

Abstract
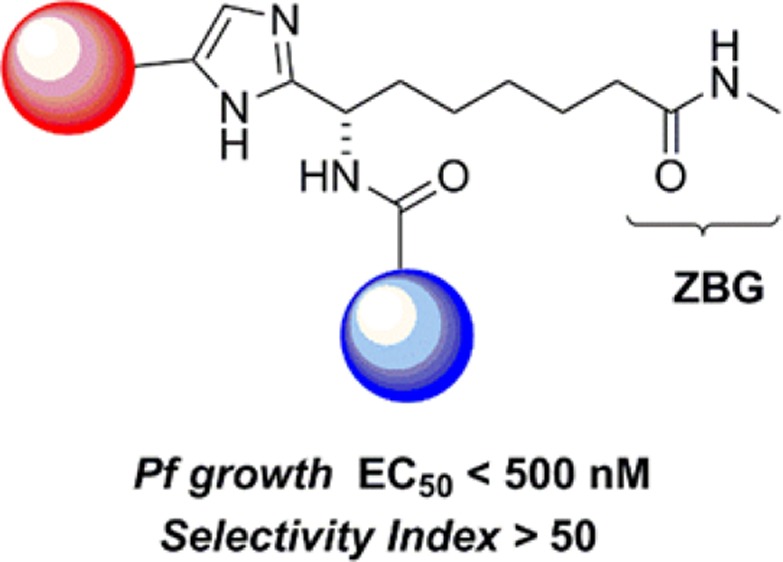
The identification of a new series of P. falciparum growth inhibitors is described. Starting from a series of known human class I HDAC inhibitors a SAR exploration based on growth inhibitory activity in parasite and human cells-based assays led to the identification of compounds with submicromolar inhibition of P. falciparum growth (EC50 < 500 nM) and good selectivity over the activity of human HDAC in cells (up to >50-fold). Inhibition of parasital HDACs as the mechanism of action of this new class of selective growth inhibitors is supported by hyperacetylation studies.
Keywords: Malaria, Plasmodium falciparum, PfHDAC1, 4-arylimidazoles
Infection with malaria parasites such as Plasmodium falciparum remains a devastating cause of death in tropical geographies with 40% of the world population at risk of acquiring the disease. There are approximately 200 million clinical cases of malaria every year leading to an estimated 600,000 deaths.1 The requirement for improved therapies to treat and to cure malaria is an evident medical and humanitarian need that is exacerbated by an alarming rise in parasite resistance to the current standard of care.2,3 Drugs that operate via novel mechanisms of action for which no innately resistant parasites are expected are therefore especially desirable.
DNA is tightly packed around histone proteins in the nucleus of eukaryotic cells with its transcription being regulated by chemical modifications to the nucleosomal histone proteins themselves. Histone deacetylases (HDACs) are zinc-dependent enzymes that play crucial roles in modulating mammalian cell chromatin structure, transcription, and gene expression.4−6 HDACs have also been identified as important regulators of transcription in P. falciparum,7−10 and inhibition of P. falciparum histone deacetylases (PfHDACs) has been reported to both effectively kill the parasites (Vorinostat, Figure 1)11−16 and lead to efficacy in animal models of malaria (compound 2).17 Such findings underscore the potential for PfHDAC inhibitors to be used for malaria therapy.18−20
Figure 1.

Structures of known human HDAC inhibitors.
Of the five HDAC encoding genes known in P. falciparum one has homology to mammalian class I isoforms (PfHDAC1), two are similar class II (PfHDAC2 and 3) mammalian HDACs, while the remaining two are class III HDACs, or silent information regulator 2 (SIR2) proteins.19 In light of the close sequence homology between PfHDAC1 and human class I HDACs21 an undesired effect of the use of HDAC inhibitors is potential toxicity resulting from induction of epigenetic changes in host mammalian cells. Approved cytotoxic anticancer agents such as Vorinostat operate via inhibition of human HDAC enzymes (hHDACs) and can cause mechanism based toxic side effects.22 Thus, the development of selective inhibitors for PfHDACs is viewed as necessary to circumvent potential toxicity issues in the host, and we report here our efforts in this direction. This work led to the development of the first selective series of inhibitors of PfHDACs reported to date.
Previous research in our laboratories led to a novel series of heterocyclic inhibitors of human class-I HDACs represented by compound 3 (Figure 1).23 This compound class features a central heterocycle that displays the pharmacophore elements common to many inhibitors of HDAC enzymes. Thus, a zinc binding group (ZBG) believed to interact with the catalytic metal ion of the HDAC enzyme is anchored by an alkyl linker. The ZBG is complemented by two surface contact groups (namely, the aryl substituent and the amide group) that are thought to interact close to the entrance of the substrate binding channel.24 With many compounds from this structural class available, our efforts toward selective inhibitors of PfHDACs logically began through screening a representative panel of human HDAC1 inhibitors against a recombinant PfHDAC1 enzyme. The PfHDAC1 enzyme required for this campaign was expressed in fusion with a flag protein tag, in either insect or mammalian cell lines, and was purified with an Anti-Flag affinity gel column.
In addition, PfHDAC1 enzyme mutated in a Tyr residue (Y301H) located in the putative catalytic site was expressed. Mutation of the corresponding Tyr-to-His in human class-I HDACs has been demonstrated to severely reduce their enzymatic activities without affecting the overall protein structure, demonstrating that Y301 is essential for catalytic turnover.25 Y301H PfHDAC1 in fusion with a flag tag was expressed and purified under analogous conditions to the wild-type enzyme. Both purified proteins showed a low, though detectable, deacetylase activity in in vitro assays with fluorogenic substrates.
Compound 3 was tested head to head on mutant and wild type PfHDAC1. No loss of enzyme activity nor inhibition was observed with the mutant enzyme, despite its (presumed) lack of catalytic efficiency. While it cannot be excluded that the role of Y301 in the catalytic mechanism of PfHDAC1 differs from that observed for the corresponding residue in human class I HDACs, these results (together with the low in vitro activities observed across a variety of expression vectors in both mammalian and insect cell lines) added to our concern that the biological activities being measured were not in fact attributable to the PfHDAC1 enzyme. This conclusion was ultimately supported by the finding that both hHDAC1 and hHDAC2 could be clearly detected by Western Blot using specific antihuman HDAC antibodies following the expression and purification of either mutant or wild-type PfHDAC1 from human HeLa cells (Figure 2). The weight of evidence indicates that PfHDAC1 is expressed as an inactive enzyme that likely needs endogenous cofactors for its biological activation, and consequently that biological results reported to date on inhibition of PfHDAC1 are potentially false positives stemming from the presence of copurified host HDACs from the cellular expression vectors, likely associated in complex with PfHDAC, at least in the expression systems used.
Figure 2.

Western Blot analysis of purified wt and Y301H mutant PfHDAC1s using anti-Flag, anti-hHDAC2, or anti-hHDAC1. Arrows indicate the position of the probed protein. M, molecular weight marker.
The apparent lack of catalytic activity following expression of PfHDAC1 posed a significant challenge for the development of selective inhibitors of this enzyme. Although cell based assays were available to measure either inhibition of P. falciparum growth in erythrocytes (Pfgrowth assay) or blockade of human class I HDACs in HeLa cells, exclusive use of the comparison of these two assays to determine reliable enzyme selectivities was viewed as precarious. Differences in cell type and assay protocol are likely to influence properties such as protein binding and cell penetration, and together with the inherently larger variability in cell-based assay data as compared to that from biochemical assays may significantly impair interpretations based on direct comparisons. Although in vitro assays measuring cell-penetration and protein binding might aid interpretation of results, issues with compound throughput under such a screening paradigm were viewed as problematic.
To alleviate the risks associated with our need to use a cell-based assay as a front-line screen for the development of selective inhibitors of PfHDACs we have undertaken an approach outlined as follows: (i) comparison of parasite growth inhibition in erythrocytes with human class I HDAC inhibition in HeLa cells was used as a preliminary readout of selectivity with multiple repetitions of the cell based assays in order to minimize interpretation errors resulting from assay variability; (ii) structure–activity relationships were focused on improving parasite growth inhibition while reducing activity against the human HDAC1 enzyme; (iii) apparent selectivities measured in cell-based assays were validated at the biochemical level by measurement of nuclear histone hyperacetylation in both human and parasite treated cells. The inclusion of histone hyperacetylation experiments, as well as screening against the hHDAC1 enzyme amounts to an approach that adds weight to data obtained from simple comparison of cell-based assay data.
Screening results for a subset of our available compound collection in both the P. falciparum growth and human class I HDAC cellular assays highlighted an apparently low degree of selectivity, with most compounds inhibiting parasite growth at concentrations comparable with their human class I HDAC inhibition EC50. However, a group of outliers that featured significantly improved selectivity for parasite growth inhibition and shared a common structural feature was clearly evident. The bulk of compounds tested (e.g., compound 4, Table 1) featured either ketone or hydroxamic acid zinc binding moieties, but the outliers in contrast were based upon a secondary amide as ZBG (e.g., compound 5). This finding was somewhat surprising based on the conserved nature of the active site across different HDACs. An often asserted view is that selectivity between P. falciparum and human HDACs is likely to be realizable by modification of the surface contact groups in the pharmacophore, while our observation was that a change to the zinc binding group was impacting apparent selectivity.21,26,27 In line with our goal of optimizing away from potent human HDAC enzyme inhibitors an attractive feature was the much lower hHDAC1 enzyme inhibition shown by compound 5 that contains a methylamide as ZBG. Compound 5 is around an order of magnitude weaker than 4 in terms of parasite growth inhibition. However, in line with the lower hHDAC enzyme inhibition for the amide, a much stronger drop in cellular inhibition of human class I HDACs was noted, resulting in an overall improvement in the apparent selectivity. That the binding orientations of 4 and 5 are similar and that the methylamide (rather than the imidazole ring) in 5 is functioning as the ZBG are supported by several pieces of unpublished data. Notably, the imidazole ring in compound 5 can be replaced with alternative ring systems that are weak Zn2+ binders without loss of parasite growth inhibition (data not shown).
Table 1. SAR of Compounds 4–8a,18.

IC50 and EC50 values in μM. Compound 3 was used as standard reference compound (see data in Supplementary Table 1).
IC50 and EC50 values are the average of at least three individual measurements ± SD.
SI: selectivity index (HeLa class I HDAC (μM)/Pfgrowth (μM)).
In view of the expected high metabolic turnover for compounds containing a naphthyl ring as the aryl substituent,23 the 2-methoxyquinoline analogue 6 was prepared.28 This structural change (naphthyl to 2-methoxyquinoline) has previously been employed to favor reduced rodent clearance for human HDACis, and pharmacokinetic studies of 5 and 6 in mouse indeed demonstrated that plasma clearance was ameliorated for the latter compound (60 vs 10 mL/min/kg). However, neither of these analogues achieved detectable plasma exposure following oral administration, with portal vein sampling studies confirming that poor intestinal absorption was an issue for the methylamide ZBG analogues. Detectable oral exposure was, however, achievable by installing neutral functionality in place of the basic amine group present in 6. Thus, both compounds 7 and 8 showed a degree of oral bioavailability (F = 10% and 20%, respectively) in mouse. The neutral amide analogues (7 and 8) showed weaker P. falciparum growth inhibition and/or failed to show any apparent selectivity, but taken as a whole, the overall characteristics of the methylamide ZBG analogues in Table 1 showed potential as a route toward selective and potentially orally available inhibitors of P. falciparum growth.
Given the detrimental effect of basic functionality on oral absorption, the electro-neutral thiazole amide fragment was retained during efforts to align potency, selectivity, and oral absorption. In order to allow efficient exploration of SAR at the aryl substituent on the imidazole core a new synthetic strategy was developed based the introduction of the aryl moiety by Suzuki–Miyaura cross-coupling chemistry as the last step of the synthesis. Starting from the known amino acid 9 (Scheme 1),28 the free carboxylic acid was transformed into an imidazole ring in three synthetic steps.29 Iodination of the imidazole was accomplished by employing a halogenation/dehalogenation protocol that furnished the desired monoiodinated compound 11. The methyl amide ZBG was installed by ester deprotection and subsequent coupling with methylamine to give the synthetically versatile intermediate 12. Removal of the Cbz protecting group in this compound and installation of the thiazole amide furnished the substrate for final Suzuki reactions. Cross couplings between 13 and boronic acids (or esters) proceeded smoothly when PdCl2 was employed as a precatalyst,30 furnishing (after HPLC purification) compounds 16–26, 28, and 29 (Scheme 1). Compounds 14,2815, 27, and 30 were synthesized using alternative procedures that are described in the Supporting Information.
Scheme 1. Chemistry for SAR at Aryl Substituent.

Reagents and conditions: (a) (i) MeNHOMe.HCl, HBTU, DIPEA, DMF, RT; (ii) LiAlH4, THF, −20 °C; (iii) glyoxal, NH3, MeOH, RT; (b) (i) NIS, CH3CN, −10 °C; (ii) Na2SO3, Bu4NHSO4, dioxane–water, 160 °C, microwave, 30 min; (c) (i) TFA, DCM, RT; (ii) MeNH2, HBTU, DMF, RT; (d) (i) HBr in AcOH, DCM, 0 °C to RT; (ii) thiazole-2-carboxylic acid, EDC.HCl, HOBt, DIPEA, DMF, RT; (e) ArB(OH)2, PdCl2(dppf), K2CO3, DME–water, 110 °C.
The results in Table 2 highlight that both Plasmodium growth potency as well as selectivity with respect to inhibition of human class I HDACs in HeLa cells were strongly influenced by the nature of the aryl substituent attached to the imidazole core. Although clear SAR trends were difficult to identify, there were indications that the aryl group attached to the imidazole ring is involved in a specific interaction with PfHDAC. Similar to the trend observed between compounds 5 and 6, the 2-naphthyl compound 14 was a marginally more potent and selective inhibitor of Plasmodium growth than 7. However, introduction of a methyl group at position 1 of the naphthyl ring (compound 15) completely abolished P. falciparum growth inhibition. Such a strong influence on activity resulting from introduction of a single methyl group likely points to a change in enzyme inhibition rather than a physicochemical effect that impacts cell-based activity. Similarly, while many 3,4-fused bicyclic heterocycles generated submicromolar P. falciparum growth inhibition (compounds 5–7, 14) no active compounds based on a 2,3-fused bicyclic ring system were found throughout the course of the program. The weak micromolar growth inhibition shown by compound 16 was illustrative of results typically obtained for 2,3-fused heterocycles. A wide variety of 3,4-fused bicyclic heterocycles were evaluated, and compounds 17–20 highlight the somewhat empirical nature of this exploration. The indole and indazole analogues (17 and 18, respectively) retained submicromolar growth inhibition and showed 10-fold improved selectivity with respect to the 2-methoxyquinoline analogue 7 that was our starting point. Furthermore, these compounds provided a first indication that P. falciparum growth inhibition in erythrocytes is stronger than activity measured in the human HDAC1 biochemical assay. In contrast, the benzimidazole analogue 19 and the benzotriazole analogue 20 were weak and poorly selective inhibitors of Plasmodium growth. Throughout the program we were unable to correlate trends between activities and measured or calculated physicochemical properties (e.g., logD) that may account for such differences in profile. Very different results were again obtained within a set of isomeric quinoline/isoquinoline analogues. Of the five isomers that were prepared, quinoline analogue 21 proved to be a very weak inhibitor, while the 4-isoquinoline 22 emerged as the most promising. Compound 22 showed submicromolar Plasmodium growth inhibition and 18-fold selectivity with respect to the human HDAC cellular assay. Substituted analogues of 22 were explored, with 23 and 24 both showing improved growth inhibition in the 100 nM range and comparable selectivity. In line with a specific enzyme interaction for the aryl group as previously described, the positioning of the quinoline substituent was crucial, with the 3-substituted analogue 25 showing weak P. falciparum growth inhibitory activity. Compounds 23 and 24 represent the most potent inhibitors that were prepared, but further improvements in selectivity with respect to human cellular HDAC inhibition were achieved.
Table 2. SAR of Compounds 7 and 14–30a.
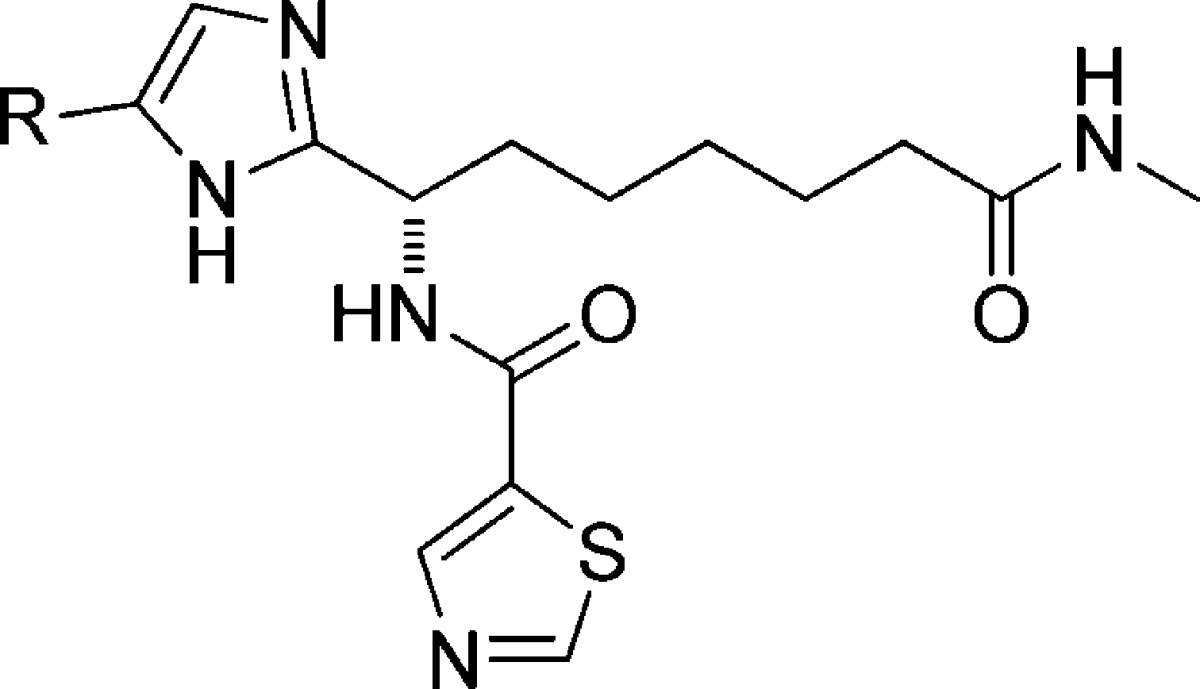

IC50 and EC50 values in μM. Compound 3 was used as standard reference compound (see data in Supplementary Table 1).
IC50 and EC50 values are the average of at least three individual measurements ± SD.
SI: selectivity index (HeLa class I HDAC(μM)/Pfgrowth(μM)).
Very weak inhibition of Plasmodium growth was measured for numerous phenyl-substituted imidazoles, including the biphenyl analogue 26, which is a 7 μM inhibitor. However, 6,5-biaryl systems turned out to show improved activity with the 4-(2,4)-thiazole analogue 27 and 4-(1,2)-pyrazole analogue 28 both showing submicromolar P. falciparum growth inhibition and selectivities around 15–20-fold. A further improvement in selectivity was achieved with the isomeric pyrazole 29, which was almost 40-fold selective for parasite growth. The most selective compound overall was the 2-hydroxy-3-quinoline analogue 30, which in multiple tests consistently failed to show activity in our human HDAC assays. The weak activity of 30 against human cellular class I HDACs meant that no IC50 value was determined; the apparent selectivity of the compound is estimated as being greater than 50-fold (based on the upper concentration at which it was tested in the assay). Notably, compounds 29 and 30 have hHDAC1 enzyme inhibition 4–5-fold weaker than activity in cell-based Plasmodium growth assay, supporting the notion that these are selective inhibitors of PfHDACs and in line with our goal of optimizing away from strong hHDAC1 enzyme inhibition.
Due to the lack of a reliable biochemical assay in which to measure enzymatic PfHDAC inhibition directly, the mechanism of action by which the optimized inhibitors elicited their antiparasitic activity was studied by histone hyperacetylation experiments. Assuming the inhibitors block the histone deacetylation process in parasites, then a phenotype that shows hyperacetylation of parasite histone proteins would be expected. Treatment of P. falciparum parasites with increasing concentrations of 29, one our most potent and selective inhibitors, and analysis of histone H4 lysine 8 clearly confirmed hyperacetylation at a concentration (EC50 = 350 nM) close to the compounds EC50 measured in the parasite growth assay (EC50 = 450 nM, Supplementary Figure 2 in the Supporting Information section). Furthermore, little histone hyperacetylation of histone H4 was observed at concentrations up to 25 μM when human HeLa cells were treated with 29, in agreement with the HDAC activity measured in this cell line (EC50 = 16.7 μM). These results, which were reproduced with several additional compounds (data not shown), support P. falciparum HDAC inhibition as the mechanism of action and also appear to corroborate the idea that comparison of P. falciparum growth inhibition and human cellular class I HDAC inhibition data can provide meaningful insight with regard to biochemical events within the cells. Further profiling of compound 29 against a panel of human HDAC enzymes (hHDAC 1–7, 9) showed that for most isoenzymes no inhibition was observed up to 5 μM, though hHDAC3 was inhibited with IC50 = 330 nM. No significant toxicity was observed for 29 in HeLa or HUVEC cells (CC50 > 25,000 nM) nor did the compound bind to hERG ion channels (IC50 > 30,000 nM). While 29 showed moderate intrinsic clearances in rat, human, and mouse liver microsomes (Clint = 27, 19, and 12 μL/min/mgP, respectively), it had clearance above liver blood flow following i.v. administration in mouse (Clp = 247 mL/min/kg). Efforts to understand the apparent extra-hepatic clearance, which does not appear to stem from instability in mouse plasma, and combine potency and selectivity with oral exposure remain under investigation in our laboratories.
In summary, we have reported findings that cast doubt on PfHDAC1 enzyme inhibition data reported to date in the literature31 and have described a screening approach that guided the discovery of what we believe to be selective inhibitors of Plasmodium falciparum HDACs. The leading compounds from this work show submicromolar inhibition of P. falciparum growth (EC50 < 500 nM), have good selectivity (>50-fold) over human HDACs in cells, and are weak inhibitors of human HDAC enzymes. Support for the mechanism of action of this compound class was provided by histone hyperacetylation studies.
Acknowledgments
The authors thank Dr. Pietro Alano and Dr. Francesco Silvestrini for the malaria parasite, Dr. Sergio Wittlin and Christian Scheurer for helping to set up the Pf growth assay, and Merck & Co. for allowing us to test a panel of HDAC inhibitors from its compound collection.
Glossary
ABBREVIATIONS
- DNA
deoxyribonucleic acid
- HDAC
histone deacetylase
- ZBG
zinc-binding group
- SAR
structure–activity relationship
- hERG
human Ether-a-go-go-Related Gene
- HUVEC
human umbilical vein endothelial cell
- Cbz
carboxybenzyl
- HBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- TFA
trifluoroacetic acid
- EDC
N-ethyl-N′-(3-(dimethylamino)propyl)carbodiimide
- HPLC
high performance liquid chromatography
- PdCl2(dppf)
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II)
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00468.
Synthetic experimental details and characterization data, description of primary biological assay protocols, and PK protocols (PDF)
Author Present Address
§ (R.L.) Teva Pharmaceutical Industries Ltd., Discovery and Product Development, P.O. Box 8077, Netanya 4250843, Israel.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
IRBM Science Park acknowledges financial support from Medicine for Malaria Venture (MMV) and National Collection of Compounds and Chemical Screening Center (CNCCS Consortium).
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. World Malaria Report 2014; World Health Organization: Geneva, 2014. [Google Scholar]
- Turschner S.; Efferth T. Drug resistance in Plasmodium: Natural products in the fight against malaria. Mini-Rev. Med. Chem. 2009, 9, 206–214. 10.2174/138955709787316074. [DOI] [PubMed] [Google Scholar]
- Hyde J. E. Drug-resistant malaria – an insight. FEBS J. 2007, 274, 4688–4698. 10.1111/j.1742-4658.2007.05999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips D. M. The presence of of acetyl groups of histones. Biochem. J. 1963, 87, 258–263. 10.1042/bj0870258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey V. G.; Faulkner R.; Mirsky A. E. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of RNA Synthesis. Proc. Natl. Acad. Sci. U. S. A. 1964, 51, 786–794. 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershey E. L.; Vidali G.; Allfrey V. G. Chemical studies of histone acetylation. The occurrence of epsilon-N-acetyllysine in the f2a1 histone. J. Biol. Chem. 1968, 243, 5018–5022. [PubMed] [Google Scholar]
- Chaal B. K.; Gupta A. P.; Wastuwidyaningtyas B. D.; Luah Y. H.; Bozdech Z. Histone deacetylases play a major role in the transcriptional regulation of the Plasmodium falciparum life cycle. PLoS Pathog. 2010, 6, e1000737. 10.1371/journal.ppat.1000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraisingh M. T.; Voss T. S.; Marty A. J.; Duffy M. F.; Good R. T.; Thompson J. K.; Freitas-Junior L. H.; Scherf A.; Crabb B. S.; Cowman A. F. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 2005, 121, 13–24. 10.1016/j.cell.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Freitas-Junior L. H.; Hernandez-Rivas R.; Ralph S. A.; Montiel-Condado D.; Ruvalcaba-Salazar O. K.; Rojas-Meza A. P.; Mancio-Silva L.; Leal-Silvestre R. J.; Marques Gontijo A.; Shorte S.; Scherf A. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 2005, 121, 25–36. 10.1016/j.cell.2005.01.037. [DOI] [PubMed] [Google Scholar]
- Hu G.; Cabrera A.; Kono M.; Mok S.; Chaal B. K.; Haase S.; Engelberg K.; Cheemadan S.; Spielmann T.; Preiser P. R.; Gillberger T.-W.; Bozdech Z. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat. Biotechnol. 2010, 28, 91–98. 10.1038/nbt.1597. [DOI] [PubMed] [Google Scholar]
- Dow G. S.; Chen Y.; Andrews K. T.; Caridha D.; Genera L.; Gettayacamin M.; Johnson J.; Li Q.; Melendez V.; Obaldia N. III; Tran T. N.; Kozikowski A. P. Antimalarial activity of phenylthiazolyl-bearing hydroxamate-based histone deacetylase inhibitors. Antimicrob. Agents Chemother. 2008, 52, 3467–3477. 10.1128/AAC.00439-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfurt J.; Chalfein F.; Prayoga P.; Wabiser F.; Kenangalem E.; Piera K. A.; Fairlie D. P.; Tjitra E.; Anstey N. M.; Andrews K. T.; Price R. N. Ex vivo activity of histone deacetylase inhibitors against multidrug-resistant clinical isolates of Plasmodium falciparum and P. vivax. Antimicrob. Agents Chemother. 2011, 55, 961–966. 10.1128/AAC.01220-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen F. K.; Sumanadasa S. D. M.; Stenzel K.; Duffy S.; Meister S.; Marek L.; Schmetter R.; Kuna K.; Hamacher A.; Mordmuller B.; Kassak M. U.; Winzeler E. A.; Avery V. M.; Andrews K. T.; Kurz T. Discovery of HDAC inhibitors with potent activity against multiple malaria parasite life cycle stages. Eur. J. Med. Chem. 2014, 82, 204–213. 10.1016/j.ejmech.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen F. K.; Skinner-Adams T. S.; Duffy S.; Marek L.; Sumanadasa S. D. M.; Kuna K.; Held J.; Avery V. M.; Andrews K. T.; Kurz T. Synthesis, Antimalarial Properties, and SAR Studies of Alkoxyurea-Based HDAC Inhibitors. ChemMedChem 2014, 9, 665–670. 10.1002/cmdc.201300469. [DOI] [PubMed] [Google Scholar]
- Giannini G.; Battistuzzi G.; Vignola D. Hydroxamic Acid Based Histone Deacetylase Inhibitors with Confirmed Activity against the Malaria Parasite. Bioorg. Med. Chem. Lett. 2015, 25, 459–461. 10.1016/j.bmcl.2014.12.051. [DOI] [PubMed] [Google Scholar]
- Engel J. A.; Jones A. J.; Avery V. M.; Sumanadasa S. D. M.; Ng S. S.; Fairlie D. P.; Adams T. S.; Andrews K. T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol.: Drugs Drug Resist. 2015, 5, 117–126. 10.1016/j.ijpddr.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumanadasa S. D. M.; Goodman C. D.; Lucke A. J.; Skinner-Adams T.; Sahama I.; Haque A.; Do T. A.; McFadden G. I.; Fairlie D. P.; Andrews K. T. Potent Antimalarial Activity of Histone Deacetylase Inhibitor Analogues. Antimicrob. Agents Chemother. 2012, 56, 3849–3856. 10.1128/AAC.00030-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews K. T.; Haque A.; Jones M. K. HDAC Inhibitors in Parasitic Diseases. Immunol. Cell Biol. 2012, 90, 66–77. 10.1038/icb.2011.97. [DOI] [PubMed] [Google Scholar]
- Andrews K. T.; Tran T. N.; Wheatley N. C.; Fairlie D. P. Targeting Histone Deacetylase Inhibitors for Anti-Malarial Therapy. Curr. Top. Med. Chem. 2009, 9, 292–308. 10.2174/156802609788085313. [DOI] [PubMed] [Google Scholar]
- Trenholme K.; Marek L.; Duffy S.; Pradel G.; Fisher G.; Hansen F. K.; Skinner-Adams T. S.; Butterworth A.; Ngwa C. J.; Moecking J.; Goodman C. D.; McFadden G. I.; Sumanadasa S. D. M.; Fairlie D. P.; Avery V. M.; Kurz T.; Andrews K. T. Lysine Acetylation in Sexual Stage Malaria Parasites Is a Target for Antimalarial Small Molecules. Antimicrob. Agents Chemother. 2014, 58, 3666–3678. 10.1128/AAC.02721-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P.; Praddhan A.; Shah F.; Tekwani B. L.; Avery M. A. Structural insights into the Plasmodium falciparum histone deacetylase 1 (PfHDAC-1): A novel target for the development of antimalarial therapy. Bioorg. Med. Chem. 2008, 16, 5254–5265. 10.1016/j.bmc.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Mann B. S.; Johnson J. R.; Cohen M. H.; Justice R.; Pazdur R. FDA Approval summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- Kinzel O.; Llauger-Bufì L.; Pescatore G.; Rowley M.; Schultz-Fademrecht C.; Monteagudo E.; Fonsi M.; Gonzalez Paz O.; Fiore F.; Steinkühler C.; Jones P. Discovery of Potent Class I Selective Ketone Histone Deacetylase Inhibitor with Antitumor Activity in Vivo and Optimized Pharmacokinetic Properties. J. Med. Chem. 2009, 52, 3453–3456. 10.1021/jm9004303. [DOI] [PubMed] [Google Scholar]
- Vannini A.; Volpari C.; Gallinari P.; Jones P.; Mattu M.; Carfì A.; De Francesco R.; Steinkühler C.; Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007, 8, 879–884. 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahm A.; Paolini C.; Pallaoro M.; Nardi M. C.; Jones P.; Neddermann P.; Sambucini S.; Bottomley M. J.; Lo Surdo P.; Carfì A.; Koch U.; De Francesco R.; Steinkühler C.; Gallinari P. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17335–17340. 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agbor-Enoh S.; Seudieu C.; Davidson E.; Dritschilo A.; Jung M. Novel Inhibitor of plasmodium Histone Deacetylase That cures P. berghei-Infected Mice. Antimicrob. Agents Chemother. 2009, 53, 1727–1734. 10.1128/AAC.00729-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews K. T.; Tran T. N.; Lucke A. J.; Kahnberg P.; Le G. T.; Boyle G. M.; Gardiner D. L.; Skinner-Adams T. S.; Fairlie D. P. Potent Antimalarial Activity of Histone Deacetylase Inhibitor Analogues. Antimicrob. Agents Chemother. 2008, 52, 1454–1461. 10.1128/AAC.00757-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compounds 4–9 and 14 were synthesized using the procedures described in Atenni B.; Ferrigno F.; Jones P.; Ingenito R.; Kinzel O.; Llauger-Bufì L.; Ontoria J. M.; Pescatore G.; Rowley M.; Scarpelli R.; Schultz-Fademrecht C.. Heterocycles Derivatives as Histone Deacetylase (HDAC) Inhibitors. WIPO Patent 061638, 2006.
- Deziel R.; Rahil J.; Wahhab A.; Allan M.; Nguyen N.. Sirtuin Inhibitors. WIPO Patent 026701, 2009.
- Zeng Q.; Chen K. X.; Anilkumar G. N.; Rosenblum S. B.; Kozlowski J. A.; Njoroge F. G.. Antiviral Compounds Composed of Three Linked Aryl Moieties to Treat Diseases such as Hepatitis C. WIPO Patent 138791, 2010.
- Patel V.; Mazitschek R.; Coleman B.; Nguyen C.; Urgaonkar S.; Cortese J.; Barker R. H. Jr.; Greenberg E.; Tang W.; Bradner J. E.; Schreiber S. L.; Duraisingh M. T.; Wirth D. F.; Clardy J. Identification and Characterization of Small Molecule Inhibitors of a Class I Histone Deacetylase from Plasmodium falciparum. J. Med. Chem. 2009, 52, 2185–2187. 10.1021/jm801654y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.