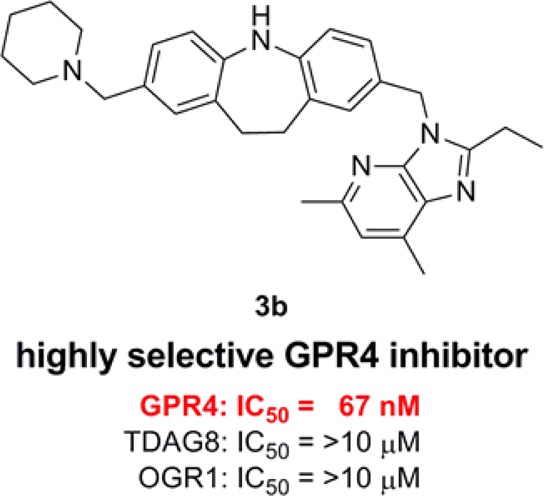
GPR4, a pH-sensing G protein-coupled receptor, is highly expressed in endothelial cells and may be activated in myocardial infarction due the decreased tissue pH. We are interested in GPR4 antagonists as potential effective pharmacologic tools and/or drug leads for the treatment of myocardial infarction. We investigated the structure–activity relationship of a known GPR4 antagonist 1 as a lead compound to identify 3b as the first potent and selective GPR4 antagonist, whose effectiveness was demonstrated in a mouse myocardial infarction model.
Keywords: GPR4, myocardial infarction, dibenzazepine derivatives, pH-sensing GPCR
Myocardial infarction, caused by occlusion of the coronary artery, results in damage to the heart due to depletion of the oxygen supply. The area downstream of the occluded artery becomes ischemic, and the cells in the ischemic area die if blood flow is not promptly restored by opening the artery. The number of cases of death by myocardial infarction in developed countries remains high. Therefore, the development of methods to effectively treat myocardial infarction to reduce the high death rate is desired.1
In myocardial infarction, not only the blood oxygen concentration in the occluded coronary artery is significantly decreased, but the extracellular environment is also dramatically altered.2 The concentrations of potassium and protons are increased in myocardial infarction.3 The proton concentration is increased due to enhanced glycolysis to produce ATP instead of oxidative phosphorylation. In addition, the intracellular and extracellular pH is decreased. The effects of these changes in the extracellular environment on myocardial infarction-induced cellular injury, however, are unclear.
Extracellular acidification regulates several groups of signaling molecules.4,5 Among them, pH-sensing G protein-coupled receptors (GPCRs) and ion channels are activated by acidification under various disease conditions.5 These GPCRs are activated when the environmental pH is decreased to less than 7.0, and ion channels are activated at a pH less than 6.0. Coronary ligation of the porcine heart leads to a decrease in the pH from 7.4 to ∼5.5,6 suggesting that pH-sensing GPCRs can be strongly activated in ischemic areas. Signaling molecules activated in myocardial infarction have not been confirmed to date. As GPCRs are effective targets of many clinically useful drugs, we have been interested in the roles of pH-sensing GPCRs in myocardial infarction-induced cellular injury from the viewpoint of drug development.
This pH-sensing GPCR family is classified as an OGR1 family.5 Four GPCRs are regulated by extracellular pH: G2A, TDAG8, OGR1, and GPR4, in which G2A is a little different from other three members, because it is fully activated when the extracellular pH is ∼7.0.7 Therefore, GPCRs activated by acidification upon myocardial infarction include TDAG8, OGR1, and GPR4. GPR4 is expressed extensively in endothelial cells,8 which play an important role in regulating heart function under physiologic and pathologic conditions.9,10 Therefore, we examined the roles of GPR4 in myocardial infarction-induced injury, where a GPR4 antagonist may be used effectively as a pharmacologic tool. GPR4 antagonists are potential drug candidates for the treatment of myocardial infarction. Based on these considerations, we planned to develop potent and selective GPR4 antagonists.
Dibenzazepine derivative 1 acts as a GPR4 antagonist in vitro, and is effective for the treatment of lung damage induced by X-ray exposure in a rodent model.11 The effects of 1 on disorders related to myocardial infarction, however, are unknown. Therefore, we performed structure–activity relationship studies of 1 as a lead compound to develop potent and selective GPR4 antagonists for the treatment of myocardial infarction-induced injury. The structure of GPR4 antagonist 1 comprises three parts, i.e., the left piperazine moiety (A), the middle dibenzazepine moiety (B), and the right imidazopyridine moiety (C), as shown in Figure 1. Thus, we examined the effects of altering each of the three moieties in 1 on GPR4 antagonist activity. Analogues of 1 were designed and synthesized as shown in Figure 1.
Figure 1.

Structures of the lead compound 1 and its analogues.
Synthesis of the analogues is outlined in Scheme 1. The left-hand moiety-modified analogues were synthesized using the previously reported Mannich reaction procedure for the synthesis of 1.12 Thus, treatment of imidazobenzyl derivative 2(12) with cyclic amines and HCHO in AcOH/CHCl3 gave the corresponding left-hand moiety-modified congeners of 1, i.e., 3a–c, respectively (Scheme 1-a). The right-hand moiety-modified analogues were prepared by nucleophilic substitution reaction of an ammonium salt 4 with various aromatic heterocycles, such as benzimidazole, indole, or imidazole derivatives, giving 5a–h (Scheme 1-b). The middle moiety-modified analogues 8a–c were prepared as shown in Scheme 1-c. Mannich reaction of 6a-c with piperidine, followed by N-methylation and a subsequent nucleophilic substitution reaction using 7 as a nucleophile provided 8a-c. The nucleophilic substitution reaction using 7 as a nucleophile to benzyl bromide or 2-(bromomethyl)naphthalene gave 9a or 9b, respectively (Scheme 1-d).
Scheme 1. Synthesis of Analogues of 1.

Reagents and conditions: (a) R1-H, 37% HCHO, AcOH/CHCl3, 60 °C; (b) R2-H, LiOH, DMF, 40 °C; (c) piperidine, 37% HCHO, AcOH/CHCl3, 60 °C; (d) MeI, AcOEt, 40 °C; (e) 7, LiOH, DMF, 40 °C; (f) R3CH2Br, LiOH, DMF, 40 °C.
The inhibitory effects of the compounds on pH-dependent GPR4 activation were evaluated using HEK293 cells expressing GPR4.13 The pH value in culture was changed from 7.4 to 7.0, where, consistent with the previous results,13 GPR4-transfected cells showed proton concentration-dependent increases in cAMP response element (CRE)-driven transcriptional activity. Antagonistic effects of the compounds against pH-dependent GPR4 activation were evaluated by measuring CRE promoter activity, and the antagonistic effects of the compounds (10 μM) are indicated as the transcriptional activity ratio relative to that in the absence of the compound (Table 1).
Table 1. GPR4 Antagonistic Activity of the Modified Analogues of 1.


CRE activity is the transcriptional activity ratio at 10 μM of compounds relative to that in the absence of compounds.
GPR4 antagonistic activity of the left-hand moiety-modified analogues is summarized in Table 1. These results revealed that replacement of the parent piperazine moiety with other six-membered cyclic amines, such as piperidine (3b) and morpholine (3c), as well as with five-membered amine pyrrolidine (3a) only slightly affected the activity. Furthermore, compound 2 in which the left-hand moiety was removed had an antagonistic effect similar to that of the parent compound 1. Therefore, we performed further structure activity relationship studies on the right-hand moiety and the middle moiety using analogues without the left-hand moiety substituent.
Subsequently, GPR4 antagonistic activity of the right-hand moiety-modified analogues was investigated. Although analogues 5a–d, in which the parent imidazopyridine was replaced with similar bicyclic aromatic heterocyles, functioned as antagonists, the effects were weaker than those of the parent compound 1 or its left-hand moiety-removed analogue 2. To elucidate the effect of the size of the right-hand moiety on the activity, the right-hand moiety was changed from a bicyclic aromatic ring to a tricyclic aromatic ring (5e) or monocyclic aromatic rings (5f-h). All of the analogues were inactive, indicating that the bicyclic aromatic ring in the right-hand moiety is essential for the antagonistic activity.
The effect of the middle moiety of the structure on the antagonistic activity was examined by adopting the imidazopyridine moiety of the lead compound 1 as the right-hand moiety. While the antagonistic activity of tricyclic aromatic compounds 8a and 8b did not surpass that of the parent compound 1, they both showed potent activity. On the other hand, diphenylamine compound 8c, which corresponds to the conformationally flexible acyclic analogue in the middle moiety of the parent compound 1, exhibited only a weak activity. The antagonistic activities of nontricyclic benzene or naphthalene analogues 9a and 9b were also weak. Accordingly, it was suggested that the tricyclic aromatic ring of the middle moiety played an important role in the activity.
The dose–response of the GPR4 antagonistic effects of selected compounds, i.e., lead compound 1 and its left-hand moiety-removed analogue 2, the left-hand moiety-modified 3b, right-hand moiety-modified 5a and 5c, and middle moiety- modified 8b and 8c, were investigated to obtain the IC50 values. As shown in Figure 2, the compounds ranked in order of their antagonistic activity as 3b > 1 ≫ 2, 8b ≫ 5a, 5c, 8c. Although the lead compound 1 (IC50 = 130 nM) was quite active, the left-hand moiety-modified analogue 3b (IC50 = 67 nM), in which the piperazine of 1 was replaced by piperidine, was approximately 2-fold more active than 1. Compounds 2 and 8b without the left-hand moiety showed a moderate IC50 value of 900 nM and 910 nM, respectively, suggesting that the presence of the left-hand six-membered cyclic amine improved the potency of the compounds. The antagonistic activity of 5a, 5c, and 8c was weak (IC50 > 10 μM), and accordingly, the middle moiety of the tricyclic aromatic ring and the right-hand moiety of the imidazopyridine ring are likely to be essential for the potent antagonistic activity.
Figure 2.
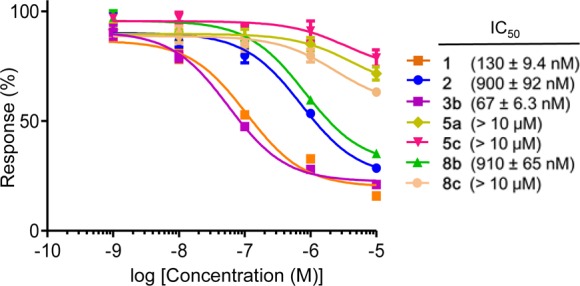
Dose response of GPR4 antagonistic effects.
As proton-sensing GPCRs, not only GPR4, but also TDAG8 and OGR1, are expressed in endothelial cells,8 which may have an important role in regulating heart function under physiologic and pathologic conditions. Therefore, we examined the effects of the two most potent compounds, 1 and 3b, on TDAG8 and OGR1. The dose–response effects of the two compounds on the CRE activity in HEK293 cells expressing TDAG8 and the NFAT activity in HEK293 cells expressing OGR1, as well as their effects on GPR4, are shown in Figure 3. Compound 1 at a higher concentration (>1 μM) had antagonistic effects on both TDAG8 and OGR1 (Figure 3a). Compound 3b was completely inactive on both TDAG8 and OGR1, even at a concentration as high as 10 μM (Figure 3b). Thus, compound 3b was identified as a highly potent and selective GPR4 antagonist.
Figure 3.
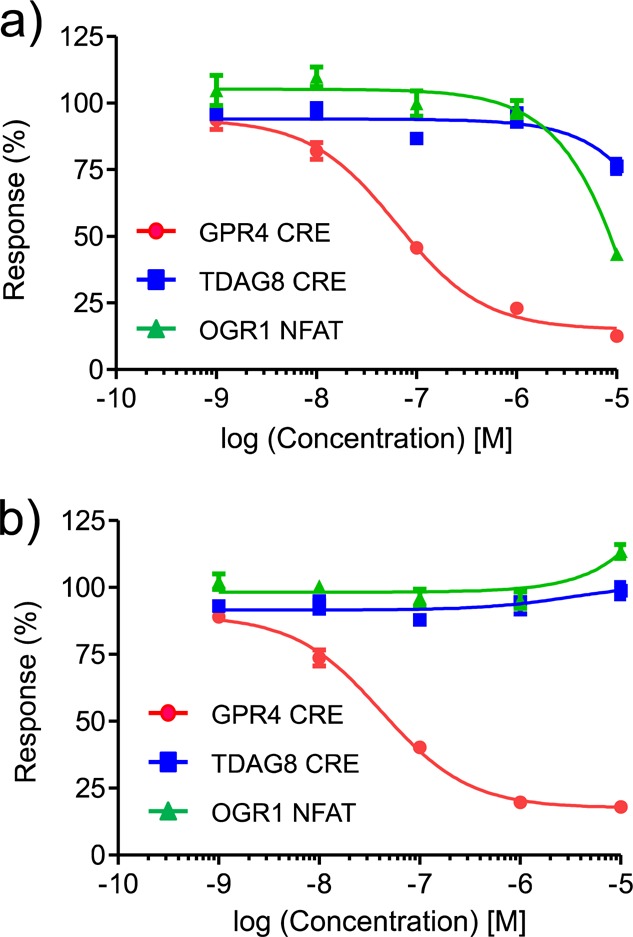
Effects of compounds 1 (a) and 3b (b) on the pH-sensing GPCRs: GPR4, TDAG8, and OGR1.
Finally, we examined the effects of the potent and selective antagonist 3b in a mouse myocardial infarction model. Mice were subjected to permanent left anterior descending coronary artery ligation under anesthetized conditions, and compound 3b (6.7 or 20 mg/kg/day) was injected intraperitoneally into the mice twice a day from 1 day before to 7 days after the operation. The survival rates of the operated mice up to day 28 are shown in Figure 4. Although most of the control mice (6/7) died within 8 days after the operation, all of the mice (7/7) treated with 3b (6.7 mg/kg/day) survived. Although the mechanism underlying survival by the GPR4 antagonist has not yet been examined in the present study, previous reports suggested that acidic pH/GPR4 stimulates inflammatory cytokine and chemokine production in endothelial cells.8,13 We therefore tentatively speculate that the involvement of inhibitory actions of the GPR4 antagonist on the inflammatory responses brings the survival of the animals.
Figure 4.
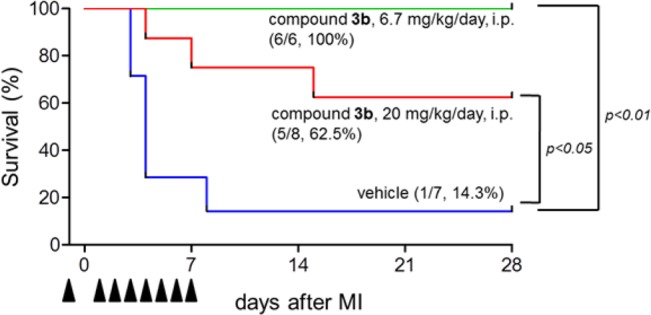
Effect of intraperitoneal administration of 3b to a mouse myocardial infarction model. Kaplan–Meier survival curves of mice treated with vehicle (n = 7) or compound 3b (6.7 mg/kg/day: n = 6, 20 mg/kg/day: n = 8) after MI operation. Vehicle or compound 3b (twice a day) was intraperitoneally injected from 1 day before MI operation until 7 days after MI operation every day, not including the operating day. The differences between mice treated with vehicle and compound 3b were evaluated by the log-rank test.
In conclusion, based on the hypothesis that pH-sensing GPR4 can be activated in ischemic areas, we investigated the structure–activity relationship of known GPR4 antagonist 1 as a lead compound to identify 3b as a potent and selective GPR4 antagonist. Treatment with 3b effectively prolonged life in the mouse myocardial infarction model in vivo. Compound 3b is the first GPR4-selective antagonist with no effect on the other proton-sensing GPCRs expressed in endothelial cells, i.e., TDAG8 and OGR1, and thus it may be a very effective tool for investigating the physiologic and pharmacologic roles of GPR4.
Glossary
Abbreviations
- GPR4
G protein-coupled receptor 4
- GPCR
G protein-coupled receptor
- OGR1
ovarian cancer G-protein-coupled receptor 1
- G2A
G2 accumulation protein
- TDAG8
T cell death associated gene-8
- HEK 293
Human Embryonic Kidney 293
- CRE
cAMP response element
- NFAT
nuclear factor of activated T-Cell
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00014.
Experimental procedures and characterization data of all synthesized compounds and assay method. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kloner R. A. Current state of clinical translation of cardioprotective agents for acute myocardial infarction. Circ. Res. 2013, 113, 451–463. 10.1161/CIRCRESAHA.112.300627. [DOI] [PubMed] [Google Scholar]
- Opie L. H.Oxygen lack: ischemia and angina. Heart: physiology, from cell to circulation; Lippincott–Raven: Philadelphia, 1998, Chapter 17, pp 515–541. [Google Scholar]
- Friedrich R.; Hirche H.; Kebbel U.; Zylka V.; Bissig R. Changes of extracellular Na+, K+, Ca2+ and H+ of the ischemic myocardium in pigs. Basic Res. Cardiol. 1981, 76, 453–456. 10.1007/BF01908341. [DOI] [PubMed] [Google Scholar]
- Krishtal O. The ASICs: Signaling molecules? Modulators?. Trends Neurosci. 2003, 26, 477–483. 10.1016/S0166-2236(03)00210-8. [DOI] [PubMed] [Google Scholar]
- Okajima F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell. Signalling 2013, 25, 2263–2271. 10.1016/j.cellsig.2013.07.022. [DOI] [PubMed] [Google Scholar]
- Hirche H.; Franz C.; Bös L.; Bissig R.; Lang R.; Schramm M. Myocardial extracellular K+ and H+ increase and noradrenaline release as possible cause of early arrhythmias following acute coronary artery occlusion in pigs. J. Mol. Cell. Cardiol. 1980, 12, 579–593. 10.1016/0022-2828(80)90016-4. [DOI] [PubMed] [Google Scholar]
- Tobo M.; Tomura H.; Mogi C.; Wang J.-Q.; Liu J.-P.; Komachi M.; Damirin A.; Kimura T.; Murata N.; Kurose H.; Sato K.; Okajima F. Previously postulated ″ligand-independent″ signaling of GPR4 is mediated through proton-sensing mechanisms. Cell. Signalling 2007, 19, 1745–1753. 10.1016/j.cellsig.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Dong L.; Li Z.; Leffler N. R.; Asch A. S.; Chi J.-T.; Yang L. V. Acidosis activation of the proton-sensing GPR4 receptor stimulates vascular endothelial cell inflammatory responses revealed by transcriptome analysis. PLoS One 2013, 8, e61991. 10.1371/journal.pone.0061991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti C. N.; Gheorghiade M.; Kalogeropoulos A. P.; Georgiopoulou V. V.; Quyyumi A. A.; Butler J. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 2012, 60, 1455–1469. 10.1016/j.jacc.2011.11.082. [DOI] [PubMed] [Google Scholar]
- Shantsila E.; Wrigley B. J.; Blann A. D.; Gill P. S.; Lip G. Y. A contemporary view on endothelial function in heart failure. Eur. J. Heart Failure 2012, 14, 873–881. 10.1093/eurjhf/hfs066. [DOI] [PubMed] [Google Scholar]
- Saki M.; Nonaka H.; Miyaji H.; Takahashi C.; Manabe H.; Hiura N.; Miki I.; Abe Y.; Sasaki K.; Kobatake C.; Ichikawa S.; Goto A.; Suda T.. Preventive and/or therapeutic agent for neutrophil inflammation disease. WO 2004/093912 A1, 2004.
- Saki M.; Nonaka H.; Miyaji H.; Hiura N.; Manabe H.; Matsumura T.; Arai H.; Sasaki K.; Kobatake C.; Iida K.; Kuboyama T.. Preventive and/or therapeutic drugs for asthma. WO 2004/017994 A1, 2004.
- Tobo A.; Tobo M.; Nakakura T.; Ebara M.; Tomura H.; Mogi C.; Im D.-S.; Murata N.; Kuwabara A.; Ito S.; Fukuda H.; Arisawa M.; Shuto S.; Nakaya M.; Kurose H.; Sato K.; Okajima F. Characterization of Imidazopyridine Compounds as Negative Allosteric Modulators of Proton-Sensing GPR4 in Extracellular Acidification-Induced Responses. PLoS One 2015, 10, e0129334. 10.1371/journal.pone.0129334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.