

Abstract

In recent years, soluble guanylate cyclase (sGC, EC 4.6.1.2) has emerged as an attractive therapeutic target for treating cardiovascular diseases and diseases associated with fibrosis and end-organ failure. Herein, we describe our design and synthesis of a series of 4-hydroxypyrimidine sGC stimulators starting with an internally discovered lead. Our efforts have led to the discovery of IWP-051, a molecule that achieves good alignment of potency, stability, selectivity, and pharmacodynamic effects while maintaining favorable pharmacokinetic properties with once-daily dosing potential in humans.
Keywords: Soluble guanylate cyclase, sGC, NO-independent stimulators, heme-dependent sGC stimulators, nitric oxide, IWP-051
Soluble guanylate cyclase (sGC, EC 4.6.1.2) is a signal-transduction enzyme that binds nitric oxide (NO) and catalyzes the conversion of guanosine-5′-triphosphate (GTP) to the secondary messenger cyclic guanosine-3′,5′-monophosphate (cGMP). The NO-sGC-cGMP signal-transduction pathway is involved in the regulation of various physiological processes, including smooth muscle relaxation, platelet inhibition, and vasodilation.1 The NO-sGC-cGMP pathway plays an important role in coordinating blood flow to tissues, providing oxygen and nutrients, and removing waste products in response to local demands.2 Impairment of sGC and/or reduced NO bioavailability has been implicated in the pathogenesis of cardiovascular, pulmonary, renal, and hepatic diseases.3 The therapeutic benefit of NO-donors such as organic nitrates is limited by lack of efficacy due to variable biometabolism4 and the development of NO tolerance.5
An alternative to NO-donors are sGC stimulators, a class of ligands that bind allosterically to the Fe(II) form of the heme-containing enzyme and stimulate the formation of cGMP.6,7 sGC stimulators can act both independently and in synergy with NO. In preclinical models, sGC stimulators have demonstrated anti-inflammatory and antifibrotic effects, as well as end-organ protections.8−11 The first marketed sGC stimulator, Bayer’s riociguat (Adempas) (Figure 1), was approved in 2013 by the FDA for the treatment of pulmonary arterial hypertension (PAH) and inoperable chronic thromboembolic pulmonary hypertension (CTEPH) based on its ability to improve exercise capacity and symptomatic profile with disease severity defined by the World Health Organization functional classification system.12 We sought to design an sGC stimulator with a sustained pharmacokinetic and pharmacodynamic profile allowing once-daily dosing and potential to minimize the risk of hypotensive side effects.
Figure 1.

Three examples of sGC stimulators.
Some known sGC stimulators, such as riociguat and BAY 41-2272,13 feature a fused ring structure (Figure 1). We have focused our discovery effort on sGC stimulators that utilize a novel, biaryl pyrazole structure, as exemplified by 1(14) (Figure 1). 1 is a potent sGC stimulator as determined by production of cGMP in a human embryonic kidney (HEK) cellular assay in the presence of the NO-donor diethylenetriamine NONOate (DETA-NO, 10 μM) (EC50 = 240 nM). When dosed at 1 and 10 mg/kg iv and po, respectively, 1 has a 26 min iv half-life and is orally bioavailable (Fpo = 53%) in rat. An off-target screen revealed that 1 binds to the κ-opioid (KOP) receptor (IC50 = 1.2 μM) and is a functional agonist (EC50 = 1.5 μM). This off-target activity, coupled with a short half-life in rat, precluded the advancement of 1 into additional preclinical models. We therefore focused on designing sGC stimulators with cellular potency comparable to or better than that of 1 with reduced KOP binding and a better pharmacokinetic profile.
With the goal of improving the selectivity and the pharmacokinetic profile of 1, we sought to introduce functionality at the pyrimidine 5-position. In the course of this effort, amidine 2a was treated with cyanoacrylate 3a (Scheme 1). In contrast to a previous report detailing a similar reaction,15 the desired 4-amino product 4 was obtained in low yield (4%) with the hydroxypyrimidine 5 predominating as a major product (38%). While 5 has attenuated cellular potency (EC50 = 500 nM) relative to 1, the compound is more stable in male rat liver microsomes (RLM, CLint = 14 versus 26 μL/min/mg protein) and comparably stable in human liver microsomes (HLM, CLint = 8 versus 10 μL/min/mg protein). When 5 was dosed orally in rats, it exhibited a significantly higher maximum plasma concentration (Cmax) and area under the curve (AUC) compared with our previously synthesized 4-amino pyrimidine compounds.16 On the basis of these encouraging data we focused our efforts on expanding the 4-hydroxypyrimidine series.
Scheme 1. Synthesis of Analogs 4 and 5.

The general synthesis of pyrimidine analogs is depicted in Scheme 2.17−20 Starting with readily accessible methyl ketones of general structure 6, Claisen condensation with diethyl oxalate in the presence of lithium hexamethyldisilazide (LHMDS) at low temperature, followed by acetic acid-mediated cyclization with benzyl and alkyl hydrazines gave intermediate pyrazole esters 7. Conversion of the pyrazole esters to amidines 2 was accomplished by treatment of the esters with excess trimethylaluminum and ammonium chloride at elevated temperatures.21 Condensation of amidines 2 with acrylates 3 in the presence of base such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or triethylamine delivered the desired pyrimidines, which could then undergo further transformations to access additional pyrazole analogs.22
Scheme 2. General Synthesis of Pyrimidine Analogs.

Using the isoxazole-substituted pyrazole as a template, various pyrimidine analogs were profiled (Table 1). The electronic nature of the substituent at the pyrimidine 5-position has a significant influence on sGC potency. A reduction in cellular potency is observed when the electron-withdrawing nitrile of 5 (EC50 = 500 nM) is replaced with a proton (8, EC50 = ND), an electron-donating benzoyl-protected amino group (9, EC50 = ND), or a carbinol (10, EC50 = ND). Alternatively, replacement of the nitrile with other electron-withdrawing groups generally results in similarly potent compounds. For instance, 5-fluoro analog 11 (EC50 = 290 nM) is more potent than 5 and is also stable in the presence of RLM (CLint = 6 μL/min/mg protein) and HLM (CLint = 3 μL/min/mg protein). Not all compounds featuring electron withdrawing groups at the 5-position are potent stimulators. While chloro-substituted derivative 13 (EC50 = 590 nM) performs comparably to the nitrile analog 5, the increased electron withdrawing nature of the trifluoromethyl group is detrimental to cellular sGC potency (14, EC50 = ND).
Table 1. Pyrimidine Ring SAR.

| Compound | R1 | R2 | R3 | EC50 (nM)a | CLint, RLM, HLM (μL/min/mg protein) |
|---|---|---|---|---|---|
| 1 | NH2 | H | H | 240 | 26, 10 |
| 5 | OH | CN | H | 500 | 14, 8 |
| 8 | OH | H | H | ND (92)b | 15, 12 |
| 9 | OH | -NHBz | H | ND (53) | 26, 14 |
| 10 | OH | -CMe2OH | H | ND (40) | 8, 8 |
| 11 | OH | F | H | 290 | 6, 3 |
| 12 | NH2 | F | H | 190 | 17, 14 |
| 13 | OH | Cl | H | 590 | 5, 7 |
| 14 | OH | CF3 | H | ND (54) | 20, 19 |
| 15 | OH | -SO2Me | H | 900 | 13, 15 |
| 16 | OH | -SO2Ph | H | 350 | 27, 18 |
| 17 | OH | -SO2(p-Cl-Ph) | H | 160 | 15, 6 |
| 18 | OH | -SO2N(Me)2 | H | ND (87) | 24, 8 |
| 19 | OH | -SO2N(Me)(Ph) | H | 730 | 58, 179 |
| 20 | OH | F | Me | ND (70) | 16,9 |
| 21 | OMe | F | H | 320 | 95, 20 |
Potency measured in human embryonic kidney (HEK) cells in the presence of 10 μM DETA-NO.
The geometric mean of at least two repeated runs.
Not determined due to incomplete concentration response. Maximal efficacy (%Emax) at 30 μM shown in parentheses.
A number of 5-substituted sulfones and sulfonamides possess moderate-to-good cellular potency. While methyl sulfone 15 is moderately potent (EC50 = 900 nM), replacing the methyl group with a phenyl moiety increases the potency (16, EC50 = 350 nM). Additionally, the 4-chlorophenyl sulfone 17 is the most potent compound in the series (EC50 = 160 nM). Sulfonamides are less potent than their sulfone counterparts (18, EC50 = ND and 19, EC50 = 730 nM) and 19 is also metabolically unstable in vitro (RLM CLint = 58 μL/min/mg protein, HLM CLint = 179 μL/min/mg protein). Introduction of the methyl group to the pyrimidine 6-position of 11 attenuates cellular potency (20, EC50 = ND). While O-methylation of 11 has no effect on potency (21, EC50 = 320 nM), the metabolic stability is decreased presumably due to oxidative O-demethylation. Finally, the addition of a fluorine atom at the pyrimidine 5-position imparts a greater improvement in potency for hydroxyprimidine 8 (8, EC50 = ND vs 11, EC50 = 290 nM) compared to aminopyrimidine 1 (1, EC50 = 240 nM vs 12, EC50 = 190 nM).
Holding the 5-fluoro-4-hydroxyprimidine moiety constant, we next explored modifications to the pyrazole 5-position (Table 2). Adjustments to the isoxazole moiety significantly affect potency in the cellular sGC assay. Introduction of nitrogen into the isoxazole framework of 11 to give 1,2,4-oxadiazole 22 (EC50 = ND) dramatically reduces sGC activity, although the metabolic stability is maintained. Oxazoles 23 (EC50 = 1500 nM) and 24 (EC50 = ND) also suffer a reduction in potency. The limited space afforded in this vector is highlighted by 5-methylisoxazole 25, in which the addition of a single methyl group to 11 drastically reduces the potency. When the isoxazole is replaced by a larger isothiazole (26, EC50 = 180 nM) cellular potency is retained, albeit at the expense of microsomal stability (RLM CLint = 129 μL/min/mg protein, HLM CLint = 169 μL/min/mg protein). Beyond 5-membered heteroarenes, 2-pyridine 27 (EC50 = ND), nitrile 28 (EC50 = ND) and cyclopropane 29 (EC50 = ND) all fail to reach full efficacy in this particular series.
Table 2. SAR of the R4 Group.


Potency measured in human embryonic kidney (HEK) cells in the presence of 10 μM DETA-NO.
The geometric mean of at least two repeated runs.
Not determined due to incomplete concentration response. Maximal efficacy (%Emax) at 30 μM shown in parentheses.
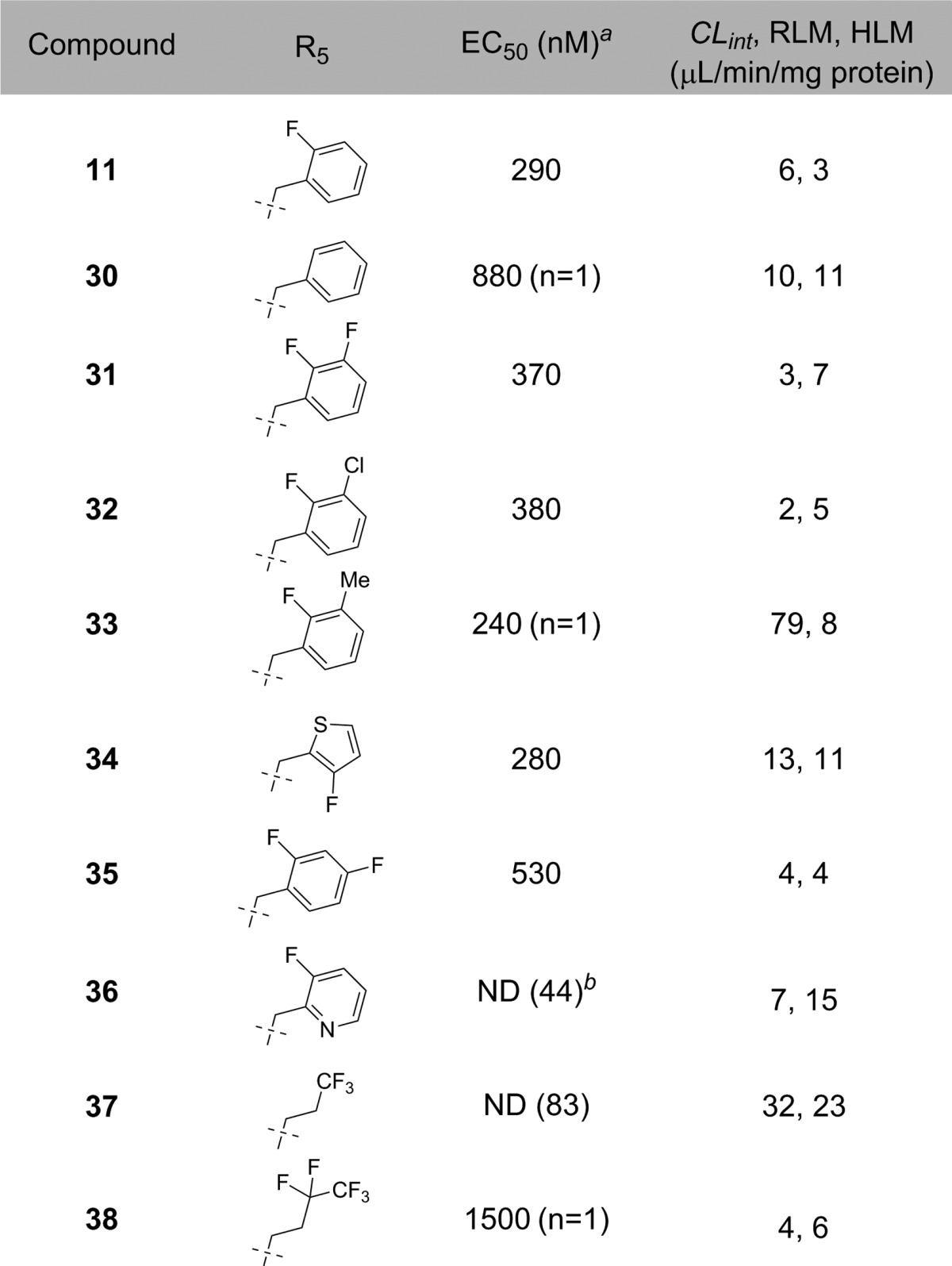
With the 5-fluoro-4-hydroxypyrimidine and isoxazole moieties established at the pyrazole 3- and 5-positions, respectively, we next optimized the pyrazole nitrogen substitution (Table 3). The 2-fluoro benzyl substituent confers sGC potency, as 2-H benzyl analog 30 (EC50 = 880 nM) is less potent than 11. sGC activity is maintained with the introduction of a halogen atom (31, EC50 = 370 nM and 32, EC50 = 380 nM) or a methyl group (33, EC50 = 240 nM) adjacent to the benzyl fluorine. However, the introduction of a methyl group introduces a metabolic site that results in decreased RLM stability (CLint = 79 μL/min/mg protein). While the sGC activity of the 3-fluorothiophene bioisostere 34 is maintained (EC50 = 280 nM), the thiophene is less stable (RLM CLint = 13 μL/min/mg protein, HLM CLint = 11 μL/min/mg protein) than the parent 11.
Table 3. SAR of the R5 Group.
Potency measured in human embryonic kidney (HEK) cells in the presence of 10 μM DETA-NO.
The geometric mean of at least two repeated runs, unless stated otherwise.
Not determined due to incomplete concentration response. Maximal efficacy (%Emax) at 30 μM shown in parentheses.
As with the pyrazole 5-position, electronic manipulations to the 2-fluorobenzyl group can result in a significant loss of potency in this particular series. Introduction of a nitrogen atom (36, EC50 = ND) is not tolerated. Incorporation of perfluorinated alkyl groups23 also adversely affects sGC activity (37, EC50 = ND and 38, EC50 = 1500 nM) in the cellular assay. Based on the exploration of the pyrazole N-1 functionality, we determined that 2-fluorobenzyl substitution was optimal for this 5-fluoro-4-hydroxypyrimidine scaffold.
In purified recombinant human sGC enzyme assay (Enzo Life Sciences) and whole cell (HEK) assays, 11 (hereafter referred to as IWP-051) as well as other compounds of this class stimulate cGMP production in the absence of NO and demonstrate synergy with NO in a concentration responsive manner. In the absence of NO, IWP-051 is less potent and it shows a lower degree of sGC stimulation as measured by maximal efficacy (%Emax). This profile is consistent with the stimulator class of sGC agonist compounds and provides evidence that IWP-051 is a direct sGC stimulator.24
IWP-051 is highly protein bound in both rat (99.9% bound) and human (99.4% bound) plasma. Albumin, a major component of plasma, binds acidic compounds,25 and IWP-051 is mildly acidic with a pKa of 5.75. The acidic hydroxypyrimidine proton also appears to affect the aqueous solubility, as the thermodynamic solubility of IWP-051 increases with increasing pH (3 μg/mL at pH = 7.4 and 142 μg/mL at pH = 9.2). IWP-051 is readily absorbed in the gut with high permeability (Papp, A-B = 21.8 × 10–6 cm/s). Further, the compound does not inhibit cytochrome P450 (CYP) isozymes (<20% inhibition at 10 μM of CYP 3A4, 2D6, 2C9, 1A2, and 2C19). With regard to off-target activity, it has minimal activity in a screen of over 60 potential targets, including the KOP receptor (21% binding at 10 μM). For a representative panel of phosphodiesterases (PDE1B, PDE2A1, PDE3A, PDE4D2, PDE5, PDE10A1) less than 30% inhibition was observed at 10 μM of IWP-051. The lack of inhibition of phosphodiesterases further supports the mechanism of the direct stimulation of sGC.
Pharmacokinetic data associated with IWP-051 in multiple species are represented in Table 4. IWP-051 has a low volume of distribution (Vdss = 180 mL/kg), low clearance (CL = 0.6 mL/min/kg), and long half-life (t1/2 = 4.1 h) in rat, all of which are consistent with high plasma protein binding and a small free fraction in plasma. When dosed orally at 1 mg/kg, IWP-051 is slowly absorbed and reaches a Cmax of over 4 μM (free drug Cmax ∼ 80 nM) at 5 h, with high bioavailability (Fpo = 96 ± 26%). In dog and mouse, the profile of IWP-051 is similar to low clearance, moderate half-life, and high Cmax.26 Standard allometric scaling using mouse, rat and dog PK parameters predicts a human elimination half-life of about 8 h, consistent with once-daily dosing.27 The slow absorption of IWP-051 may be related to its pH-dependent solubility profile with solubility-limited absorption in the upper GI.
Table 4. Pharmacokinetic Parameters of IWP-051 in Mouse, Rat, and Doga.
| parameter | mouse | rat | dog |
|---|---|---|---|
| CL (mL/min/kg) | 5.0 ± 0.1 | 0.6 ± 0.1 | 0.2 ± 0.01 |
| t1/2 iv (h) | 3.4 ± 0.5 | 4.1 ± 0.5 | 5.1 ± 0.9 |
| Vdss (mL/kg) | 1300 ± 100 | 180 ± 60 | 95 ± 12 |
| Tmax (h) | 1.7 ± 1.0 | 5.3 ± 1.2 | 3.3 ± 1.2 |
| Cmax (ng/mL) | 1000 ± 300 | 1500 ± 400 | 3500 ± 600 |
| AUC0-∞ (min·μg/mL) | 270 ± 75 | 1500 ± 280 | 2500 ± 290 |
| Fpo (%) | >100 | 96 ± 26 | 45 ± 3 |
Oral doses of compound administered as solutions in PEG400 at 1 mg/kg; iv doses administered in 10% DMI, 35% propylene glycol, 15% EtOH, 40% D5W (5% dextrose in water) at 0.1 mg/kg. Please see the Supporting Information for additional detail.
When dosed orally as a PEG400 solution in normotensive Sprague–Dawley rats, IWP-051 elicits a dose dependent lowering of mean arterial pressure (MAP), with a 10 mmHg drop in MAP at the minimum effective dose of 1 mg/kg (Figure 2).28 The reduction in MAP approaches −30 mmHg at the 30 mg/kg dose. At the 10 mg/kg dose and above, the reduction in MAP is maintained through 24 h.
Figure 2.
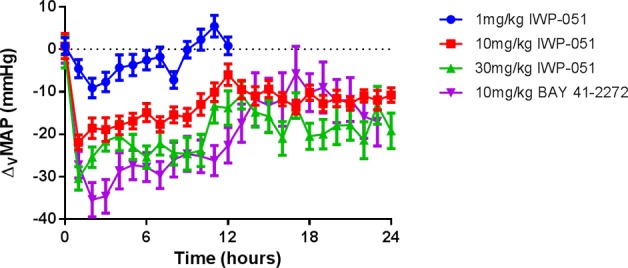
IWP-051 (1, 10, and 30 mg/kg in PEG400) and Bay 41-2272 (10 mg/kg in 0.5% methylcelluolose, Sigma-Aldrich) were dosed orally to male, normotensive Sprague–Dawley rats. Vehicle-subtracted mean arterial pressure (ΔvMAP) in mmHg was monitored for 12–24 h via a conscious, tethered rat model. Data is represented as the change from their own vehicles as 1 h averages + SEM, n = 5–9.
In summary, IWP-051 is a novel, potent, orally bioavailable sGC stimulator that was discovered through the structural manipulation of a 1,3,5-substituted pyrazole scaffold. IWP-051 exhibit minimal off-target liabilities, including CYP and phosphodiesterase activities. Across species, IWP-051 has high protein binding, low clearance, long half-life, and slow absorption. It also exhibits sustained and dose-dependent reduction in MAP in rats. Pharmacokinetic modeling suggests that IWP-051 may have once-daily dosing potential in humans. Based on its in vitro and in vivo profile, IWP-051 is a useful molecule for probing pharmacologic utility of sustained sGC stimulation in preclinical models of human disease.
Acknowledgments
We thank our colleagues at Ironwood Pharmaceuticals, especially, Dr. Vishnu Karnati and Dr. Song Xu, for providing starting materials and intermediates. We thank Dr. James Sheppeck II and Dr. Paul Renhowe for helpful discussions. We also thank Sam Rivers for his technical assistance.
Glossary
Abbreviations
- sGC
soluble guanylate cyclase
- PAH
pulmonary arterial hypertension
- CTEPH
chronic thromboembolic pulmonary hypertension
- HEK
human embryonic kidney
- DETA-NO
diethylenetriamine NONOate
- KOP
κ-opioid
- Cmax
maximum plasma concentration
- AUC
area under the curve
- EtOH
ethanol
- LHMDS
lithium hexamethyldisilazide
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- RLM
rat liver microsome
- HLM
human liver microsome
- CYP
cytochrome P450 isozymes
- MAP
mean arterial pressure
- SEM
the standard error of the mean
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00479.
Synthetic procedures, compound characterization data, description of the cGMP generation assay, determination of cGMP concentrations using LC-MS/MS, error propagation for potency, description of the rat liver microsomal and human liver microsomal stability assays, pharmacokinetic data of select compounds, and procedures for measuring hemodynamic parameters. (PDF)
The authors declare the following competing financial interest(s): All authors own stock/stock options in Ironwood Pharmaceuticals, Inc.
Supplementary Material
References
- Ignarro L. J.; Buga G. M.; Wood K. S.; Byrns R. E.; Chandhuri G. Endothelium derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 9265–9269. 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl U.; de Wit C. A unique role of NO in the control of blood flow. News in Physiological Sciences 1999, 14, 74–80. [DOI] [PubMed] [Google Scholar]
- Stasch J.-P.; Hobbs A. J.. NO-independent, haem-dependent soluble guanylate cyclase stimulators. In Handbook of Experimental Pharmacology; Schmidt H. H. H. W., Hofmann F., Stasch J.- P., Eds.; Springer: Berlin, 2009; Vol. 199, pp 277–308. [DOI] [PubMed] [Google Scholar]
- Li Y.; Zhang D.; Jin W.; Shao C.; Yan P.; Xu C.; Sheng H.; Liu Y.; Yu J.; Xie Y.; Zhao Y.; Lu D.; Nebert D. W.; Harrison D. C.; Huang W.; Jin L. Mitochondrial aldehyde dehydrogenase-2 (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin. J. Clin. Invest. 2006, 116, 506–511. 10.1172/JCI26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzel T.; Daiber A.; Mülsch A. Explaining the phenomenon of nitrate tolerance. Circ. Res. 2005, 97, 618–628. 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- Evgenov O. V.; Pacher P.; Schmidt P. M.; Haskõ G.; Schmidt H. H. H. W.; Stasch J.-P. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat. Rev. Drug Discovery 2006, 5, 755–768. 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe A.; Koesling D. Mechanism of yc-1-induced activation of soluble guanylate cyclase. Mol. Pharmacol. 1998, 53, 123–127. [DOI] [PubMed] [Google Scholar]
- Ahluwalia A.; Foster P.; Scotland R. S.; McLean P. G.; Mathur A.; Perretti M.; Moncada S.; Hobbs A. J. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of P-selectin expression and leukocyte recruitment. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 1386–1391. 10.1073/pnas.0304264101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer C.; Reich N.; Schindler S. C.; Akhmetshina A.; Dees C.; Tomcik M.; Hirth-Dietrich C.; von Degenfeld G.; Sandner P.; Distler O.; Schett G.; Distler J. H. W. Stimulation of soluble guanylate cyclase reduces experimental dermal fibrosis. Ann. Rheum. Dis. 2012, 71, 1019–1026. 10.1136/annrheumdis-2011-200862. [DOI] [PubMed] [Google Scholar]
- Beyer C.; Zenzmaier C.; Palumbo-Zerr K.; Mancuso R.; Distler A.; Dees C.; Zerr P.; Huang J.; Maier C.; Pachowsky M. L.; Friebe A.; Sandner P.; Distler O.; Schett G.; Berger P.; Distler J. H. W. Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non-canonical TGFβ signaling. Ann. Rheum. Dis. 2015, 74, 1408–1416. 10.1136/annrheumdis-2013-204508. [DOI] [PubMed] [Google Scholar]
- Masuyama H.; Tsuruda T.; Sekita Y.; Hatakeyama K.; Imamura T.; Kato J.; Asada Y.; Stasch J.-P.; Kitamura K. Pressure-independent effects of pharmacological stimulation of soluble guanylate cyclase on fibrosis in pressure-overloaded rat heart. Hypertens. Res. 2009, 32, 597–603. 10.1038/hr.2009.64. [DOI] [PubMed] [Google Scholar]
- Conole D.; Scott L. J. Riociguat: first global approval. Drugs 2013, 73, 1967–75. 10.1007/s40265-013-0149-5. [DOI] [PubMed] [Google Scholar]
- Mittendorf J.; Weigand S.; Alonso-Alija C.; Bischoff E.; Feurer A.; Gerisch M.; Kern A.; Knorr A.; Lang D.; Muenter K.; Radtke M.; Schirok H.; Schlemmer K.-H.; Stahl E.; Straub A.; Wunder F.; Stasch J.-P. Discovery of riociguat (BAY 63–2521): A potent, oral stimulator of soluble guanylate cyclase for the treatment of pulmonary hypertension. ChemMedChem 2009, 4, 853–865. 10.1002/cmdc.200900014. [DOI] [PubMed] [Google Scholar]
- Additional pharmacokinetic parameters of 1 are provided in the Supporting Information.
- Straub A.; Benet-Buchholz J.; Fröde R.; Kern A.; Kholsdorfer C.; Schmitt P.; Schwarz T.; Siefert H.-M.; Stasch J.-P. Metabolites of orally active NO-independent pyrazolopyridine stimulators of soluble guanylate cyclase. Bioorg. Med. Chem. 2002, 10, 1711–1717. 10.1016/S0968-0896(02)00034-2. [DOI] [PubMed] [Google Scholar]
- Additional pharmacokinetic parameters of 5 are provided in the Supporting Information. Also:Nakai T.; Perl N. R.; Im G-Y. J.; Lee T. W.-H.; Rohde J. M.; Kim C.; Moore J.; Barden T. C.; Fretzen A.; Butler C.; Long K.; Sarno R.; Germano P.; Jin H.; Carvalho A.; Solberg E. O.; Zimmer D.. Discovery of a novel, orally bioavailable soluble guanylate cyclase stimulator (IWP-051). Presented at 12th Winter Conference on Medicinal & Bioorganic Chemistry, Steamboat Springs, Colorado, January 24–29, 2015.
- Kim C.; Nakai T.; Moore J.; Perl N. R.; Im G-Y. J.; Barden T. C.; Iyengar R. R.; Zimmer D. P.; Fretzen A.; Renhowe P. A.. 2-Benzyl,3-(pyrimidin-2-yl) substituted pyrazoles useful as sgc stimulators. WO 2013/101830A1.
- Kim C.; Nakai T.; Lee T. W.-H.; Moore J.; Perl N. R.; Rhode J.. SGC stimulators. WO 2012/064559 A1.
- Im G-Y. J.; Iyengar R.; Moore J.; Fretzen A.. SGC stimulators. WO 2014/047111A1.
- Nakai T.; Moore J.; Perl N. R.; Iyengar R. R.; Mermerian A.; Im G-Y. J.; Lee T. W.-H.; Hudson C.; Rennie G. R.; Jia J.; Renhowe P. A.; Barden T. C.; Yu X. Y.; Sheppeck J. E.; Iyer K.; Jung J.. SGC stimulators. WO 2014/144100 A2.
- Gielen H.; Alonso-Alija C.; Hendrix M.; Niewöhner U.; Schauss D. A novel approach to amidines from esters. Tetrahedron Lett. 2002, 43, 419–421. 10.1016/S0040-4039(01)02162-1. [DOI] [Google Scholar]
- Detailed procedures for the synthesis of all analogs including 14 and 26 are described in the Supporting Information.
- Raghavan S.; Stelmach J. E.; Smith C. J.; Li H.; Whitehead A.; Waddell S. T.; Chen Y.-H.; Miao S.; Ornoski O. A.; Garfunkle J.; Liao X.; Chang J.; Han X.; Guo J.; Groeper J. A.; Brockunier L. L.; Rosauer K.; Parmee E. R.. Soluble guanylate cyclase activators. WO 2011/149921A1.
- Milne T.; Butler C.; Long K.; Miyashiro J.; Bernier S.; Jacobson S.; Tobin J.; Solberg E.; Shea C.; Germano P.; Moore J.; Chien Y.-t.; Zimmer D.. Iwp-051, a novel, orally available small molecule soluble guanylate cyclase (sgc) stimulator with once-daily dosing potential for the treatment of cardiovascular diseases. Presented at 8th International Nitric Oxide Conference & 6th International Nitrite/Nitrate Conference, Cleveland, OH, June 16–20th, 2014.
- Kerns E. H.; Di Li. Drug-like properties: concepts, structure design and methods: from ADME to toxicity optimization; Elsevier: Oxford, 2008; pp 188. [Google Scholar]
- Urine collected from dogs following the PO dose contained <5% of the circulating IWP-051 excreted as parent. The metabolites in urine were not quantified nor feces samples collected. Due to its low metabolic turnover and low observed in life clearance, IWP-051 is most likely cleared as parent through an undetermined route.
- The interspecies scaling approach to predict clearance in humans from mouse, rat and dog data was performed using simple allometry as previously described:Mahmood I.; Balian J. D. Interspecies scaling: predicting clearance of drugs in humans. Three different approaches. Xenobiotica 1996, 26 (9), 887–95. 10.3109/00498259609052491. [DOI] [PubMed] [Google Scholar]
- The method for measuring hemodynamic parameters is provided in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.