

Abstract

In our efforts to develop second generation DPP-4 inhibitors, we endeavored to identify distinct structures with long-acting (once weekly) potential. Taking advantage of X-ray cocrystal structures of sitagliptin and other DPP-4 inhibitors, such as alogliptin and linagliptin bound to DPP-4, and aided by molecular modeling, we designed several series of heterocyclic compounds as initial targets. During their synthesis, an unexpected chemical transformation provided a novel tricyclic scaffold that was beyond our original design. Capitalizing on this serendipitous discovery, we have elaborated this scaffold into a very potent and selective DPP-4 inhibitor lead series, as highlighted by compound 17c.
Keywords: Diabetes, dipeptidyl peptidase IV (DPP-4), inhibitor, tricyclic heterocycles, crystal structure, OGTT
Diabetes mellitus remains a challenging health problem that affects 415 million adults worldwide as of 2015, with an alarming growth predicted over the next quarter of century.1 Type 2 diabetes (T2DM), a chronic disease characterized by elevated blood sugar levels and insulin resistance, is the most prevalent form. Dipeptidyl peptidase IV (DPP-4), a serine protease in blood, regulates glucose metabolism through degradation of the incretins glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) that are secreted following a meal. Inhibition of DPP-4 thus increases the levels of GLP-1 and GIP, which inhibits glucagon release and in turn stimulates insulin secretion thereby reducing blood glucose levels.2 Indeed, the usefulness of DPP-4 inhibitors for treatment of T2DM has been demonstrated by the successful launch of JANUVIA (sitagliptin, 1, Figure 1), the first DPP-4 inhibitor approved by the U.S. Food and Drug Administration.3 Systematic structure–activity relationship (SAR) studies of conformationally restricted analogues of sitagliptin culminated in discovery, development, and launch of MARIZEV (omarigliptin, 2) as a once-weekly treatment for T2DM.4 Over the past decade, several other DPP-4 inhibitors with distinct structures have also been developed, such as 3 (alogliptin)5 and 4 (linagliptin).6
Figure 1.

Representative DPP-4 inhibitors.
In our continued efforts to develop second generation DPP-4 inhibitors, we endeavored to identify distinct structures with long-acting (once weekly, QW) potential. Taking advantage of available X-ray cocrystal structures of sitagliptin and other DPP-4 inhibitors under late-stage development, such as alogliptin and linagliptin bound to DPP-4, and assisted by molecular modeling, we designed several series of heterocyclic compounds as initial synthetic targets (Figure 2).7 Over the course of the synthesis, an unexpected chemical transformation provided a novel tricyclic scaffold. Capitalizing on this serendipitous discovery, we elaborated the scaffold into a very potent and selective DPP-4 inhibitor lead series. In this communication, we report our preliminary findings and results of initial SAR exploration.
Figure 2.

Design of cyclic guanine scaffold.
As depicted in Scheme 1, we planned to construct the tricyclic guanine scaffold starting from the readily available 2,6-dichloropurine derivative 5.8 Compound 5 was hydrolyzed selectively to give 6, which in turn was alkylated under basic conditions to afford intermediate 7. At this stage, we reasoned that substitution of a β-aminoalcohol for the chlorine should give intermediate 8, and activation of the hydroxy of 8 would result in ring closure to provide the tricyclic guanine scaffold. However, treatment of 7 with 2-aminoethanol in DMF under microwave heating conditions did not give any expected product 8 as judged by LC–MS, although the starting material was consumed. Instead, an amidine compound 9 was isolated and its structure has been verified rigorously.9
Scheme 1. Synthesis of Cyclic Guanines.

Reagents and conditions: (a) NaOH, 90%; (b) BrCH2CN, DIEA, DMF, 55%; (c) HOCH2CH2NH2, DMF, 120 °C, 65%.
Although our plan did not work out, we were curious to explore the reaction at lower temperatures using simple methylamine, as outlined in Scheme 2. Mixing compound 7 with an excess amount of methylamine as a solution in 1,4-dioxane at room temperature, or even at 0 °C, brought about a fast reaction (in less than 10 min) to give amidine 10 as the major product along with some lactam 11. This result was quite surprising because amidine formation from a nitrile usually occurs under harsh conditions or in the presence of catalysts.10 What was more striking was identification of lactam 11 as a minor product, presumably through hydrolysis of 10, hinting that the amidine moiety might be easily converted to the more stable lactam. To our great pleasure, a mixture of compounds 10 and 11 was refluxed in 6 N HCl to give compound 12, with migration of the N-PMB group from N3 to N1.11 Lactams 11 and 12 can be differentiated by LC–MS and TLC, but their difference in protection regiochemistry is inconsequential because the PMB group was removed by TFA in the next step to give tricyclic core 13.
Scheme 2. Identification of a Novel Scaffold and Elaboration to Tricyclic DPP-4 Inhibitors.

Reagents and conditions: (a) MeNH2 or EtNH2, 1,4-dioxane, room temperature; (b) 6 N HCl, reflux, ∼100% over 2 steps; (c) TFA, ∼90%; (d) Br2, 50–60%; (e) R1Br, NaH, 40–60%; (f) (R)-3-(Boc-amino)piperidine; (g) TFA, DCM, ∼20–40% over two steps.
With identification of the compelling novel tricyclic scaffold of compound 13, we endeavored to scale up this structure and elaborate it to generate tricyclic targets as DPP-4 inhibitors. The stability of the tricyclic core under the above harsh acidic hydrolysis conditions further augmented our confidence in its usefulness. After bromination at the C2 position of intermediate 13, N-alkylation of the imidazole moiety of 14 gave a mixture of substituted regioisomers 15aa–15ea and 15ab–15eb. Finally, displacement of the C2-bromide of the isolated N1-alkylated compound 15ca with the (R)-3-aminopiperidine moiety, an optimized structural feature in both alogliptin and linagliptin, gave intermediate 16c, and removal of N-Boc group furnished target 17c. Analogues 17a, 17b, 17d, and 17e were prepared from the corresponding mixtures of regioisomers 15 using the above two-step procedures. Ethyl substituted analogues 25a–25c were prepared accordingly using ethylamine instead of methylamine in the first step of Scheme 2.
Analogues were evaluated for their in vitro inhibition of human DPP-4, and the results are summarized in Table 1. Analogue 17a (IC50 = 123 nM) bearing a but-2-ynyl group on N1 is much less potent than linagliptin (4, IC50 = 0.10 nM), which has an extra quinazoline substituent at N1. Replacement of the buty-2-nyl group with a 2-cyanobenzyl moiety improved potency significantly (17b, IC50 = 10.6 nM), comparable to that of alogliptin (3, IC50 = 7.6 nM). Further improvement was achieved with an additional 5-fluoro substituent on the benzyl ring (17c, IC50 = 1.7 nM). Electron withdrawing replacements of the cyano group (17d and 17e) decreased potency. N5-Ethyl substituted analogues are similarly potent (25b vs 17d) or less potent (25a vs 17c and 25c vs 17e) than the corresponding N-methyl substituted counterparts. The most active analogues 17b, 17c, 17d, 25a, and 25b (IC50 < 100 nM) were also counterscreened against other related enzymes12 such as QPP,13 DPP8,14 DPP9,15 and FAP.16 All analogues except 25a showed no activity against QPP, DPP-8, and DPP-9 (>100 μM, data not reported in Table 1) and great selectivity against FAP. Compound 25a showed no activity against QPP but modest activity against DPP-8 and DPP-9 (IC50 = 21163 and 15828 nM, respectively).
Table 1. In Vitro Biological Dataa.
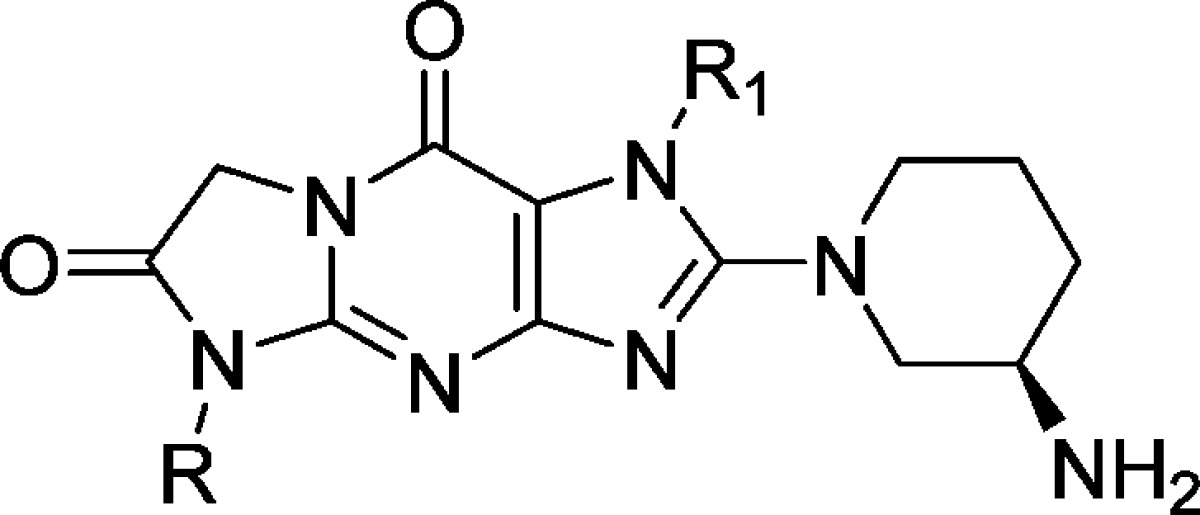
Two internal standard compounds were included in all the assay runs to track the robustness of the assays (n = 3), and data were within 3-fold of the reported values. Standard deviation was within 10%. Literature compounds 3 and 4 were also tested for comparison.
The most potent analogue 17c in this new series of DPP-4 inhibitors was further characterized. It showed no significant CYP450 inhibition (>50 μM for CYP3A4, 2C8, 2C9, and 2D6) or time-dependent CYP3A4 inhibition. The compound displayed weak activity in the MK-499 displacement binding assay for hERG activity with an IC50 of 40 μM. It had no PXR inhibition (IC50 > 30 μM). Compound 17c also exhibited low hepatocyte clearance across species (human, rat, cyno monkey, dog = 0.1, 0.3, <1, <1 μL min–1 106 cell–1, respectively).17
On the basis of its favorable in vitro profile, compound 17c was evaluated in lean mice for its ability to improve glucose tolerance (Figure 3). In an oral glucose tolerance test (OGTT), a single dose of compound 17c (0.3, 1, 3, and 10 mg/kg) was given orally to C57BL/6 mice 1 h prior to dextrose challenge. Compound 17c significantly reduced blood glucose excursion in a dose-dependent manner from 0.3 to 3 mg/kg with a plateau between 3 and 10 mpk. The observed reduction of glucose in AUC was consistent with the measured plasma inhibitor concentration (0.3 mg/kg, 38% reduction in glucose AUC, plasma concentration 2 nM; 1 mg/kg, 52% reduction, 10 nM; 3 mg/kg, 64% reduction, 70 nM; 10 mg/kg, 63% reduction, 235 nM). The maximum efficacy of glucose lowering in this model was similar to that achieved with alogliptin at oral dose of 3 mg/kg.
Figure 3.
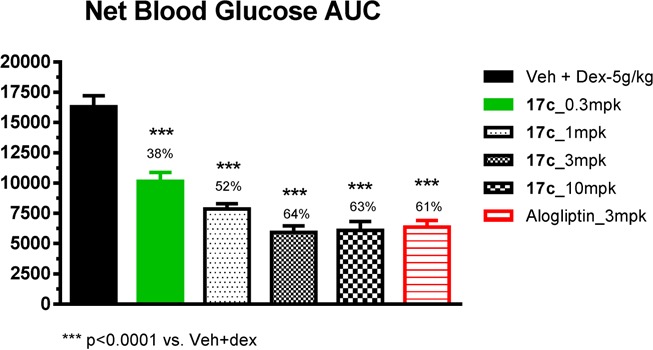
OGTT data of compound 17c.
In the corresponding ex vivo pharmacodynamic differentiating assay, compound 17c was dosed orally at 1, 3, and 10 mg/kg to C57BL/6N mice, and a time course of plasma DPP-4 inhibition was measured at 2, 6, and 24 h. As shown in Figure 4, at doses of 3 and 10 mg/kg, plasma DPP-4 activity was inhibited by >80% (uncorrected for assay dilution) at 2 h and maintained at 6 h. The effect had returned to near baseline at 24 h. The relative high ratio of inhibition at 6 h to that at 2 h (≥0.8) for all the three doses suggested long-acting potential for 17c (ratios >0.7 are typically a good predictor of extended duration of action), which is consistent with what the low in vitro clearance would indicate.
Figure 4.
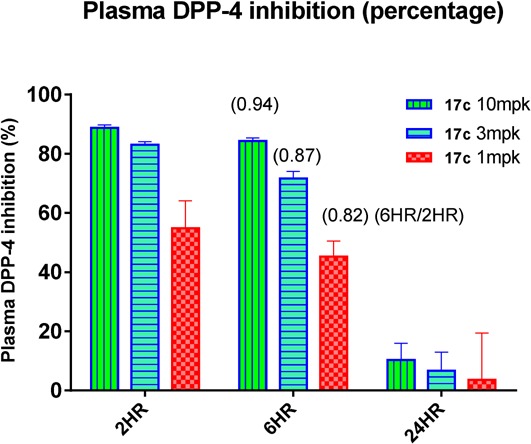
Ex vivo pharmacodynamic data of compound 17c.
As revealed in Figure 5, compound 17c (yellow) binds in the DPP-4 binding site in a very similar way to that observed for alogliptin (3, blue). The amino group binds to the conserved glutamic acids (E205 and E206), and the CN group is within hydrogen bonding distance of Arg125. The tricyclic core is stacked against Y547, forming a more extensive interaction than that observed with alogliptin. Elements of the tricyclic core also engage in hydrogen bonds to the backbone NH of Tyr631, to the backbone NH of Trp629, to the side-chain of Lys544 (through a water molecule), and to two other ordered water molecules.
Figure 5.
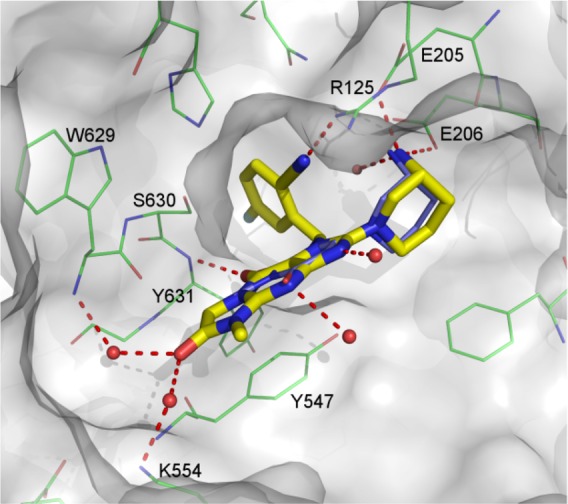
Superposition of compound 17c (yellow) and alogliptin (3, blue) in the DPP-4 active site using their cocrystal structures of DPP-4 (PDB codes 5I7U and 3G0B). The image was generated using PyMol.
In summary, we have identified a series of potent and selective DPP-4 inhibitors with a novel tricyclic core. Among them, compound 17c demonstrated robust glucose reduction efficacy after oral administration in mice, as well as potential for a long duration of action. The successful identification of this novel class of DPP-4 inhibitors was made possible by serendipity as well as careful analytical work and critical medicinal chemistry follow-up of the observed biology.
Acknowledgments
The authors would like to thank Dr. Tse-Ming Chan for NMR analytical support and helpful discussion. We also thank Mr. Wilfredo Pinto for HRMS analysis.
Glossary
ABBREVIATIONS
- T2DM
type 2 diabetes mellitus
- DPP-4
dipeptidyl peptidase 4
- DPP-8
dipeptidyl peptidase 8
- DPP-9
dipeptidyl peptidase 9
- FAP
seprase or fibroblast activation protein
- GIP
glucose-dependent insulinotropic polypeptide
- GLP-1
glucagan like peptide-1
- QPP
quiescent cell proline dipeptidase
- OGTT
oral glucose tolerance test
- QW
once-weekly
- SAR
structure–activity relationship
- PMB
para-methoxybenzyl
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00027.
Experimental details for synthetic procedures and compound characterization for compounds 6–25 as well as biological assay conditions (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- International Diabetes Federation. IDF Diabetes Atlas, 7th ed.; International Diabetes Federation: Brussels, Belgium, 2015. http://www.diabetesatlas.org. [Google Scholar]
- Flatt P. R.; Bailey C. J.; Green B. D. Dipetidyl peptidase IV (DPP IV) and related molecules in type 2 diabetes. Front. Biosci., Landmark Ed. 2008, 13, 3648–60. 10.2741/2956. [DOI] [PubMed] [Google Scholar]
- Kim D.; Kowalchick J. E.; Brockunier L. L.; Parmee E. R.; Eiermann G. J.; Fisher M. H.; He H.; Leiting B.; Lyons K.; Scapin G.; Patel S. B.; Petrov A.; Pryor K. D.; Roy R. S.; Wu J. K.; Zhang X.; Wyvratt M. J.; Zhang B. B.; Zhu L.; Thornberry N. A.; Weber A. E. Discovery of Potent and Selective Dipeptidyl Peptidase IV Inhibitors Derived from β-Aminoamides Bearing Substituted Triazolopiperazines. J. Med. Chem. 2008, 51, 589–602. 10.1021/jm070330v. [DOI] [PubMed] [Google Scholar]
- Biftu T.; Sinha-Roy R.; Chen P.; Qian X.; Feng D.; Kuethe J. T.; Scapin G.; Gao Y. D.; Yan Y.; Krueger D.; Bak A.; Eiermann G.; He J.; Cox J.; Hicks J.; Lyons K.; He H.; Salituro G.; Tong S.; Patel S.; Doss G.; Petrov A.; Wu J.; Xu S.; Sewell C.; Zhang X.; Zhang B.; Thornberry N. A.; Weber A. E. Omarigliptin (MK-3102): a novel long-acting DPP-4 inhibitor for once-weekly treakment of type-2 diabetes. J. Med. Chem. 2014, 57, 3205–3212. 10.1021/jm401992e. [DOI] [PubMed] [Google Scholar]
- Feng J.; Zhang Z.; Wallace W. B.; Stafford J. A.; Kaldor S. W.; Kassel D. B.; Navre M.; Shi L.; Skene R. J.; Asakawa T.; Takeuchi K.; Xu R.; Webb D. R.; Gwaltney S. L. Discovery of Alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV. J. Med. Chem. 2007, 50, 2297–2300. 10.1021/jm070104l. [DOI] [PubMed] [Google Scholar]
- Eckhardt M.; Langkopf E.; Mark M.; Tadayyon M.; Thomas L.; Nar H.; Pfrengle W.; Guth B.; Lotz R.; Sieger P.; Fuchs H.; Himmelsbach F. 8-(3-(R)-Aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2007, 50, 6450–6453. 10.1021/jm701280z. [DOI] [PubMed] [Google Scholar]
- Burnett D. A.; Cole D.; Domalski M.; Josien H.; Pissarnitski D. A.; Sasikumar T.; Wu W. L.; Zhao Z. U.S. Patent WO 2012078448, A1, 2012.
- Kelley J. L.; Linn J. A.; Krochmal M. P.; Selway J. W. T. 9-Benzyl-6-(dimethylamino)-(H-purines with antihinovirus activity. J. Med. Chem. 1988, 31, 2001–2004. 10.1021/jm00118a025. [DOI] [PubMed] [Google Scholar]
- The structure of compound 9 has been assigned on the basis of extensive NMR techniques including proton, carbon, HSQC, HMBC, and NOESY (see Supporting Information).
- Wang J.; Xu F.; Cai T.; Shen Q. Addition of Amines to Nitriles Catalyzed by Ytterbium Amides: An Efficient One-Step Synthesis of Monosubstituted N-Arylamidines. Org. Lett. 2008, 10, 445–448. 10.1021/ol702739c. [DOI] [PubMed] [Google Scholar]
- A similar migration of p-methoxybenzyl (PMB) has been reported.Jones M. I.; Froussios C.; Evans D. A. A short, versatile synthesis of Porphobilinogen. J. Chem. Soc., Chem. Commun. 1976, 472–473. 10.1039/c39760000472. [DOI] [Google Scholar]
- Rosenblum J. S.; Kozarich J. W. Prolyl peptidases: a serine protease subfamily with high potential for drug discovery. Curr. Opin. Chem. Biol. 2003, 7, 496–504. 10.1016/S1367-5931(03)00084-X. [DOI] [PubMed] [Google Scholar]
- Collins P. J.; McMahon G.; O’Brien P.; O’Connor B. Purification, identification and characterization of seprase from bovine serum. Int. J. Biochem. Cell Biol. 2004, 36, 2320–2333. 10.1016/j.biocel.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Abbott C. A.; Yu D. M.; Woollatt E.; Sutherland G. R.; McCaughan G. W.; Gorrell M. D. Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPP8. Eur. J. Biochem. 2000, 267, 6140–6150. 10.1046/j.1432-1327.2000.01617.x. [DOI] [PubMed] [Google Scholar]
- Olsen C.; Wagtmann N. Identification and characterization of human DPP9, a novel homologue of dipeptidyl peptidase IV. Gene 2002, 299, 185–195. 10.1016/S0378-1119(02)01059-4. [DOI] [PubMed] [Google Scholar]
- Scanlan M. J.; Raj B. K.; Calvo B.; Garin-Chea P.; Sanz-Moncasi M. P.; Healey J. H.; Old L. J.; Retting W. J. Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts ofepithelial cancers. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 5657–5661. 10.1073/pnas.91.12.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information for more details and references on CYP, PXR, hERG, and clearance assays.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.