

Abstract

CBP and EP300 are highly homologous, bromodomain-containing transcription coactivators involved in numerous cellular pathways relevant to oncology. As part of our effort to explore the potential therapeutic implications of selectively targeting bromodomains, we set out to identify a CBP/EP300 bromodomain inhibitor that was potent both in vitro and in cellular target engagement assays and was selective over the other members of the bromodomain family. Reported here is a series of cell-potent and selective probes of the CBP/EP300 bromodomains, derived from the fragment screening hit 4-methyl-1,3,4,5-tetrahydro-2H-benzo[b][1,4]diazepin-2-one.
Keywords: Bromodomain, CREBBP, CBP, EP300, benzodiazepinone
Interest in exploring the biological function and therapeutic relevance of bromodomain inhibition has increased in recent years, especially following the progression of several BET bromodomain inhibitors into clinical trials.1 Among other bromodomain-containing proteins implicated in disease pathways, cyclic-AMP response element binding protein (CBP) and adenoviral E1A binding protein of 300 kDa (EP300), a highly homologous2 pair of bromodomain-containing transcriptional coactivators,3 were of interest to us as potential drug targets because of their reported involvement in a number of disease states. Both are multidomain polypeptides with intrinsic protein lysine acetyltransferase activity and have been shown to acetylate histone tails as well as a number of oncology-relevant transcription factors, including p53, c-MYC, and c-MYB.4−8 The single bromodomain of both CBP and EP300 (located on the N-terminus relative to the acetyltransferase domain) is known to be required for the acetylation of nucleosomal histones and transactivation function.9
Both CBP and EP300 play a key role in several disease pathways. For instance, loss of function mutations or heterozygous loss of CBP/EP300 have been linked to the neurological disorder Rubenstein–Taybi syndrome10 as well as late onset lymphomas in mice.11,12 Additionally, both CBP and EP300 are involved in the regulation of critical surveillance mechanisms that ensure appropriate cell proliferation and tissue growth,13 are part of oncogenic fusion proteins in rare forms of leukemia, and are also required for the oncogenic activity of additional fusion proteins.3 Overexpression of CBP and EP300 has been identified in several tumor types and has been directly correlated with aggressiveness.14,15 Finally, recent sequencing efforts have demonstrated recurrent loss-of-function mutations in both CBP and EP300 in a number of tumor types.16−20
As part of our bromodomain inhibition platform, we sought to design a selective and cell-potent small molecule probe of the CBP/EP300 bromodomain in order to characterize the pharmacological impact of disrupting acetyllysine recognition without perturbing the scaffolding or enzymatic functions of the proteins. Specifically, we targeted a compound with submicromolar potency in a CBP/EP300 cellular target engagement assay and 100-fold biochemical selectivity over other bromodomains.21 At the outset of our investigations, no inhibitors that met those criteria were available. Since our probe optimization, a handful of potent CBP bromodomain inhibitors have been reported.22−33
Our effort began by screening a fragment collection of roughly 2000 low molecular weight compounds in a thermal shift assay.25 Compounds that increased the CBP bromodomain melting temperature by at least one degree Celsius were further characterized by dose–response in a time-resolved fluorescence resonance energy transfer (TR-FRET) assay, and a subset of those that showed a dose–response were evaluated by 15N HSQC NMR, isothermal titration calorimetry (ITC), and X-ray crystallography. While potency was of obvious consideration in evaluating hits, of equal importance was selectivity over off-target bromodomains, especially the BET family (using BRD4 BD-1 as a surrogate) because of the pronounced cellular phenotypes associated with BET inhibition.26 Out of this effort, 4-methyl-1,3,4,5-tetrahydro-2H-benzo[b][1,4]diazepin-2-one (Figure 1, Table 1, 1) emerged as a promising validated hit, demonstrating a TR-FRET IC50 of 32 μM, which corresponded to a lipophilic ligand efficiency of 3.4.27,28 Most importantly, however, 1 had a BRD4 BD-1 TR-FRET IC50 of 220 μM, representing a 7-fold selectivity for CBP, which figured prominently in its choice as a starting point for further optimization.
Figure 1.
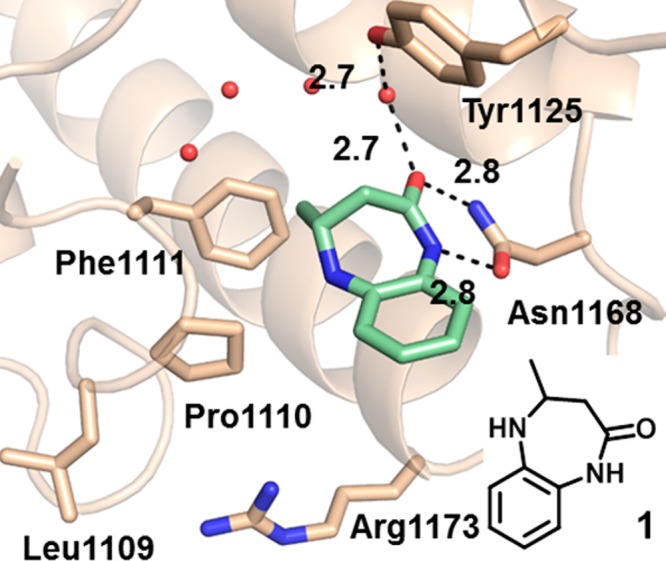
Cocrystal structure of 1 bound to the CBP bromodomain (1.6 Å resolution). PDB code: 4YK0. Hydrogen bond interactions between 1, Asn1168, water1, and Tyr1125 are highlighted by dashed lines (distances in Å). The compound makes hydrophobic interactions with Leu1120, Ile1122, and Val1174 (not depicted for clarity). The BRD4 BD-1 binding site differs primarily at residues Leu1109 and Arg1173 (Trp81 and Asp145, respectively, in BRD4 BD-1).
Table 1. Impact of Various Modifications on 1 CBP Potency.
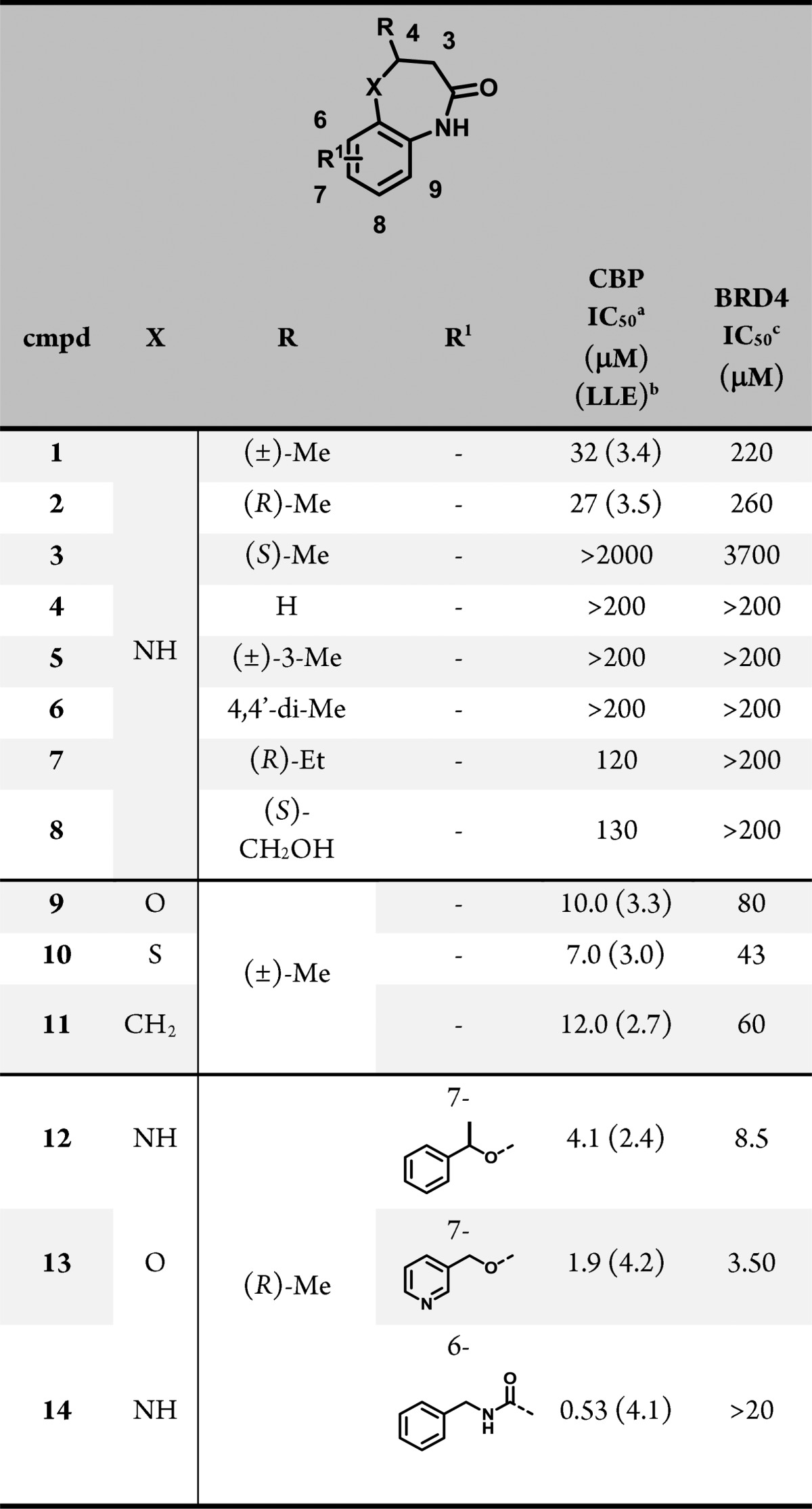
Time-resolved fluorescence resonance energy transfer (TR-FRET) assay with the isolated CBP bromodomain. N = 2 or 3.
LLE for key compounds = pIC50 – clogP.
TR-FRET assay with the isolated BRD4 BD-1 bromodomain. N = 2 or 3.
The binding mode of this series was established with a 1.6 Å resolution cocrystal structure of 1 bound to the isolated CBP bromodomain. As seen in Figure 1, 1 is positioned in the acetyl lysine binding pocket, where the benzodiazepinone carbonyl satisfies the critical hydrogen bonding interaction with Asn1168 observed in the crystal structures of all reported bromodomain inhibitors. A second, solvent-mediated interaction between the carbonyl of 1, water1 (labeled according to Hewings et al.29), and Tyr1125 was also observed. The lactam NH of 1 contributes a further H-bonding interaction with Asn1168, and the 4-methyl group is positioned in the pocket where the acetyl methyl of the natural substrate binds, making favorable van der Waals interactions with Val1174. The benzene ring of 1 fills some of the remaining space in the binding site, making hydrophobic contacts with the Leu1120 and Ile1122 side chains (not depicted in Figure 1 for clarity). Finally, since only the R enantiomer of 1 was observed in the electron density, the individual enantiomers of 1 were separated and tested. Consistent with the binding model, the R enantiomer was >50-fold more potent than the S analogue (Table 1, 2 and 3), a trend that held true for elaborated analogues as well (data not shown).
As expected, many features of the starting fragment were essential to binding. For example, disruption of either the H-bonding interaction with Asn1168 through substitution at the lactam NH (data not shown) or alteration of the van der Waals interaction by removal of the methyl group (Table 1, 4) caused a drastic loss in potency. Likewise, shifting the 4-methyl group to the 3-position (5) or geminal dimethyl substitution at the 4-position (6) resulted in a dramatic loss in binding affinity. Modifications that extended (7) the 4-methyl group or attempted to engage the buried waters at that site (8) were somewhat tolerated but were not advantageous. Various replacements for the NH linker resulted in analogues that bound the bromodomain with potency and selectivity comparable to 1 (9, 10, 11) but which did not represent an improvement in lipophilic ligand efficiency (LLE). Although our primary aim was to generate useful probes for cellular studies, we aimed to keep the physicochemical properties of compounds in this series (as captured by LLE) within a range that would be consistent with moderate clearance in animals, in order to be able to progress compounds as in vivo tools, should a meaningful phenotype be discovered.
Next, we substituted the benzene ring to modulate potency and physicochemical properties, and we determined that these substituents had an impact not only on potency but also on selectivity. Groups at the 8-position did not improve CBP potency and dramatically decreased selectivity over BRD4, while only very small substituents at the 9-position were tolerated and did not improve potency (data not shown). A variety of substitutions at the 7-position (e.g., 12 and 13) improved affinity substantially but reduced selectivity over BRD4. A cocrystal structure with the CBP bromodomain (Figure 2A) showed that the α-methylbenzyl substituent of 12 positioned the pendant phenyl group into a pocket formed between Pro1110 and the Arg1173 side chain. The binding mode of the core benzodiazepinone was otherwise unchanged from the parent compound. Based on an analysis of this and other cocrystal structures, we hypothesized that shifting substitution to the 6-position would orient groups along the LPF shelf (residues L1109–P1110-F1111), increasing binding affinity through close contacts with the protein surface while also introducing potential steric clashes with the bromodomains of the BET family. To that end, compound 14, which demonstrated submicromolar potency against CBP, improved selectivity over BRD4 BD-1 and higher lipophilic ligand efficiency provided an early validation for this approach, which became the focus of our subsequent efforts. A cocrystal structure of 14 and the bromodomain of CBP (Figure 2B) highlighted the proximity of the N-benzyl carboxamide moiety to the LPF shelf and the propensity for lipophlic substituents to cover Pro1110. Interestingly, the amide carbonyl of the side chain was positioned 2.7 Å away from the NH of the diazepinone ring, suggesting that the intramolecular hydrogen bond stabilizes the observed conformation. In order to determine whether the improvements in CBP bromodomain inhibition that we observed in our biochemical assay would translate into a cellular context, a cell-based, target-engagement assay was employed.30 Specifically, a bioluminescence resonance energy transfer (BRET, Promega) assay was used, in which a small molecule CBP/EP300 bromodomain inhibitor disrupted the interaction between a HaloTag-labeled histone and the bromodomain conjugated to NanoLuc luciferase. Most compounds demonstrated a >10-fold shift in potency between the biochemical and cellular assays, which led us to target bromodomain inhibitors with biochemical potencies well below 0.1 μM.
Figure 2.
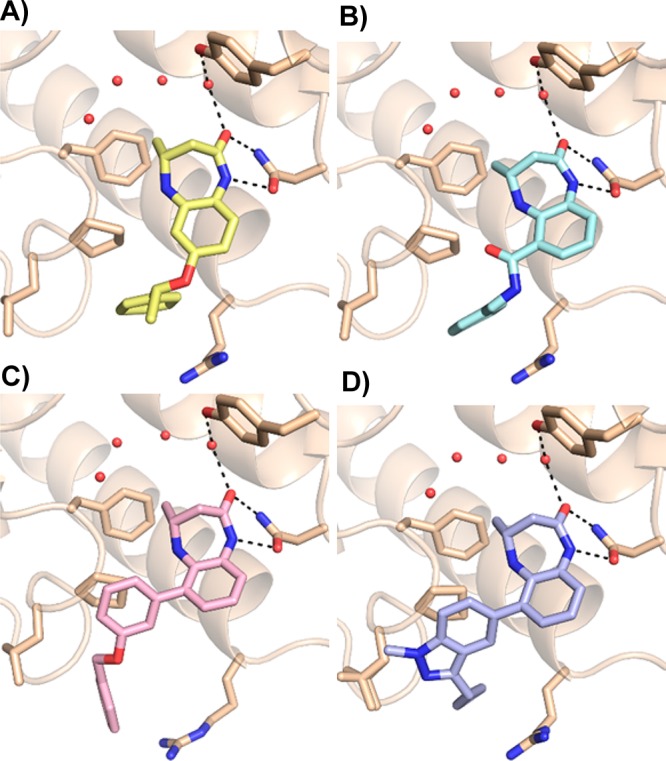
Cocrystal structures of benzodiazepinones bound to the CBP bromodomain. (A) Compound 12 (1.4 Å resolution). The 7-benzyloxy side chain is oriented toward solvent and adopts a favorable conformation that positions the phenyl group into the cleft between Pro1110 and Arg1173. PDB code: 5I83. (B) Compound 14 (1.1 Å resolution). A substituent at the 6-position is proximal to the LPF shelf and still provides a vector to the Pro/Arg cleft. PDB code: 5I86. (C) Compound 23 (2.3 Å resolution). The substituted phenyl side chain fills space over Pro1110 and positions its benzyloxy group into the Pro/Arg cleft. PDB code: 5I8B. (D) Compound 27 (1.1 Å resolution). Indazoles orient 3-position substituents into the cleft between Pro1110 and Arg1173. PDB code: 5I89.
Our exploration of 6-substitution focused on improving the interaction between the compounds and the residues of the LPF shelf of CBP and on introducing clashes with nonconserved residues in BRD4. To that end, α-methyl benzyl compounds 15 and 16 were prepared to introduce a conformational constraint to 14, which reduced BRD4 activity in both diastereomers but failed to improve potency against CBP. Subsequent efforts to modify the N-benzyl ring (17), extend the α-substituent (18), or cyclize the amide substituent (19) did not improve CBP affinity, reduced selectivity over BRD4, or both. Anilide derivatives such as 20 demonstrated the possibility of identifying potent CBP inhibitors, but this subseries was not pursued because of concern about extended conjugation and lower solubility (Table 2).
Table 2. Structure–Activity Relationship of 6-Substituted Benzodiazepinones.
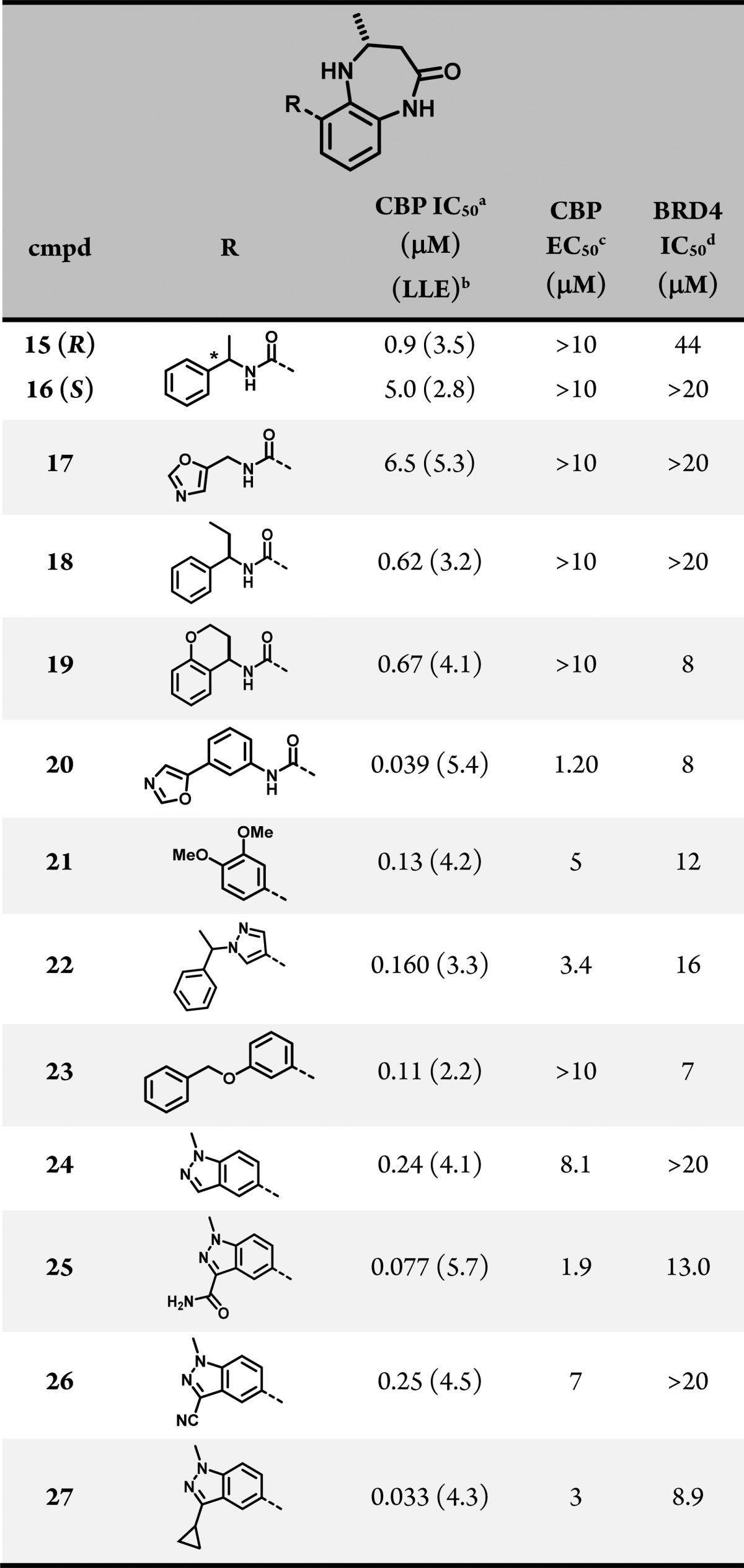
Time-resolved fluorescence resonance energy transfer (TR-FRET) assay with the isolated CBP bromodomain. N = 2.
LLE = pIC50 – clogP.
Bioluminesence resonance energy transfer cellular target engagement assay. N = 3.
TR-FRET assay with the isolated BRD4 BD-1 bromodomain. N = 3.
A breakthrough came with the discovery that potency and selectivity could also be achieved with a variety of directly linked 6-aryl groups, as evidenced by 21, 22, and 23. A crystal structure of 23 (Figure 2C) showed that the directly linked phenyl group stacked on top of Pro1110, with the benzyloxy side chain sitting in the Pro/Arg cleft. Encouraged by this finding, we prepared several 3-substituted 5-linked indazoles, which we intended to orient substituents into the same space while maintaining a more favorable clogP.31 A range of groups at the indazole 3-position led to meaningful improvements in CBP inhibition relative to unsubstituted parent indazole 24 (i.e., 25 and 27). Most importantly, the biochemical potency of these indazoles translated into low-micromolar cellular potency. A cocrystal structure of 27 in the CBP bromodomain (Figure 2D) confirmed that the indazoles filled space over Pro1110 as expected and oriented their 3-substituent into the Pro/Arg cleft.
Further exploration of the indazole substituents resulted in 28 (CPI-637), which demonstrated substantial biochemical potency that was confirmed by isothermal titration calorimetry. A cocrystal structure of 28 in the CBP bromodomain indicated that the compound recapitulated the key hydrogen bonding interactions observed with the parent compound, 1, with the substituted indazole filling space above Pro1110 and the Pro/Arg cleft (Figure 4). As expected, 28 was also potent against EP300 (Table 3), and its opposite enantiomer displayed a >200-fold loss in potency. The biochemical potency of 28 translated well into cells (CBP BRET EC50 = 0.3 μM), and the compound demonstrated a >700-fold selectivity over the BET family of bromodomains (BRD4 IC50 = 11.0 ± 0.6 μM). Compound 28 was also highly selective against other bromodomains (detailed list in the Supporting Information), displaying substantial biochemical activity only against BRD9, which we deemed acceptable, since inhibition of the BRD9 bromodomain has not been shown to produce a pronounced cellular phenotype (data not shown). Recently, we have reported that the bromodomain of CBP/EP300 regulates MYC, a transcription factor widely expressed in human cancer.32 In a cellular assay, we found that 28 inhibits the expression of MYC with an EC50 of 0.60 μM, providing an orthogonal measure of the target engagement of the compound (Figure 3). In our hands, the reported CBP bromodomain inhibitors I-CBP112 and SGC-CBP30 demonstrated EC50 values of >20 and 2.7 μM, respectively. The inactive enantiomer of 28 displayed an EC50 in the same assay of >10 μM. In summary, 28 met our criteria for biochemical potency and off-target bromodomain selectivity and cellular target engagement. Together with its enantiomer, this compound represents a useful probe for the investigation of the biological implications of CBP/EP300 bromodomain inhibition. Further developments of the chemical biology application of these and other bromodomain inhibitors will be reported in due course.
Figure 4.
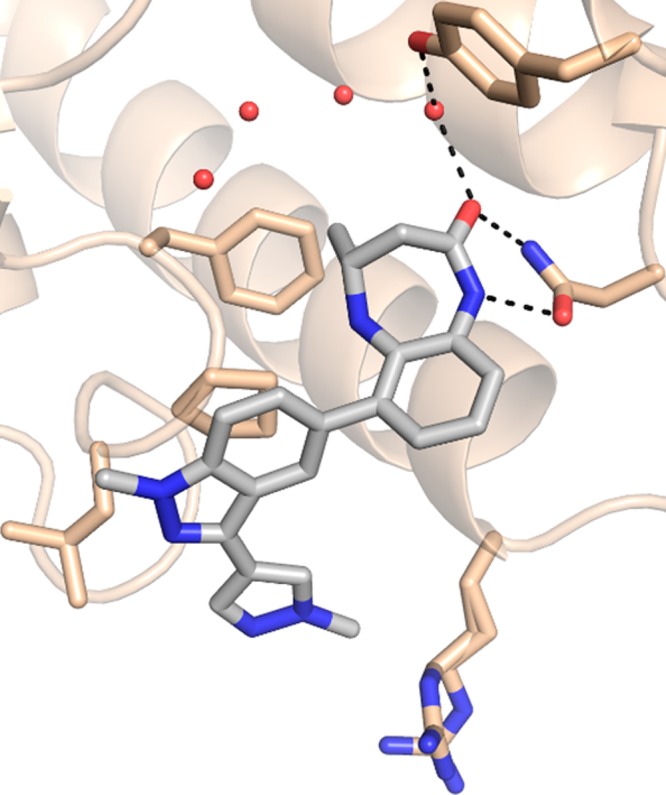
Cocrystal structure of 28 in the CBP bromodomain (1.1 Å resolution). PDB code: 5I8G. The benzodiazepinone core recapitulates the key hydrogen bonding interactions observed with the starting fragment, 1, while the 3-pyrazolylindazole substituent improves potency by filling space over Pro1110 and the Pro/Arg cleft.
Table 3. Profile of CBP/EP300 Probe Compound 28 (CPI-637) and Its Enantiomer.
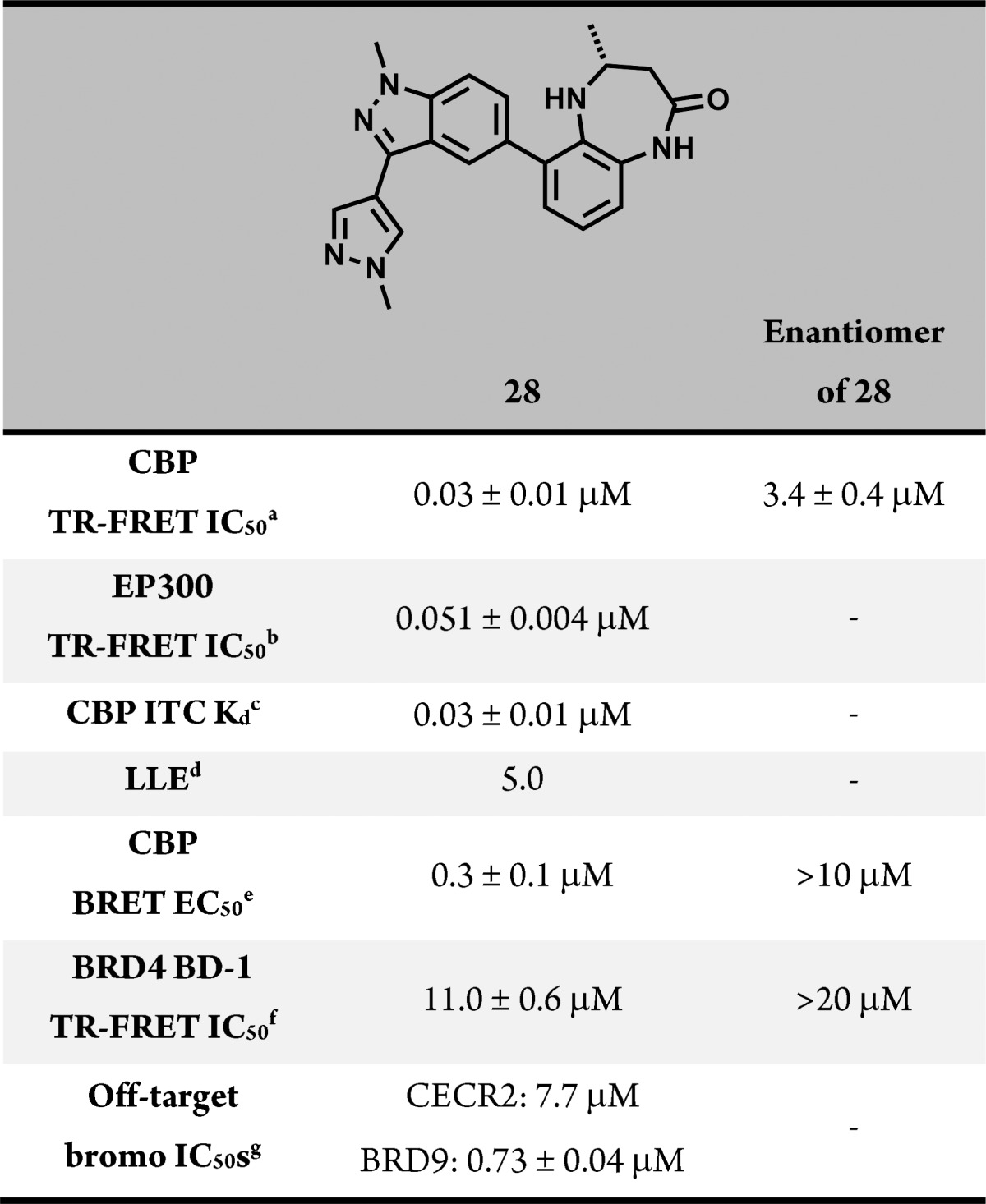
Time-resolved fluorescence energy transfer (TR-FRET) assay with the isolated CBP bromodomain. N > 3.
TR-FRET assay with the isolated EP300 bromodomain. N = 3.
Kd = 0.031 ± 0.012 μM, N = 0.968, ΔH = 7007 ± 115.5 cal/mol, ΔS = 10.9 cal/mol/deg.
LLE = pIC50 – clogP.
Bioluminescence resonance energy transfer cellular assay. N = 2.
TR-FRET assay with the isolated BRD4 BD-1 bromodomain. N > 3.
TR-FRET assays with the respective isolated bromodomains; inhibition is considered off-target if IC50 < 10 μM. See Supporting Information for details.
Figure 3.
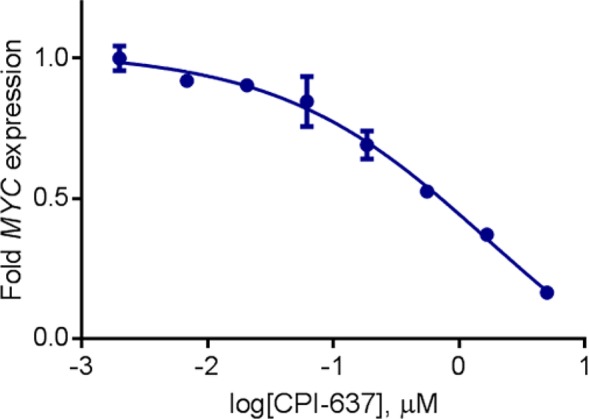
Compound 28 inhibits MYC expression in AMO-1 cells (EC50 = 0.60 μM). Its enantiomer displays an EC50 > 10 μM. Gene expression normalized to GAPDH/PPIB.
Acknowledgments
We are grateful to Ted Peters and Christina Lee in the Lead Discovery group for plating compounds used in these studies and to Custom NMR Services for compound characterization.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00075.
Descriptions of the biochemical and cellular assays and results; synthetic and characterization data for the compounds reported; crystallographic methods and data; off-target bromodomain potency data for 28; and author present addresses (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Romero F. A.; Taylor A. M.; Crawford T. D.; Tsui V.; Cote A.; Magnuson S. Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors. J. Med. Chem. 2016, 59, 1271. 10.1021/acs.jmedchem.5b01514. [DOI] [PubMed] [Google Scholar]
- CBP and EP300 are approximately 60% identical at the amino acid level, but within these functional domains their homology approaches 100%, suggesting that any small molecule inhibitor will target both proteins.
- Goodman R. H.; Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [PubMed] [Google Scholar]
- Jin Q.; Yu L.-R.; Wang L.; Zhang Z.; Kasper L. H.; Lee J.-E.; Wang C.; Brindle P. K.; Dent S. Y. R.; Ge K. Distinct Roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C.; Lucia M. S.; Hansen K. C.; Tyler J. K. CBP/p300-Mediated Acetylation of Histone H3 on Lysine 56. Nature 2009, 459, 113–117. 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A.; Lai C. H.; Zhao X.; Saito S.; Hamilton M. H.; Appella E.; Yao T. P. p300/CBP-Mediated p53 Acetylation is commonly Induced by p53-activating Agents and Inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. 10.1093/emboj/20.6.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J.; Lüscher-Firzlaff J. M.; Rottmann S.; Lilschkis R.; Walsemann G.; Dohmann K.; Austen M.; Lüscher B. Stimulation of c-MYC Transcriptional Activity and Acetylation by Recruitment of the Cofactor CBP. EMBO Rep. 2003, 4, 484–490. 10.1038/sj.embor.embor821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita A.; Towatari M.; Tsuzuki S.; Hayakaw F.; Kosugi H.; Tamai K.; Miyazaki T.; Kinoshita T.; Saito H. C-Myb Acetylation at the Carboxy-Terminal Conserved Domain by Transcriptional Co-Activator P300. Oncogene 2000, 19, 444–451. 10.1038/sj.onc.1203329. [DOI] [PubMed] [Google Scholar]
- Kim J. H.; Cho E. J.; Kim S. T.; Youn H. D. CtBP Represses p300-Mediated Transcriptional Activation by Direct Association with its Bromodomain. Nat. Struct. Mol. Biol. 2005, 12, 423–428. 10.1038/nsmb924. [DOI] [PubMed] [Google Scholar]
- Petrij F.; Giles R. H.; Dauwerse H. G.; Saris J. J.; Hennekam R. C.; Masuno M.; Tommerup N.; van Ommen G. J.; Goodman R. H.; Peters D. J.; Breuning M. H. Rubinstein-Taybi Syndrome Caused by Mutations in the Transcriptional Co-Activator CBP. Nature 1995, 376, 348–351. 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Kung A. L.; Rebel V. I.; Bronson R. T.; Ch’ng L.-E.; Sieff C. A.; Livingston D. M.; Yao T.-P. Gene Dose-Dependent Control of Hematopoisis and Hematologic Tumor Suppression by CBP. Genes Dev. 2000, 14, 272–277. [PMC free article] [PubMed] [Google Scholar]
- Rebel V. I.; Kung A. L.; Tanner E. A.; Yang H.; Bronson R. T.; Livingston D. M. Distinct Roles for CREB-Binding Proetin and P300 in Hematopoietic Stem Cell Self-Renewal. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 14789–14794. 10.1073/pnas.232568499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles R. H.; Peters D. J.; Breuning M. H. Conjunction Dysfunction: CBP/P300 in Human Disease. Trends Genet. 1998, 14, 178–183. 10.1016/S0168-9525(98)01438-3. [DOI] [PubMed] [Google Scholar]
- Li Y.; Yang H. X.; Luo R.-Z.; Zhang Y.; Li M.; Wang X.; Jia W. H. High Expression of P300 Has An Unfavorable Impact on Survival in Resectable Esophageal Squamous Cell Carcinoma. Ann. Thorac. Surg. 2011, 91, 1531–1538. 10.1016/j.athoracsur.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Li M.; Luo R.-Z.; Chen J.-W.; Cao Y.; Lu J.-B.; He J.-H.; Wu Q.-L.; Cai M.-Y. High Expression of Transcriptional Coactivator P300 Correlates with Aggressive Features and Poor Prognosis of Hepatocellular Carcinoma. J. Transl. Med. 2011, 9, 5. 10.1186/1479-5876-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeno K.; Yoshida H.; Pan L.; Luo J. M.; Fujisawa S.; Naito K.; Nakamura S.; Shinjo K.; Takeshita A.; Ohno R.; Ohnishi K. Disease-Related Potential of Mutations in Transcriptional Cofactors CREB-Binding Protein and P300 in Leukemias. Cancer Lett. 2004, 213, 11–20. 10.1016/S0304-3835(03)00442-7. [DOI] [PubMed] [Google Scholar]
- Mullighan C. G.; Zhang J.; Kasper L. H.; Lerach S.; Payne-Turner D.; Phillips L. A.; Heatley s. L.; Holmfeldt L.; Collines-Underwood J. R.; Ma J.; Buetow K. H.; Pui C. H.; Baker S. D.; Brindle P. K.; Downing J. R. CREBBP Mutations in Relapsed Acute Lymphoblastic Leukaemia. Nature 2011, 471, 235–239. 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto M.; Kohno T.; Okudela K.; Otsuka A.; Sasaki H.; Tanabe C.; Sakiyama T.; Hirama C.; Kitabayashi I.; Minna J. D.; Takenoshita S.; Yokota J. Clin. Cancer Res. 2005, 11, 512–510. [PubMed] [Google Scholar]
- Pasqualucci L.; Dominquez-Sola D.; Chiarenz A.; Fabbri G.; Grunn A.; Trifonov V.; Kasper L. H.; Lerach S.; Tang H.; Ma J.; Rossi D.; Chadburn A.; Murty V. V.; Mullighan C. G.; Gaidano G.; Rabadan R.; Brindle P. K.; Dalla-Favera R. Inactivating Mutations of Acetyltransferase Genes in B-Cell Lymphoma. Nature 2011, 471, 189–195. 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huether R.; Dong L.; Chen X.; Wu G.; Parker M.; Wei L.; Ma J.; Edmonson M. N.; Hedlund E. K.; Rusch M. C.; Shurtleff S. A.; Mulder H. L.; Boggs K.; Vadordaria B.; Cheng J.; Yergeau D.; Song G.; Becksfort J.; Lemmon G.; Weber C.; Cai Z.; Dang J.; Walsh M.; Gedman A. L.; Faber Z.; Easton J.; Gruber T.; Kriwacki R. W.; Partridge J. F.; Ding L.; Wilson R. K.; Mardis E. R.; Mullighan C. G.; Gilbertson R. J.; Baker S. J.; Zambetti G.; Ellison D. W.; Zhang J.; Downing J. R. The Lanscape of Somatic Mutations in Epigenetic Regulators across 1,000 Paediatric Cancer Genomes. Nat. Commun. 2014, 5, 3630. 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnage M. E.; Piatnitski Chekler E. L.; Jones L. H. Target Validation Using Chemical Probes. Nat. Chem. Biol. 2013, 9, 195–199. 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- Picaud S.; Fedorov O.; Thanasopoulou A.; Leonards K.; Jones K.; Meier J.; Olzscha H.; Monteiro O.; Martin S.; Philpott M.; Tumber A.; Filippakopoulos P.; Yapp C.; Wells C.; Che K. H.; Bannister A.; Robson S.; Kumar U.; Parr N.; Lee K.; Lugo D.; Jeffrey P.; Taylor S.; Vecellio M. L.; Bountra C.; Brennan P. E.; O’Mahony A.; Velichko S.; Müller S.; Hay D.; Daniels D. L.; Urh M.; La Thangue N. B.; Kouzarides T.; Prinjha R.; Schwaller J.; Knapp S. Generation of a Selective Small Molecule Inhibitor of the CBP/p300 Bromodomain for Leukemia Therapy. Cancer Res. 2015, 75, 5106–5119. 10.1158/0008-5472.CAN-15-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatnitski Chekler E. L.; Pellegrino J. A.; Lanz T. A.; Denny R. A.; Flick A. C.; Coe J.; Langille J.; Basak A.; Liu S.; Stock I. A.; Sahasrabudhe P.; Bonin P. D.; Lee K.; Pletcher M. T.; Jones L. H. Transcriptional Profiling of a Selective CREB Binding Protein Bromodomain Inhibitor Highlights Therapeutic Opportunities. Chem. Biol. 2015, 22, 1588–1596. 10.1016/j.chembiol.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Unzue A.; Xu M.; Dong J.; Wiedmer L.; Spiliotopoulos D.; Caflish A.; Nevado C. Fragment-Based Design of Selective Nanomolar Ligands of the CREBBP Bromodomain. J. Med. Chem. 2015, 59, 1350. 10.1021/acs.jmedchem.5b00172. [DOI] [PubMed] [Google Scholar]
- Rooney T. P. C.; Filippakopoulos P.; Fedorov O.; Picaud S.; Cortopassi W. A.; Hay D.; Martin S.; Tumber A.; Rogers C. M.; Philpott M.; Wang M.; Thompson A. L.; Heightman T. D.; Pryde D. C.; Cook A.; Paton R. S.; Müller S.; Knapp S.; Brennan P. E.; Conway S. J. A Series of Potent CREBBP Bromodomain Ligands Reveals an Induced-Fit Pocket Stabilized by a Cation−π Interaction. Angew. Chem., Int. Ed. 2014, 53, 6126–6130. 10.1002/anie.201402750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantoliano M. W.; Petrella E. C.; Kwasnoski J. D.; Lobanov V. S.; Myslik J.; Graf E.; Carver T.; Asel E.; Springer B. A.; Lane P.; Salemme F. R. High-Density Miniaturized Thermal Shift Assays as a General Strategy for Drug Discovery. J. Biomol. Screening 2001, 6, 429–440. 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- Mertz J. A.; Conery A. R.; Bryant A. M.; Sandy P.; Balasubramanian S.; Mele D. A.; Bergeron L.; Sims R. J. 3rd. Targeting MYC Dependence in Cancer by Inhibiting BET Bromodomains. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16669. 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LLE = pIC50 – clogPShultz M. D. The Thermodynamic Basis for the Use of Lipophilic Efficiency (LipE) in Enthalpic Optimizations. Bioorg. Med. Chem. Lett. 2013, 23, 5992–6000. 10.1016/j.bmcl.2013.08.030. [DOI] [PubMed] [Google Scholar]
- Interestingly, a benzodiazepinone was recently reported to inhibit the first bromodomain of BRD4. Although that compound and compound 1 both bind through a hydrogen-bonding interaction with the highly conserved Asn residue, their binding modes are flipped, relative to each other. For details, seeZhao H.; Gartenmann L.; Dong J.; Caflisch A. Discovery of BRD4 Bromodomain Inhibitors by Fragment-Based High-throughput Docking. Bioorg. Med. Chem. Lett. 2014, 24, 2493–2496. 10.1016/j.bmcl.2014.04.017. [DOI] [PubMed] [Google Scholar]
- Hewings D. S.; Rooney T. P. C.; Jennings L. E.; Hay D. A.; Schofield C. J.; Brennan P. E.; Knapp S.; Conway S. J. Progress in the Development and Application of Small Molecule Inhibitors of Bromodomain-acetyl-lysine interactions. J. Med. Chem. 2012, 55, 9393–9413. 10.1021/jm300915b. [DOI] [PubMed] [Google Scholar]
- Machleidt T.; Woodroofe C. C.; Schwinn M. K.; Mendez J.; Robers M. B.; Zimmerman K.; Otto P.; Daniels D. L.; Kirkland T. A.; Wood K. V. NanoBRET -- A Novel BRET Platform for the Analysis of Protein-Protein Interactions. ACS Chem. Biol. 2015, 10, 1797–1804. 10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
- Our interest in pursuing more polar compounds was driven by our consistent observation that, between two compounds of comparable CBP potency, increased polarity correlated with better selectivity over BRD4 (data not shown).
- Conery A. R.; Centore R. C.; Neiss A.; Keller P. J.; Joshi S.; Spillane K. L.; Sandy P.; Hatton C.; Pardo E.; Zawadzke L.; Bommi-Reddy A.; Gascoigne K. E.; Bryant B. M.; Mertz J. A.; Sims R. J. 3rd. Bromodomain Inhibition of the Transcriptional Coactivators CBP/EP300 as a Therapeutic Strategy to Target the IRF4 Network in Multiple Myeloma. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.