

Abstract
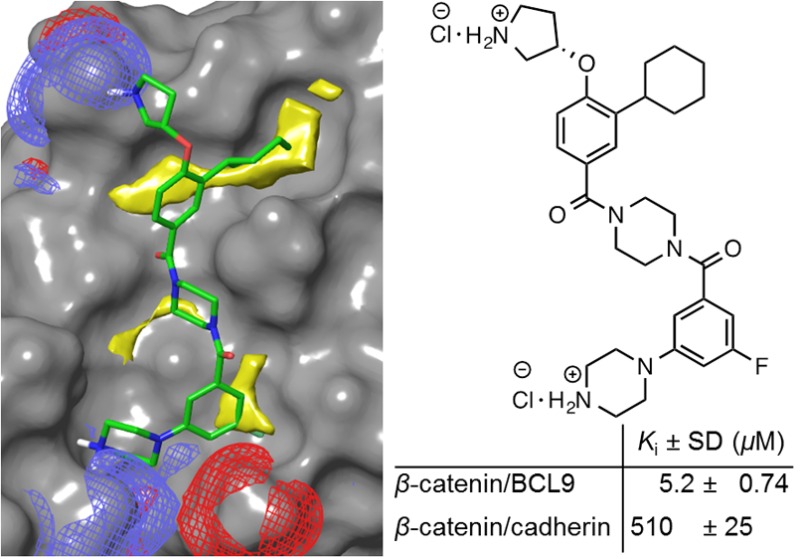
A small-molecule inhibitor with a 1,4-dibenzoylpiperazine scaffold was designed to match the critical binding elements in the β-catenin/B-cell lymphoma 9 (BCL9) protein–protein interaction interface. Inhibitor optimization led to a potent inhibitor that can disrupt the β-catenin/BCL9 interaction and exhibit 98-fold selectivity over the β-catenin/cadherin interaction. The binding mode of new inhibitors was characterized by structure–activity relationships and site-directed mutagenesis studies. Cell-based studies demonstrated that this series of inhibitors can selectively suppress canonical Wnt signaling and inhibit growth of Wnt/β-catenin-dependent cancer cells.
Keywords: Wnt signaling, β-Catenin, B-cell lymphoma 9, protein−protein interactions, inhibitor, selectivity
The Wnt/β-catenin signaling pathway is instrumental in embryonic development, stem cell maintenance, and tissue homeostasis.1 The protein β-catenin is the central hub of this pathway. In the absence of Wnt signaling, most intracellular β-catenin is complexed with cadherin to stabilize cell–cell junctions. Free cytosolic β-catenin enters a destruction complex that consists of adenomatous polyposis coli (APC), Axin, glycogen synthase kinase 3β, casein kinase 1α, and protein phosphatase 2A. This destruction complex phosphorylates β-catenin, which further leads to β-catenin ubiquitination and degradation. Wnt signaling is initiated extracellularly by Wnt proteins that bind to the membrane receptors Frizzled and LRP 5/6.2 Upon binding, the β-catenin destruction complex is recruited to the cell membrane where the ubiquitination of β-catenin is blocked. β-Catenin is then translocated to the cell nucleus, displaces the repressor protein Groucho/TLE, and binds T-cell factor (Tcf)/lymphoid enhancer-binding factor (Lef), B-cell lymphoma 9 (BCL9)/BCL9-like (B9L), CREB (cAMP response element-binding protein)-binding protein (CBP)/p300, etc., to activate transcription of Wnt target genes.3,4
Inactivating mutations of the β-catenin destruction complex, the epigenetic mutations of Wnt antagonist genes, or the autocrine/paracrine activation of Wnt proteins, Frizzled, and LRP5/6 allows β-catenin to escape degradation, translocate to the cell nucleus, bind with BCL9/B9L, etc., and initiate transcription of Wnt target genes.4,5 The unintended hyperactivation of canonical Wnt signaling causes many cancers and fibroses including triple negative breast cancer and idiopathic pulmonary fibrosis. Furthermore, the initiation, metastasis, and recurrence of cancers are believed driven by a subpopulation of cancer cells, called cancer stem cells.6−8 These cancer stem cells exhibit two distinct characteristics: the ability to self-renew and the ability to regenerate the phenotypic heterogeneity of the parental tumor. Canonical Wnt signaling is highly activated in cancer stem cells.9,10 Significant efforts have been made to discover inhibitors for the key effectors of the canonical Wnt signaling pathway.5 Small-molecule inhibitors of the enzymes Porcupine11−13 and tankyrase11,14 have been discovered. Unfortunately, both enzymes act upstream, preventing them from being effective targets to combat cancers involving mutations in the β-catenin destruction complex.15 Inhibitors for the effectors downstream of the destruction complex are more desired.16−18 These efforts have resulted in a β-catenin crystal structure in complex with an Axin-derived stapled peptide.17
An alternate downstream target is the β-catenin/BCL9 protein–protein interaction (PPI). BCL9 and its paralogue B9L connect β-catenin with the chromatin remodeler Pygopus (Pygo).19,20 BCL9 and B9L were observed highly overexpressed in Wnt-dependent cancer cells (but not in normal tissues).19,21,23,24 The individual knockdown of BCL9 and B9L19,21−25 or the use of dominant negative BCL9 or B9L21,23 inhibited the activity of canonical Wnt signaling in cell-based reporter assays, downregulated transcription of Wnt target genes, inhibited cancer cell migration, and induced an epithelial-like phenotype in colon cancer and multiple myeloma cells. The knockdown of BCL9 also enhanced survival of xenograft mouse models of cancer by reducing cancer load, metastasis, and host angiogenesis24 and inhibited invasion of breast cancer in in vivo ductal carcinoma in situ progression models.25 The conditional ablation of BCL9 and B9L in mice reduced Wnt responsive genes that promote cancer stem cells and epithelial-to-mesenchymal transition.19,20,26 BCL9/B9L-ablation in murine oncogenic intestinal organoids provoked differentiation and abrogated tumorigenicity.26 However, the conditional gene ablation of BCL9 and B9L showed normal cell lineage commitment and proliferation,19,20 indicating the selective disruption of the β-catenin/BCL9 PPI might not affect normal tissue homeostasis. Two other merits that make the β-catenin/BCL9 PPI appealing for inhibitor design are (1) the binding site of β-catenin for BCL9 partially overlaps with that for cadherin but has no overlap with that for Axin and APC; and (2) the interaction between β-catenin and BCL9 or B9L is relatively weak with a dissociation constant (Kd) of 0.465 μM.27−29
Previous attempts to inhibit the β-catenin/BCL9 PPI have resulted in little to mild success; with one small-molecule inhibitor, carnosic acid, being identified from high-throughput screening30 and three different stapled α-helices being designed.31,32 The stapled α-helix, SAH-BCL9, was able to disrupt the β-catenin/BCL9 PPI and suppress tumor growth, angiogenesis, invasion, and metastasis in mouse xenograft models.32 MicroRNA miR-30–50p was reported to downregulate BCL9 and Wnt transcriptional activity and reduce tumor burden and metastatic potential of multiple myeloma in vivo.33
Recently, we designed a generic scaffold, 4′-fluoro-N-phenyl-[1,1′-biphenyl]-3-carboxamide, that itself can mimic the binding mode of the side chains of hydrophobic projecting hot spots at positions i, i + 3, and i + 7 of an α-helix. The decoration of this scaffold offered a small-molecule inhibitor, 1 in Figure 2, that can disrupt the β-catenin/BCL9 PPI and exhibit selectivity for β-catenin/BCL9 over β-catenin/cadherin PPIs.34 This study indicates the binding mode mimicry for hydrophobic side chains of α-helical hot spots in PPI structures can provide a new starting point to design small-molecule inhibitors for α-helix-mediated PPIs. To validate this strategy, in this study the structural features for PPI inhibition, which were explored by 1,34 were employed to design a second scaffold that has better drug-like properties.
Figure 2.

Design of new β-catenin/BCL9 inhibitors. (A) Chemical structure and the AlphaScreen Ki values of 1. (B) Chemical structures of 2–9. (C) AutoDock result of 3 with β-catenin (PDB id, 2GL7(27)). A stick model of this docking result is shown in Figure S2. (D) AlphaScreen Ki values of 2–9 and carnosic acid. Each set of data was expressed as mean ± standard deviation (n = 3). Details are in Figure S3.
Mutational and crystallographic studies have identified that residues L366, I369, and L373 of BCL9 are the projecting hot spots that form close contact with a hot spot pocket that contains L156, L159, and L178 of β-catenin.23,27,28 SiteMap35 was used to calculate the three-dimensional energy maps around the BCL9 L366/I369/L373 binding site and highlight favorable sites for a specific functional group. The molecular interaction fields (MIFs) for hydrophobic interactions were mainly from the upper pocket that is lined with the side chains of A152, L156, L159, L160, V167, K170, A171, and M174 of β-catenin, as shown in Figure 1A. SiteMap also identified additional hydrophobic MIFs generated from the side chains of L148, A149, A152, M174, L178, and K181 in the bottom pocket. The hydrophobic side chains of these residues were extracted as critical binding elements for inhibitor design. The SiteMap MIFs for H-bond acceptors were determined by the side chain carboxylic oxygen atoms of β-catenin D145, E155, D162, and S184 (Figure 1B). The SiteMap MIFs for H-bond donors were from the side chain NH3 of β-catenin K181. These functional groups were also extracted as critical binding elements for H-bond and charge–charge interactions.
Figure 1.
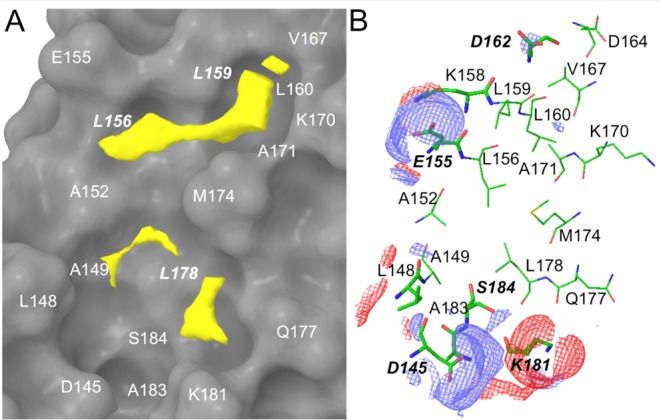
SiteMap results at the β-catenin/BCL9 PPI interface (PDB id, 2GL727). (A) Hydrophobic map. β-Catenin is shown as a surface model. The threshold for the hydrophobic contour in yellow was set to −0.5 kcal/mol. A stick model for the hydrophobic SiteMap is shown in Supporting Information Figure S1. (B) H-bond map. The threshold for the H-bond donor (red) and acceptor (blue) contours was set to −8 kcal/mol. The β-catenin residues are colored green.
Starting from fragment 2 from our previous study,34 compound 3 in Figure 2B was designed to meet the derived critical binding elements. The AutoDock model of 3 with β-catenin is shown in Figure 2C. The 4-fluorobiphenyl substructure was designed to match the hydrophobic critical binding elements in the upper pocket. The phenyl group of the 3-fluoro-5-(piperazin-1-yl) benzamide substructure was designed to match the hydrophobic critical binding elements in the lower pocket, as shown in Figure 1A. The positively charged pyrrolidin-3-yl and piperazin-1-yl groups aimed to form salt bridge interactions with β-catenin E155 and D145, respectively. After the synthesis (Scheme S1), the AlphaScreen assay showed that 3 can disrupt the β-catenin/BCL9 PPI with a Ki value of 19 ± 6.4 μM. The introduction of a hydrophilic pyridine nitrogen atom to 3 afforded 4, which was designed to evaluate the importance of hydrophobic interactions in the upper pocket. The synthetic route of 4 is shown in Scheme S2. The AlphaScreen Ki value of 4 is 4-fold higher than 3 (Figure 2D), indicating the importance of hydrophobicity of this pocket. Compound 5 with a shorter ethylamine side chain exhibited no obvious inhibition at the highest concentration tested (500 μM). It is likely the combination of the pyridine and the ethylamine is deleterious to potency. The pyridine is too hydrophilic for the hydrophobic β-catenin/BCL9 pocket, while the flexibility of the ethylamine results in an entropic penalty. Compounds 6–9 were designed to further explore the bottom pocket. The synthetic route for 6–9 is shown in Scheme S3. A comparison of the Ki values of 3 and 6–8 indicated that the piperazine substructure worked best for this series of inhibitors. The piperazine nitrogen atom attaching to the phenyl ring does not provide additional contribution to the inhibitory potency, as the Ki value of 9 is similar to that of 3. The different Ki values between 5 and 6 again suggested the importance of hydrophobic interactions in the upper pocket for inhibitor binding.
In lieu of the structure–activity relationships (SARs) of 2–9 and the proposed binding mode of 3 with β-catenin in Figure 2C, a second series of potential inhibitors was designed to explore the BCL9 L366 binding pocket, as shown in Figure 3A. Compound 10 was a fairly conservative alteration from 3. One reason to synthesize this compound was because the 3,4-difluoro substituted derivative exhibited the best activity for the previous scaffold.34 The synthetic route for 10 is shown in Scheme S4. Unfortunately, the AlphaScreen assay showed the activity of 10 was approximately 3-fold worse than 3. Since the upper hydrophobic pocket was evolved to accept aliphatic L366, the modification of aromatic to aliphatic ring was envisioned. The choice to use a ring over a chain (to more directly mimic leucine) was made in an effort to keep the number of rotatable bonds and the entropic penalty of binding low. Compound 11 with an unsubstituted cyclohexyl ring was designed and synthesized. The Ki value of this compound is 5.2 ± 0.74 μM and comparable with that of carnosic acid. The introduction of a methyl group to the cyclohexyl group decreased the activity. The replacement of the methyl group with a trifluoromethyl group afforded 13 with better potency. The bulkier 4-ethyl, 4-dimethyl, and 4-difluoro derivatives all were worse than 11 (Figure 3). Compound 17 with a cyclopentyl substitution and 18 with a methyl group exhibited lower activities than 11, suggesting the importance of van der Waals contacts in the BCL9 L366 binding pocket. The synthetic routes for 11–14 and 15–18 are shown in Schemes 1 and S4, respectively. A further optimization was centered on evaluation of the R2 group as shown in Figure 3D. The synthetic route for 19–22 is shown in Scheme S5. The results of the AlphaScreen competitive inhibition assay indicated that the fluorine atom was the best substituent for this position.
Figure 3.

Optimization of new β-Catenin/BCL9 inhibitors. Each set of data was expressed as mean ± standard deviation (n = 3). (A) Chemical structures of 10–18. (B) AutoDock result of 11 with β-catenin (PDB id, 2GL7(27)). A stick model of this docking result is shown in Figure S4. (C) AlphaScreen Ki values of 10–18. Details are in Figure S5. (D) Chemical structures and AlphaScreen Ki values of 19–22. (E) In-parallel AlphaScreen competitive inhibition assay of 11 to disrupt wild-type β-catenin/wild-type BCL9 and mutant β-catenin/wild-type BCL9 PPIs. Details are in Figure S7.
Scheme 1.

Site-directed mutagenesis studies were performed to evaluate the binding mode of new β-catenin/BCL9 inhibitors. Previous crystallographic analysis indicated that D145 and E155 of β-catenin did not interfere with the β-catenin/BCL9 PPI.34 Three β-catenin mutants, D145A, E155A, and D145A/E155A, were made. Native gel electrophoresis, thermal shift, and CD experiments confirmed the homogeneity, the thermal stability, and the secondary structure integrity of the purified proteins. The AlphaScreen competitive binding assays and the fluorescence polarization binding assays demonstrated that D145A, E155A, and D145A/E155A of β-catenin had the same Kd values as wild-type β-catenin when binding with wild-type BCL9, as shown in Figure S6. The AlphaScreen competitive inhibition assay was performed to evaluate the roles of D145 and E155 in inhibitor binding. As shown in Figure 3D, the Ki values of 11 for β-catenin D145A/BCL9 and β-catenin E155A/BCL9 PPIs were 6–8-fold higher than that for the wild-type β-catenin/BCL9 PPI. Further, the Ki value of 11 for the β-catenin D145A/E155A double mutant/BCL9 PPI was 53 ± 4.2 μM and higher than that for the β-catenin D145A/BCL9 or β-catenin E155A/BCL9 PPI, indicating that the carboxylate side chains of D145 and E155 of β-catenin were important for the inhibitory potency of 11. The AlphaScreen selectivity assay was used to quantify inhibitor selectivity. As shown in Figure 4A, compound 11 exhibited 98-fold selectivity for β-catenin/BCL9 over β-catenin/cadherin PPIs.
Figure 4.

Inhibitor selectivity of 11 for β-catenin/BCL9 over β-catenin/cadherin PPIs and cell-based characterization of 11 as a new β-catenin/BCL9 inhibitor. (A) In-parallel AlphaScreen selectivity assay results for 11. Each set of data is expressed as mean ± standard deviation (n = 3). (B) TOPFlash and FOPFlash luciferase reporter assay results of 11 using pcDNA3.1−β-catenin transfected HEK293 cells. The data are expressed as mean ± standard deviation (n = 2). (C) MTs assay to monitor the inhibitory effects of 3, 11, and 13 on growth of SW480, HCT116, A549, and HEK293 cells. n.d., not determined.
The Wnt-responsive luciferase reporter assays were performed with pcDNA3.1−β-catenin transfected human embryonic kidney 293 (HEK293) cells for 11, 13, and carnosic acid. As shown in Figures 4B and S8, compounds 11 and 13 suppressed the TOPFlash (with wild-type Tcf binding sites) luciferase activity in a dose-dependent manner. This compound did not affect the FOPFlash (with mutant Tcf binding sites) luciferase activity even at 100 μM. MTs cell viability assays were performed to assess the inhibitory effects of 3, 11, and 13 on growth of colorectal cancer cell lines SW480 and HCT116 that have hyperactivated Wnt signaling. Compound 11 inhibited growth of Wnt-activated cancer cells with the IC50 values of 22 ± 4.0 and 26 ± 6.6 μM for SW480 and HCT116 cells, respectively (Figure 4C). Compounds 11 and 13 exhibited >10-fold selectivity for Wnt signaling-activated cancer cells over Wnt signaling-latent cancer cells, such as lung adenocarcinoma A549 cells. These two compounds also exhibited 3–4-fold selectivity for Wnt signaling-activated cancer cells over HEK293 cells.
The β-catenin/BCL9 PPI, a key downstream effector of canonical Wnt signaling, represents an appealing therapeutic target for treatment of cancer and fibrosis and eradication of cancer stem cells. In this study, the critical binding elements at the BCL9 binding site for hydrophobic, H-bond, and charge–charge interactions were extracted from the crystal structure of the β-catenin/BCL9 PPI (PDB ids, 2GL7(27) and 3SL9(30)). A small-molecule inhibitor with a novel scaffold was designed to match the proposed critical binding elements. A further structural optimization resulted in 11 and 13. Compound 11 exhibited a Ki value of 5.2 ± 0.74 μM for disruption of the β-catenin/BCL9 PPI and 98-fold selectivity for β-catenin/BCL9 over β-catenin/cadherin PPIs. The SARs and site-directed mutagenesis results are in agreement with the proposed binding mode of this series of inhibitors. The cell-based studies demonstrated that 11 and 13 can suppress transactivation of canonical Wnt signaling and inhibit growth of cancer cells with hyperactivated Wnt signaling. Compound 11 is similar to 1 from our previous study34 in that both are designed to form salt bridges with E155 and D145 of β-catenin, and both have hydrophobic moieties intended to interact with the hydrophobic pockets of β-catenin near L156 and L159 (Figure 1A). However, compared to 1, compound 11 is more hydrophilic giving it better drug-like properties. In addition, the piperazine moiety in the middle of the molecule can be functionalized, allowing future optimizations to access more chemical space.
Acknowledgments
We thank the Center for High-Performance Computing at the University of Utah for computer time.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00284.
Figures S1–S8, Schemes S1–S5, experimental details, HPLC conditions, HPLC tracers, and NMR spectra for 3–22 (PDF)
This work was supported by the Department of Defense CDMRP BCRP breakthrough award W81XWH-14-1-0083 (to H.J).
The authors declare no competing financial interest.
Supplementary Material
References
- Clevers H.; Loh K. M.; Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. 10.1126/science.1248012. [DOI] [PubMed] [Google Scholar]
- Zeng X.; Huang H.; Tamai K.; Zhang X.; Harada Y.; Yokota C.; Almeida K.; Wang J.; Doble B.; Woodgett J.; Wynshaw-Boris A.; Hsieh J.-C.; He X. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development 2008, 135, 367–375. 10.1242/dev.013540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann C.; Hausmann G.; Basler K. β-Catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 2009, 10, 276–286. 10.1038/nrm2654. [DOI] [PubMed] [Google Scholar]
- Clevers H.; Nusse R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Anastas J. N.; Moon R. T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- Singh S. K.; Hawkins C.; Clarke I. D.; Squire J. A.; Bayani J.; Hide T.; Henkelman R. M.; Cusimano M. D.; Dirks P. B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- O’Brien C. A.; Pollett A.; Gallinger S.; Dick J. E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani L.; Lombardi D. G.; Pilozzi E.; Biffoni M.; Todaro M.; Peschle C.; De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- Malanchi I.; Peinado H.; Kassen D.; Hussenet T.; Metzger D.; Chambon P.; Huber M.; Hohl D.; Cano A.; Birchmeier W.; Huelsken J. Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature 2008, 452, 650–653. 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]
- Yeung J.; Esposito M. T.; Gandillet A.; Zeisig B. B.; Griessinger E.; Bonnet D.; So C. W. E. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010, 18, 606–618. 10.1016/j.ccr.2010.10.032. [DOI] [PubMed] [Google Scholar]
- Chen B.; Dodge M. E.; Tang W.; Lu J.; Ma Z.; Fan C.-W.; Wei S.; Hao W.; Kilgore J.; Williams N. S.; Roth M. G.; Amatruda J. F.; Chen C.; Lum L. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Moon J.; Dodge M. E.; Pan X.; Zhang L.; Hanson J. M.; Tuladhar R.; Ma Z.; Shi H.; Williams N. S.; Amatruda J. F.; Carroll T. J.; Lum L.; Chen C. The development of highly potent inhibitors for porcupine. J. Med. Chem. 2013, 56, 2700–2704. 10.1021/jm400159c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Pan S.; Hsieh M. H.; Ng N.; Sun F.; Wang T.; Kasibhatla S.; Schuller A. G.; Li A. G.; Cheng D.; Li J.; Tompkins C.; Pferdekamper A.; Steffy A.; Cheng J.; Kowal C.; Phung V.; Guo G.; Wang Y.; Graham M. P.; Flynn S.; Brenner J. C.; Li C.; Villarroel M. C.; Schultz P. G.; Wu X.; McNamara P.; Sellers W. R.; Petruzzelli L.; Boral A. L.; Seidel H. M.; McLaughlin M. E.; Che J.; Carey T. E.; Vanasse G.; Harris J. L. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20224–20229. 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.-M.; Mishina Y. M.; Liu S.; Cheung A.; Stegmeier F.; Michaud G. A.; Charlat O.; Wiellette E.; Zhang Y.; Wiessner S.; Hild M.; Shi X.; Wilson C. J.; Mickanin C.; Myer V.; Fazal A.; Tomlinson R.; Serluca F.; Shao W.; Cheng H.; Shultz M.; Rau C.; Schirle M.; Schlegl J.; Ghidelli S.; Fawell S.; Lu C.; Curtis D.; Kirschner M. W.; Lengauer C.; Finan P. M.; Tallarico J. A.; Bouwmeester T.; Porter J. A.; Bauer A.; Cong F. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- Dancey J. E.; Bedard P. L.; Onetto N.; Hudson T. J. The genetic basis for cancer treatment decisions. Cell 2012, 148, 409–420. 10.1016/j.cell.2012.01.014. [DOI] [PubMed] [Google Scholar]
- Emami K. H.; Nguyen C.; Ma H.; Kim D. H.; Jeong K. W.; Eguchi M.; Moon R. T.; Teo J.-L.; Kim H. Y.; Moon S. H.; Ha J. R.; Kahn M. A small molecule inhibitor of β-catenin/CREB-binding protein transcription. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12682–12687. 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann T. N.; Yeh J. T.-H.; Bowman B. R.; Chu Q.; Moellering R. E.; Verdine G. L. Inhibition of oncogenic Wnt signaling through direct targeting of β-catenin. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 17942–17947. 10.1073/pnas.1208396109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Zhang M.; Burton S. D.; Katsakhyan L. N.; Ji H. Targeting the Tcf4 G13ANDE17 binding site to selectively disrupt β-catenin/T-cell factor protein-protein interactions. ACS Chem. Biol. 2014, 9, 193–201. 10.1021/cb400795x. [DOI] [PubMed] [Google Scholar]
- Brembeck F. H.; Wiese M.; Zatula N.; Grigoryan T.; Dai Y.; Fritzmann J.; Birchmeier W. BCL9–2 promotes early stages of intestinal tumor progression. Gastroenterology 2011, 141, 1359–1370. 10.1053/j.gastro.2011.06.039. [DOI] [PubMed] [Google Scholar]
- Deka J.; Wiedemann N.; Anderle P.; Murphy-Seiler F.; Bultinck J.; Eyckerman S.; Stehle J.-C.; André S.; Vilain N.; Zilian O.; Robine S.; Delorenzi M.; Basler K.; Aguet M. Bcl9/Bcl9l are critical for Wnt-mediated regulation of stem cell traits in colon epithelium and adenocarcinomas. Cancer Res. 2010, 70, 6619–6628. 10.1158/0008-5472.CAN-10-0148. [DOI] [PubMed] [Google Scholar]
- Adachi S.; Jigami T.; Yasui T.; Nakano T.; Ohwada S.; Omori Y.; Sugano S.; Ohkawara B.; Shibuya H.; Nakamura T.; Akiyama T. Role of a BCL9-related β-catenin-binding protein, B9L, in tumorigenesis induced by aberrant activation of Wnt signaling. Cancer Res. 2004, 64, 8496–8501. 10.1158/0008-5472.CAN-04-2254. [DOI] [PubMed] [Google Scholar]
- Brembeck F. H.; Schwarz-Romond T.; Bakkers J.; Wilhelm S.; Hammerschmidt M.; Birchmeier W. Essential role of BCL9–2 in the switch between β-catenin’s adhesive and transcriptional functions. Genes Dev. 2004, 18, 2225–2230. 10.1101/gad.317604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche M.; Worm J.; Bienz M. The function of BCL9 in Wnt/β-catenin signaling and colorectal cancer cells. BMC Cancer 2008, 8, 199. 10.1186/1471-2407-8-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani M.; Carrasco D. E.; Zhang Y.; Takada K.; Gatt M. E.; Dutta-Simmons J.; Ikeda H.; Diaz-Griffero F.; Pena-Cruz V.; Bertagnolli M.; Myeroff L. L.; Markowitz S. D.; Anderson K. C.; Carrasco D. R. BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res. 2009, 69, 7577–7586. 10.1158/0008-5472.CAN-09-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsarraj H. S.; Hong Y.; Valdez K. E.; Michaels W.; Hook M.; Smith W. P.; Chien J.; Herschkowitz J. I.; Troester M. A.; Beck M.; Inciardi M.; Gatewood J.; May L.; Cusick T.; McGinness M.; Ricci L.; Fan F.; Tawfik O.; Marks J. R.; Knapp J. R.; Yeh H.-W.; Thomas P.; Carrasco D. R.; Fields T. A.; Godwin A. K.; Behbod F. Expression profiling of in vivo ductal carcinoma in situ progression models identified B cell lymphoma-9 as a molecular driver of breast cancer invasion. Breast Cancer Res. 2015, 17, 128. 10.1186/s13058-015-0630-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moor A. E.; Anderle P.; Cantù C.; Rodriguez P.; Wiedemann N.; Baruthio F.; Deka J.; André S.; Valenta T.; Moor M. B.; Győrffy B.; Barras D.; Delorenzi M.; Basler K.; Aguet M. BCL9/9L-β-catenin signaling is associated with poor outcome in colorectal cancer. EBioMedicine 2015, 2, 1932–1943. 10.1016/j.ebiom.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampietro J.; Dahlberg C. L.; Cho U. S.; Hinds T. R.; Kimelman D.; Xu W. Crystal structure of a β-catenin/BCL9/Tcf4 complex. Mol. Cell 2006, 24, 293–300. 10.1016/j.molcel.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Kawamoto S. A.; Thompson A. D.; Coleska A.; Nikolovska-Coleska Z.; Yi H.; Wang S. Analysis of the interaction of BCL9 with beta-catenin and development of fluorescence polarization and surface plasmon resonance binding assays for this interaction. Biochemistry 2009, 48, 9534–9541. 10.1021/bi900770z. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Wisniewski J. A.; Ji H. AlphaScreen selectivity assay for β-catenin/B-cell lymphoma 9 inhibitors. Anal. Biochem. 2015, 469, 43–53. 10.1016/j.ab.2014.09.018. [DOI] [PubMed] [Google Scholar]
- de la Roche M.; Rutherford T. J.; Gupta D.; Veprintsev D. B.; Saxty B.; Freund S. M.; Bienz M. An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat. Commun. 2012, 3, 680. 10.1038/ncomms1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto S. A.; Coleska A.; Ran X.; Yi H.; Yang C.-Y.; Wang S. Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein–protein interaction. J. Med. Chem. 2012, 55, 1137–1146. 10.1021/jm201125d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K.; Zhu D.; Bird G. H.; Sukhdeo K.; Zhao J.-J.; Mani M.; Lemieux M.; Carrasco D. E.; Ryan J.; Horst D.; Fulciniti M.; Munshi N. C.; Xu W.; Kung A. L.; Shivdasani R. A.; Walensky L. D.; Carrasco D. R. Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med. 2012, 4, 148ra117. 10.1126/scitranslmed.3003808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.-J.; Lin J.; Zhu D.; Wang X.; Brooks D.; Chen M.; Chu Z.-B.; Takada K.; Ciccarelli B.; Admin S.; Tao J.; Tai Y.-T.; Treon S.; Pinkus G.; Kuo W. P.; Hideshima T.; Bouxsein M.; Munshi N.; Anderson K.; Carrasco R. miR-30–5p functions as a tumor suppressor and novel therapeutic tool by targeting the oncogenic Wnt/β-catenin/BCL9 pathway. Cancer Res. 2014, 74, 1801–1813. 10.1158/0008-5472.CAN-13-3311-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggard L. R.; Zhang Y.; Zhang M.; Panic V.; Wisniewski J. A.; Ji H. Rational design of selective small-molecule inhibitors for catenin/B-cell lymphoma 9 protein–protein interactions. J. Am. Chem. Soc. 2015, 137, 12249–12260. 10.1021/jacs.5b04988. [DOI] [PubMed] [Google Scholar]
- Halgren T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.