

Abstract
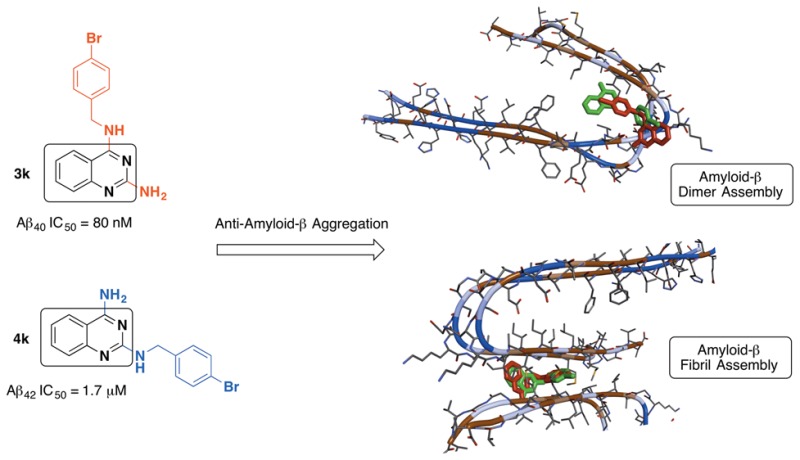
A library of isomeric 2,4-diaminoquinazoline (DAQ) derivatives were synthesized and evaluated for antiaggregation potential toward Aβ40/42. Structure–activity relationship data identified compound 3k (N4-(4-bromobenzyl)quinazoline-2,4-diamine) with a 4-bromobenzyl substituent as the most potent inhibitor (Aβ40 IC50 = 80 nM) and was almost 18-fold more potent compared to the reference agent curcumin (Aβ40 IC50 = 1.5 μM). The corresponding N2-isomer 4k (N2-(4-bromobenzyl)quinazoline-2,4-diamine) was also able to prevent Aβ aggregation (Aβ40 IC50 = 1.7 μM). However, compound 4k exhibited superior inhibition of Aβ42 aggregation (Aβ42 IC50 = 1.7 μM) compared to compound 3k (Aβ42 IC50 = 14.8 μM) and was ∼1.8-fold more potent compared to curcumin (Aβ42 IC50 = 3.1 μM). These results were supported by Aβ aggregation kinetics investigations and transmission electron microscopy studies, which demonstrate the suitability of DAQ ring system to develop antiamyloid agents as pharmacological tools to study Aβ aggregation.
Keywords: Quinazolines, amyloid, Aβ aggregation, Alzheimer’s disease
One of the most influential hallmarks of Alzheimer’s pathology is the collapse of cellular amyloid management, leading to the progressive accumulation of neurotoxic Aβ-deposits.1−4 While the mechanisms intertwining the amyloid pathway are vast and complex, a number of strategies to combat its neuronal insults have been proposed, researched, and evaluated in various preclinical and clinical settings.5−7 An essential tool utilized by researchers to further understand the aggregation mechanisms of amyloidogenic peptides, such as Aβ, is to develop and evaluate aggregation modulators and inhibitors.8,9 Not only is this beneficial in understanding the kinetics of amyloid aggregation but it may reveal potential therapeutic candidates.
When it comes to developing drug candidates, nature plays an important role in highlighting ideas for potential core templates and scaffolds. Honing in on the case of Aβ with respect to aggregation modulators and inhibitors, compounds such as curcumin, a component of the spice turmeric, and resveratrol, a phytoalexin found in grapes and berries (Figure 1), are considered as model compounds in this context.10−13 The synthetic compound, orange G is a commonly used stain/dye and, like curcumin and resveratrol, is used as a pharmacological tool in drug discovery due to its excellent activity against amyloid aggregation.14,15
Figure 1.

Structures of naturally occurring and synthetic amyloid aggregation inhibitors and the DAQ scaffold.
From a chemical standpoint, these small molecules share structural features including aromaticity, conjugation, and planarity resulting in their ability to intercalate and disrupt the backbone hydrogen bonding interactions in the beta-sheet assembly thereby providing a framework to design small molecules as pharmacological tools to study Aβ aggregation and inhibition.16,17 As part of our research program aimed at discovering and developing novel small-molecules as potential pharmacological tools to study Alzheimer’s disease (AD) and design novel anti-AD agents, we embarked on the development of isomeric 2,4-diamino-quinazolines (DAQ) library as a novel class of compounds that exhibit anti-Aβ aggregation properties. The planar, bicyclic quinazoline ring template can be considered as a bioisostere of the naphthalene ring present in orange G (Figure 1). A library of 34 isomeric DAQs were synthesized and their anti-Aβ aggregation activity was evaluated by monitoring the Aβ40 and Aβ42 aggregation kinetics using ThT-fluorescence and by transmission electron microscopy (TEM) measurements. Computational experiments were used to propose their binding interactions with Aβ-aggregates. These studies show that the anti-Aβ activity was sensitive to isomeric placement of substituents either at the 2 or 4-position of the quinazoline amine template and that they represent a novel class of compounds that can be useful to design small molecules with antiamyloid aggregation properties.
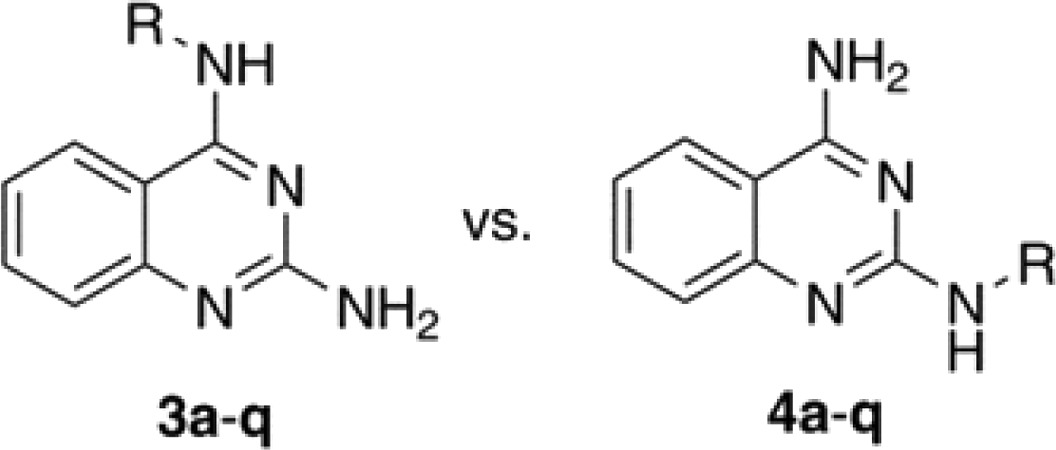
The DAQ template (2, Scheme 1) was synthesized starting from 2-fluoro (1a) or 2-aminobenzonitrile (1b) by heating with excess guanidine carbonate in dimethylacetamide (DMA) under pressure (Scheme S1, Supporting Information).18,19 The reaction with 2-fluorbenzonitrile provided superior yield (∼70%) of 2 compared to 2-aminobenzonitrile (∼40%). The most challenging aspect of this synthesis was developing selective alkylation methods to prepare either N2- or N4-substituted quinazoline amines. After investigating a variety of base, solvent, time, and temperature combinations (Table S1, Supporting Information), two selective conditions were identified. Using NaH and DMSO provided the N4-isomer exclusively, whereas combination of potassium carbonate and DMA favored the N2-isomer exclusively (Scheme 1). Their chemical structures were confirmed by 1H, 13C NMR, and 2D COSY NMR studies. These studies show that the N4-NHs are more acidic (δ 7.74 ppm) compared to the N2-NH protons (δ 6.53 ppm).
Scheme 1.

Reagents and conditions: (a) Alkyl- or aromatic halides, NaH, DMSO, 0 °C to r.t. 14 h. (b) Alkyl- or aromatic halides, potassium carbonate, DMA, r.t. to 85 °C, 5 h.
The conditions used for selective alkylation provided low yield (14–31%). The TLC examinations showed that the reactions did not go to completion. Attempts to increase the yield by increasing the base equivalence along with that of the R-groups resulted in mixed isomers and/or double substitutions as confirmed by NMR studies. Based on these observations, selective alkylation at either N2 or N4-position was achieved using K2CO3/DMA and NaH/DMSO, respectively, using various substrates (Scheme 1, R = benzyl, substituted benzyl, phenethyl, n-Pr, i-Pr, or cyclohexylmethyl). Attempts at synthesizing the N2- or N4-methylpyridyl based DAQ compounds using the same conditions were not successful. The assessment of these isomeric DAQ derivatives against the self-induced aggregation mechanisms of both Aβ40 and Aβ42 revealed a combination of structure–activity relationships (SARs). As shown in Table 1, the aggregation IC50 values are listed for each derivative and were compared with standard controls (orange G, curcumin, resveratrol) and the DAQ template itself. Examining the SAR data obtained based on a thioflavin T (ThT) based fluorescence spectroscopy method (Table 1) demonstrates the overall ability of these N2- and N4-substituted DAQ derivatives to inhibit the aggregation of amyloid peptides. In contrast, the DAQ template alone (Table 1) was promoting the aggregation process. Generally, N4-substituted DAQ derivatives (3a–q) were more effective at inhibiting Aβ40 compared to their N2-substituted isomers (4a–q). Interestingly, this observation is completely reversed with respect to Aβ42. The addition of a benzyl substituent either at N4- or N2-position in compounds 3a and 4a provided Aβ40 aggregation inhibition (Table 1). However, the N4-regioisomer 3a did not exhibit any inhibition of Aβ42 unlike the N2-regioisomer 4a. The N2-placement in 4a was more effective against Aβ42 (IC50 = 8.4 μM). Replacing the benzyl substituent with a more lipophilic 3- or 4-methylbenzyl substituent in 3b, 3c and 4b, 4c modified the biological profiles significantly. When compared to 3a, the N4-placement of a methylbenzyl substituent, regardless of meta- or para-positioning, slightly improved activity against Aβ40 (no more than ∼1.3-fold improvement) with no change toward Aβ42. This was not the case with the N2-isomers, where the methyl-substituted benzyl group resulted in loss of activity against both Aβ40 and Aβ42 (4b, Aβ40/42 IC50 > 25 μM) or a much weaker profile (4c, Aβ40 IC50 = 13.1 μM, Aβ42 IC50 = 22.5 μM) compared to 4a (Table 1). The addition of an electron-donating methoxybenzyl group, in 3d, 3e and 4d, 4e, yielded mixed outcomes. The N4-placement of a 3-methoxybenzyl group (3d, Aβ40 IC50 = 20.6 μM, Aβ42 IC50 = inactive at 25 μM) was detrimental to antiaggregation activity compared to the benzyl derivative 3a, while the 4-methoxybenzyl substituent (3e, Aβ40 IC50 = 1.1 μM, Aβ42 IC50 > 25 μM) enhanced the activity against Aβ40 by ∼4–4.5-fold (compared to 3a and 3c). However, N2-placement of the 3-methoxy-substituted benzyl group (4d, Aβ40/42 IC50 > 25 μM) did not offer any benefits compared to 4a and was ineffective in providing antiaggregation activity. The 4-methoxy compound (4e, Aβ40 IC50 = 6.8 μM, Aβ42 IC50 > 25 μM) was ∼6-fold less potent compared to its N4-isomer (3e) toward Aβ40.
Table 1. Inhibition Data for DAQ Isomeric Derivatives 3a–q and 4a–q against Self-Induced Aβ40 and Aβ42 Aggregation.
| IC50 (μM)a,b |
IC50 (μM)a,b |
|||||
|---|---|---|---|---|---|---|
| N4-isomer | Aβ40 | Aβ42 | R-group | Aβ40 | Aβ42 | N2-isomer |
| 3a | 4.8 | >25 | benzyl | 2.2 | 8.4 | 4a |
| 3b | 3.6 | >25 | 3-Me-benzyl | >25 | >25 | 4b |
| 3c | 3.9 | >25 | 4-Me-benzyl | 13.1 | 22.5 | 4c |
| 3d | 20.6 | n.a | 3-OMe-benzyl | >25 | >25 | 4d |
| 3e | 1.1 | >25 | 4-OMe-benzyl | 6.8 | >25 | 4e |
| 3f | 3.5 | 21.0 | 3-CF3-benzyl | 12.5 | 13.8 | 4f |
| 3g | 1.9 | >25 | 4-CF3-benzyl | 2.3 | 5.3 | 4g |
| 3h | 1.9 | >25 | 3-Cl-benzyl | 5.9 | 13.4 | 4h |
| 3i | 0.62 | >25 | 4-Cl-benzyl | 0.93 | 8.1 | 4i |
| 3j | 0.58 | 22.3 | 3-Br-benzyl | 1.5 | 2.7 | 4j |
| 3k | 0.08 | 14.8 | 4-Br-benzyl | 1.7 | 1.7 | 4k |
| 3l | 2.8 | >25 | 3-F-benzyl | 11.3 | 14.7 | 4l |
| 3m | 3.1 | >25 | 4-F-benzyl | >25 | >25 | 4m |
| 3n | 7.8 | >25 | phenethyl | 14.9 | >25 | 4n |
| 3o | 14.9 | n.a | n-Pr | >25 | >25 | 4o |
| 3p | 10.2 | n.a | i-Pr | >25 | >25 | 4p |
| 3q | 13.6 | >25 | cyclohexylmethyl | >25 | >25 | 4q |
| DAQ | P.A | P.A | 1.5 | 3.1 | curcumin | |
| orange G | 3.0 | 8.7 | 2.6 | 4.1 | resveratrol | |
IC50 values are calculated based on the ThT-based fluorescence spectroscopy assay (excitation = 440 nm, emission = 490 nm).
Values are mean values of triplicate readings for three independent experiments. n.a = not active. P.A = promotes aggregation.
The effect of electron-withdrawing trifluoromethylbenzyl substituent was investigated in compounds 3f, 3g and 4f, 4g. Interestingly, its presence at N4-position in compound 3f (3-CF3-benzyl) provided similar inhibition profile compared to 3b (3-Me) with IC50 values around 3.6 μM for Aβ40 and over 20 μM for Aβ42. This was not the case with 3g (4-CF3), which was ∼2-fold more potent toward Aβ40 compared to its methylbenzyl bioisostere (3c). However, this modification did not provide better inhibition of Aβ42. With N2-placement, however, the modification from (3/4-Me) to (3/4-CF3) was positive across the board. While 4b was ineffective against both Aβ40/42 (IC50 > 25 μM), 4f (3-CF3) was active toward both species (IC50 ≈ 12–13 μM). Derivative 4g, however, was more potent compared to 4c (4-Me), equipotent to 4a (Aβ40), and ∼1.6-fold more potent toward Aβ42 compared to 4a. It exhibited equipotent/comparable activity to the reference compound resveratrol (Table 1). In the next set of compounds, the effect of benzyl halides at N2 and N4-position was explored. Generally, the presence of benzyl halides increased overall inhibition potency (with the exception of fluorobenzyl). Starting with the N4-placement of 3-chloro (3h) or 4-chlorobenzyl (3i) substituents, both were more potent toward Aβ40 compared to 3a (∼2.5-fold and ∼8-fold, respectively). In this regard 3i exhibited potent inhibition of Aβ40 with an IC50 = 620 nM and was approximately 2.4-fold more potent compared to the reference agent curcumin (Aβ IC50 = 1.5 μM). However, both 3i and 3h were ineffective against Aβ42 (IC50 > 25 μM). Interestingly, the N2-placement of these chlorobenzyl groups (4h/4i) exhibit inhibition of both Aβ40 and Aβ42 aggregation. They were not as potent as 3i and 3h against Aβ40 (Table 1). The bromine-based bioisosteres (3j, 3k and 4j, 4k) were all more potent against both Aβ40/42 compared to their chlorine-based counterparts with one exception (4k on Aβ40, Table 1). Derivative 3k (4-bromobenzyl) was identified as the most potent Aβ40 aggregation inhibitor (IC50 ≈ 80 nM). It was ∼7.5-fold more potent compared to its 3-bromobenzyl isomer (3j, Aβ40 IC50 = 580 nM, Aβ42 IC50 = 22.3 μM) and ∼21-fold more potent compared to its N2-isomer (4k, Aβ40 and Aβ42 IC50 = 1.7 μM). Both 3k and 3j were much more potent inhibitors compared to reference agents orange G, curcumin, and resveratrol. Furthermore, placing the 3-bromobenzyl substituent at N2-position, significantly enhances the Aβ42 aggregation inhibition in compound 4j (Aβ40 IC50 = 2.7 μM) with almost 8-fold gain in activity compared to the corresponding N4-isomer 3j (Aβ42 IC50 = 22.3 μM, Table 1). In contrast, the presence of an electronegative fluorobenzyl substituent at either N2- or N4-position was detrimental suggesting that smaller size of a fluorine atom compared to either a bromine or chlorine atom was not favorable. The N4-derivatives 3l and 3m were ineffective against Aβ42. Although they lost considerable activity toward Aβ40, they did maintain moderate levels of inhibition (IC50 ≈ 3 μM) comparable to the methyl- (3b, 3c) and trifluoromethyl-based derivatives (3f, 3g). The extent of activity loss was greater with N2-placements, where derivative 4l exhibited weak activity toward both Aβ40/42 (IC50 ≈ 11–15 μM) and 4m was ineffective against both Aβ40/42 (Table 1). In other SAR modifications, the addition of a methylene group (phenethyl vs benzyl) in compounds 3n and 4n, did not improve the Aβ inhibitory activity compared to 3a and 4a (Table 1). Compound 3n was ∼1.6-fold less potent toward Aβ40 compared to 3a and not very effective toward Aβ42 either. This modification also caused a loss of Aβ42 inhibitory activity for 4n along with a ∼ 6.7-fold decrease in potency toward Aβ40. Similarly, the addition of alkyl substituents (n-Pr and i-Pr) either at N2- or N4-position, as seen in compounds 3o, 3p and 4o, 4p resulted in either weak or no inhibition of aggregation (Table 1). Furthermore, the addition of a cyclic nonaromatic substituent such as a cyclohexylmethyl group (compound 3q and 4q) was detrimental to antiaggregation activity clearly displaying the importance of having planar aromatic substituents to exhibit antiaggregation properties.
In summary, based on this SAR study, we identified a total of nine DAQ based compounds (3e, 3g–k, and 4i–k) that exhibited superior or equipotent anti-Aβ aggregation activity profile compared to reference agents orange G, curcumin, and resveratrol (Table 1). Compounds 4j and 4k met or surpassed the Aβ42 activity seen with curcumin (the most potent among the reference agents used), while a total of 9 derivatives (3e, 3g–k and 4i–k) closely met or surpassed the Aβ40 activity shown by the same reference agents. Derivative 3k, N4-(4-bromobenzyl)quinazoline-2,4-diamine, was identified as the most potent Aβ40 aggregation inhibitor (IC50 ≈ 80 nM), while its N2-isomer, N2-(4-bromobenzyl)quinazoline-2,4-iamine (4k), was identified as the most potent and dual Aβ40/42 aggregation inhibitor (IC50 ≈ 1.7 μM).
To better understand the modes of inhibition exerted by 3k and 4k (and their regioisomers 3j and 4j), we investigated the full aggregation kinetic plots (Figure 2a, panels A–D, and Figure S1, Supporting Information) derived from the ThT based aggregation assay used to determine the IC50 values listed in Table 1. The assessment of the kinetic plots provided greater insight into the potential mode of action for this class of compounds. The aggregation kinetic plots for compounds 3j and 3k (Figure 2a, panels A and B) show that both of these derivatives achieve their superior inhibitory activity by stabilizing the monomeric Aβ40 structures. The half time (t50) that defines the time at which the fluorescence intensity reaches the midpoint between pre- and postaggregation baseline growth phase in the absence of test compounds was approximately 12.5 h for Aβ40 control curve. In this regard, both compounds 3j and 3k were able to prevent the nucleation-dependent aggregation process significantly, with a relatively weak growth phase seen toward the end of the 24 h aggregation period. Significantly, both of these compounds were exhibiting potent antiamyloid aggregation properties at all the tested concentrations (1, 5, and 25 μM, Figure 2a, panels A and B). The aggregation kinetics plot of the corresponding N2-isomers 4j and 4k (Figure S1, panels A and B, Supporting Information) displayed concentration-dependent inhibition via improving monomer stability and reducing the rate of aggregation; however, the antiaggregation effect was not as drastic as compounds 3j and 3k. Compound 4j at 25 μM was able to delay the aggregation compared to Aβ control curve (t50 ≈ 17 h vs 12.5 h for Aβ control with no test compound), whereas 4k at 25 μM was able to exhibit complete inhibition of Aβ40 aggregation. These studies demonstrate that at 25 μM, N4-isomers, compounds 3j, 3k, and the N2-isomer 4k were capable of preventing the formation of Aβ aggregates including dimers, trimers, oligomers, protofibrils, and fibrils.
Figure 2.

(a) Amyloid aggregation kinetics study. Panels A and B: 5 μM Aβ40 with varying concentrations (1, 5, or 25 μM) of DAQ derivatives 3j and 3k, respectively. Panels C and D: 5 μM Aβ42 with varying concentrations (1, 5, or 25 μM) of DAQ derivatives 4j and 4k, respectively. Aggregation kinetics were monitored by ThT-fluorescence spectroscopy (excitation = 440 nm, emission = 490 nm) for 24 h at 37 °C in phosphate buffer at pH 7.4. Results are based on three independent experiments in triplicate measurements. (b) Amyloid morphology analysis using transmission electron microscopy (TEM) after 24 h incubation at 37 °C in phosphate buffer at pH 7.4. Panel A, 25 μM Aβ40 control; Panel B, 25 μM Aβ40 with 25 μM of DAQ derivative 3k; Panel C, 25 μM Aβ42 control; Panel D, 25 μM Aβ42 with 25 μM of DAQ derivative 4k. Scale: black/white bars represent 500 nm.
With respect to Aβ42, the dominance of the N2-isomers (4j and 4k, Figure 2a, panels C and D) became evident when compared to their N4-counterparts (3j and 3k, Figure S1, panels C and D, Supporting Information). The half time t50 was approximately 2 h for Aβ42 in the absence of test compounds. In this regard, compounds 3j and 3k were able to delay the lag phase to a great extent (t50 values of approximately 2.6 and 3.5 h, respectively, at 25 μM compared to Aβ42 control. In contrast, the N4-isomers 4j and 4k exhibited better inhibition profile with compound 4j showing a significant delay in aggregation (t50 ≈ 6 h), whereas 4k was able to show complete inhibition at 25 μM (Figure 2a, panels C and D). While all the kinetic plots for Aβ42 demonstrated concentration-dependent inhibition, none of these derivatives were capable of completely halting the saturation phase except for the N2-isomer 4k at the highest concentration tested.
To further validate these observations, Aβ40/42 morphology was assessed using transmission electron microscopy (TEM, Figure 2b, Figure S2 and S3, Supporting Information) in the presence and absence of most potent DAQ derivatives (3j, 3k, 4j, and 4k, Figure 2b and Figure S2, Supporting Information). As observed with TEM, both Aβ40/42 control samples (25 μM) displayed high levels of dense and full aggregate species. Coincubation experiments with DAQ derivatives (3j, 3k, 4j and 4k), at 25 μM, were able to drastically hinder the aggregation process with the N4-isomer 3k (Figure 2b, panel B) and N2-isomer 4k (Figure 2b, panel D) exhibiting almost complete inhibition of aggregation. These results correlate with the ThT aggregation kinetics data (Figure 2a and Figure S1, Supporting Information).
In order to gain some insights into the mechanism of anti-Aβ aggregation properties of DAQ derivatives (3j, 3k, 4j, and 4k), we investigated their binding interactions in both the Aβ dimer and Aβ fibril model (Figures S4 and S5, Supporting Information). In the dimer model (Figure S4, panels A and B, Supporting Information), featuring the Aβ9–40 fragment,20 each of the docked DAQ derivatives interacted with the dimeric structure in a unique fashion, although these interactions were generally localized in the dimer-turn region (Asp23-Gly29). This might explain the trends seen in the kinetic plots, where monomer stability and substantial delays in the aggregation process were observed. The DAQ ring scaffold of 3j was oriented between Phe20 (perpendicular to the phenyl ring) and Val24 (distance ∼5–7 Å), where the 3-bromophenyl ring was aligned, in a parallel fashion, toward Ser26 and Asn27 (distance ≈ 5 Å). The bromine atom was oriented toward Ile32-Leu34 pocket at the C-terminal end (Figure S4, panel A). The C2-amine was positioned across from Val24-Gly25 on the adjacent monomer (distance ≈ 5 Å). However, the corresponding N2-isomer 4j had its DAQ ring scaffold between Asp23 and Lys28 (distance ≈ 5 Å), where the C4-amine was undergoing hydrogen-bonding interaction with Asp23 (distance < 3 Å). The 3-bromophenyl ring was oriented toward Asp23 and Val24 (distance ≈ 5 Å), where the C2–NH was undergoing hydrogen-bonding with the Asp23 side-chain and backbone carbonyl of Val24 (distance < 3 Å). The 4-bromobenzyl isomers (3k and 4k) exhibited nearly opposite binding modes in the dimer model (Figure S4, panel B). The DAQ ring scaffold of 3k was oriented parallel to the Ser26-Lys28 turn region, where the C2-amine was in contact with the Asp23 side-chain and backbone carbonyl of Val24 (distance = 2.7–3.1 Å). The 4-bromophenyl group was between Asp23 and Ile31 (distance ≈ 5–6 Å). In contrast, the corresponding N2-isomer 4k had its DAQ ring scaffold oriented between Ile31/32 and Ala21-Asp23 in a perpendicular fashion, where the C4-amine was hydrogen-bonding with the carbonyl backbone of Ile32 (distance ≈ 2.7 Å). The C2–NH was oriented toward Ala30 and was in contact with its carbonyl backbone (distance ≈ 3 Å), while the 4-bromophenyl group was stacked, in a parallel orientation, between Asp23 and Gly29 (distance ≈ 5 Å). These studies suggest that DAQ ring system serves as a suitable template to design small molecule probes to study Aβ aggregation and inhibition.
In conclusion, we investigated the selective alkylation of the 2,4-diaminoquinazoline (DAQ) template, a privileged scaffold, to generate a library of N2 and N4-substituted DAQ derivatives. These compounds were then screened for antiaggregation properties toward Aβ40/42 by monitoring their aggregation kinetics, which revealed that halogen-substituted benzyl groups generally exhibited superior anti-Aβ aggregation effect with N4-isomers providing better selectivity for Aβ40, whereas the N2-isomers exhibited better inhibition of Aβ42 aggregation. The N4-isomer 3k with a 4-bromobenzyl substituent was identified as the most potent Aβ40 aggregation inhibitor (IC50 = 80 nM), whereas the corresponding N2-isomer (4k) yielded our most potent Aβ42 aggregation inhibitor (IC50 = 1.7 μM), which also exhibited dual Aβ40/42 aggregation inhibition. The outcomes of this study demonstrates the usefulness of quinazoline diamine template to design novel antiamyloid agents. These small molecules serve as valuable pharmacological tools to study and develop potential therapies to treat AD.
Acknowledgments
The authors would like to thank the Faculty of Science, Office of Research, the School of Pharmacy at the University of Waterloo, Ontario Mental Health Foundation (graduate scholarship for T.M.), NSERC-Discovery (RGPIN: 03830-2014), Canada Foundation for Innovation (CFI-JELF), Ontario Research Fund (ORF), and Early Researcher Award, Ministry of Research and Innovation, Government of Ontario, Canada (PR) for financial support of this research project.
Glossary
ABBREVIATIONS
- AD
Alzheimer’s disease
- Aβ
amyloid-beta
- DAQ
diaminoquinazoline
- DMA
dimethylacetamide
- DMSO
dimethyl sulfoxide
- NaH
sodium hydride
- SAR
structure–activity relationship
- DMAP
4-dimethylaminopyridine
- DBU
1,8-diazabicycloundec-7-ene
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00039.
Synthetic and biological methods along with characterization and analytical data (PDF)
Author Contributions
P.P.N.R. and T.M. conceived the project and designed the experiments. T.M., A.S., and G.T., performed the experiments. T.M., A.S., and P.P.N.R. analyzed and interpreted the data. T.M. wrote the manuscript. T.M., A.S., G.T., and P.P.N.R. revised the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Selkoe D. J. Cell biology of the amyloid β protein precursor and the mechanism of Alzheimer’s disease. Annu. Rev. Cell Biol. 1994, 10, 373–403. 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- LaFerla F. M.; Green K. N.; Oddo S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Price D. L.; Sisodia S. S.; Gandy S. E. Amyloid beta amyloidosis in Alzheimer’s disease. Curr. Opin. Neurol. 1995, 8, 268–274. 10.1097/00019052-199508000-00004. [DOI] [PubMed] [Google Scholar]
- Guela C.; Wu C.; Saroff D.; Lorenzo A.; Yuan M.; Yanker B. A. Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nat. Med. 1998, 4, 827–831. 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- Hock C.; Konietzko U.; Streffer J. R.; Tracy J.; Signorell A.; Muller-Tillmanns B.; Lemke U.; Henke K.; Moritz E.; Garcia E.; Wollmer M. A.; Umbricht D.; de Quervain D. J. F.; Hofmann M.; Maddalena A.; Papassotiropoulos A.; Nitsch R. M. Antibodies against β-amyloid slow cognitive decline in Alzheimer’s disease. Neuron 2003, 38, 547–554. 10.1016/S0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- Menendez-Gonzalez M. Biomarkers in neurodegenerative disorders: translating research into clinical practice. Front. Aging Neurosci. 2014, 6, 1–2. 10.3389/fnagi.2014.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godyn J.; Jonczyk J.; Panek D.; Malawska B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2016, 68, 127–138. 10.1016/j.pharep.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Masliah E.; Rockenstein E.; Price D.; Bonhaus D.; Meier D.; Moesler H.; Konrat R.; Tsigelny I.; Wrasidlo W. Small molecules targeting the formation of neurotoxic oligomers for the treatment of AD/PD: a second look. Neurobiol. Aging 2014, 35, S15. 10.1016/j.neurobiolaging.2014.01.087. [DOI] [Google Scholar]
- Doig A. J.; Derreumaux P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015, 30, 50–56. 10.1016/j.sbi.2014.12.004. [DOI] [PubMed] [Google Scholar]
- Ono K.; Hasegawa K.; Naiki H.; Yamanda M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. J. Neurosci. Res. 2004, 75, 742–750. 10.1002/jnr.20025. [DOI] [PubMed] [Google Scholar]
- Hu S.; Maiti P.; Ma Q.; Zuo X.; Jones M. R.; Cole G. M.; Frautschy S. A. Clinical development of curcumin in neurodegenerative disease. Expert Rev. Neurother. 2015, 15, 629–637. 10.1586/14737175.2015.1044981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzotto A.; Zatta P. Resveratrol and Alzheimer’s disease: message in a bottle on red wine and cognition. Front. Aging Neurosci. 2014, 6, 95–102. 10.3389/fnagi.2014.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastianetto S.; Menard C.; Quirion R. Neuroprotective action of resveratrol. Biochim. Biophys. Acta, Mol. Basis Dis. 2015, 1852, 1195–1201. 10.1016/j.bbadis.2014.09.011. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Liu C.; Leibly D.; Landau M.; Zhao M.; Hughes M. P.; Eisenberg D. S. Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. eLife 2013, 2, e00857. 10.7554/eLife.00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P. P. N.; Mohamed T.; Teckwani K.; Tin G. Curcumin binding to beta amyloid: A computational study. Chem. Biol. Drug Des. 2015, 86, 813–820. 10.1111/cbdd.12552. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. The Amyloid beta peptide: A chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev. 2012, 112, 5147–5192. 10.1021/cr3000994. [DOI] [PubMed] [Google Scholar]
- Soto C.; Kindy M. S.; Baumann M.; Frangione B. Inhibition of Alzhemier’s amyloidosis by peptides that prevent β-sheet conformation. Biochem. Biophys. Res. Commun. 1996, 226, 672–680. 10.1006/bbrc.1996.1413. [DOI] [PubMed] [Google Scholar]
- Chao B.; Tong X.; Tang W.; Li D.; He P.; Garcia J.; Zeng L.; Gao A.; Yang L.; Li J.; Nan F.; Jacobs M.; Altmeyer R.; Zuo J.; Hu Y. Discovery and optimization of 2,4-diaminoquinazoline derivatives as a new class of potent Dengue virus inhibitors. J. Med. Chem. 2012, 55, 3135–3143. 10.1021/jm2015952. [DOI] [PubMed] [Google Scholar]
- Thurmond J.; Butchbach M. E. R.; Palomo M.; Pease B.; Rao M.; Bedell L.; Keyvan M.; Pai G.; Mishra R.; Haraldsson M.; Andresson T.; Bragason G.; Thosteinsdottir M.; Bjornsson J. M.; Coovert D. D.; Burghes A. H. M.; Gurney M. E.; Singh J. Synthesis and biological evaluation of novel 2,4-diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy. J. Med. Chem. 2008, 51, 449–469. 10.1021/jm061475p. [DOI] [PubMed] [Google Scholar]
- Petkova A. T.; Yau W. M.; Tycko R. Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry 2006, 45, 498–512. 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.