

Abstract
Background
The familial inherited genetic disorder of lipoprotein metabolism affects more than 10 million individuals around the world. Lebanon is one of the several endemic areas for familial hypercholesterolemia (FH) with a founder mutation in the low‐density lipoprotein cholesterol receptor (LDLR) gene, responsible for most of the cases. We have previously shown that 16% of all familial cases with hypercholesterolemia do not show genotype segregation of LDLR with the underlying phenotype.
Methods
We used Sanger sequencing to genotype 25 Lebanese families with severe FH for the gene encoding the LDLR‐associated protein (LDLRAP1), responsible for the recessive form of the disease starting with the four families that did not show any genotype‐phenotype correlation in our previous screening.
Results
We showed that the previously reported p.Q136* variant is linked to the hypercholesterolemia phenotype in the four families. In addition, we showed a variable phenotype between families and between members of the same family. One family exhibits mutations in both LDLR and LDLRAP1 with family members showing differential phenotypes unexplained by the underlying genotypes of the two genes.
Conclusion
The p.Q136* variant in LDLRAP1 is yet another founder mutation in Lebanon and coupled with the LDLR p.C681* variant explains all the genetic causes of FH in Lebanon.
Keywords: Familial Hypercholesterolemia, LDLRAP1, LDLR, founder mutation
Introduction
Familial hypercholesterolemia (FH) (MIM#143890) is an inherited disorder caused by a defect that impairs the normal uptake in the liver of low‐density lipoprotein cholesterol (LDL‐C) from the blood (Khachadurian 1964; Pullinger et al. 2003; Soutar and Naoumova 2007; Kolovou et al. 2011). High LDL‐C in the blood deposits in tissues leads to a 20‐fold increase in the risk of coronary artery disease (Knowles et al. 2015). When the circulating LDL‐C levels are twice the normal values, the disease is called “heterozygous,” whereas the term “homozygous” is linked to levels that are at least fourfold higher than the normal levels (Singh and Bittner 2015). We have suggested previously that the number of mutant alleles does not always correlate with the severity of the clinical phenotype (Fahed and Nemer 2011). Based on the clinical phenotype, the prevalence of the disease has been estimated worldwide to be 1/500 for the “heterozygous form” and 1/1,000,000 for the “homozygous form” (Goldberg et al. 2011; Knowles et al. 2015; Lahtinen et al. 2015; Singh and Bittner 2015). The number of affected individuals is estimated to be more than 10 millions worldwide, with a preponderant prevalence of the “heterozygous form” in some populations like the French Canadians, Afrikaners in South Africa, Indians in South Africa, Finns, and Lebanese reaching up to 1/67 in the Danish population (Benn et al. 2012; Singh and Bittner 2015).
Mutations in three genes have been so far linked to the autosomal dominant form of the disease (ADH): the first encoding the LDL receptor (LDLR) (Brown and Goldstein 1986), the second the apolipoprotein ApoB‐100 (Soria et al. 1989), and the third the pro‐protein convertase subtilin/kexin 9 (PCSK9) (Abifadel et al. 2003). Mutations in the LDLR gene account for nearly 70% of all ADH cases with more than 1600 mutations reported so far (http://www.ucl.ac.uk/ldlr), and with a clinical phenotype directly linked to gene dosage, albeit not uniform even among siblings (Fahed and Nemer 2011; Singh and Bittner 2015). Those in the APOB and PCSK9 account for only 2–5% of the cases in Europe, and <5% of all cases worldwide, respectively (Nissen et al. 1998; Rabes et al. 2000; Abifadel et al. 2003; Lahtinen et al. 2015; Singh and Bittner 2015). The autosomal recessive form of the disease is caused by mutations in the gene encoding the LDLR‐associated protein 1 (LDLRAP1), which is an adaptor protein that binds directly to the LDLR protein and mediates its cellular internalization via the clathrin machinery (Wilund et al. 2002; Cohen et al. 2003; Tietge et al. 2003; Pisciotta et al. 2006; Robles‐Osorio et al. 2006; Quagliarini et al. 2007; Soutar and Naoumova 2007; Soutar 2010). Similar to the case of the LDLR, some mutations in LDLRAP1 are geographically linked mainly in Europe with a pronounced founder effect (Garcia et al. 2001; Wilund et al. 2002; Cohen et al. 2003; Pisciotta et al. 2006; Quagliarini et al. 2007).
The Lebanese National FH Database has recruited so far 124 patients clinically diagnosed with FH through identification of severe index cases undergoing or referred to LDL apheresis, and subsequent cascade screening of family members (Fahed and Nemer 2011; Fahed et al. 2011, 2012). The patients belong to 25 families from different parts of the country. We previously reported major findings from this database based on sequencing of LDLR, PCSK9, and APOB on 80 patients, and we noted that 27.5% of patients belonging to four families did not have a genetic explanation for the phenotype. In order to close this gap, we hereby report the sequencing results of all the three genes in the remaining four families in addition to those of the LDLRAP1 genes in all patients of the database. We explain the genetic cause of FH in 100% of patients. We subsequently focus on understanding the phenotype in 44 patients belonging to four families with mutations in LDLRAP1 gene in light of the concomitant presence of mutations for both LDLR and LDLRAP1 within members of the same family.
Methodology
Families and subjects
The study was approved by the Institutional Review Board of the American University of Beirut. A national database of Lebanese FH patients was established at the American University of Beirut starting in 2008. Patients were recruited from the National LDL Apheresis Center, which is the only center for treatment of severe FH in Lebanon. This was subsequently followed by cascade recruitment and screening of family members. Community visits aided in recruitment of multiplex families. Blood was collected from all subjects for DNA extraction and for fasting lipid level testing. For patients on LDL apheresis, fasting lipid profile testing was collected before apheresis sessions. Clinical data were also extracted from existing medical records.
Genetic studies
DNA extraction from blood was performed using a Qiagen kit (27220 Turnberry Lane Suite 200. Valencia, CA 91355, USA), following the manufacturer's protocol. Primers flanking all exons of the LDLR, LDLRAP1, and PCSK9, and exon 26 of APOB were sequenced in all subjects. Amplicons flanked exons by at least 50 bp from each side to identify splicing mutations. Sequencing was performed using the ABI3100A sequencer at the Molecular Core Facility at the American University of Beirut, following manufacturer's protocol, and as described previously (Nemer et al. 2006; Yehya et al. 2006).
Results
Families and subjects
A total of 25 families (a total of 124 individuals) with FH were recruited as part of the Lebanese National FH Database between 2008 and 2012. Twenty‐one families carried mutations in LDLR gene and whose phenotype was explained by these mutations. Sanger sequencing of the remaining four families (A, B, C, and D) showed a previously reported mutation in LDLRAP1 (Figs. 1, 2, 3) and are the focus of this study. Out of 44 subjects recruited from these families, 18 had normal LDL‐C, 9 had mild FH, and 17 had severe FH (Table 1). The median age of subjects was 39.5 years (range 7–70). Their average LDL‐C level was 270.5 mg/dL. Twelve (27%) subjects had xanthomas and 4 (11%) had xanthelasmas.
Figure 1.
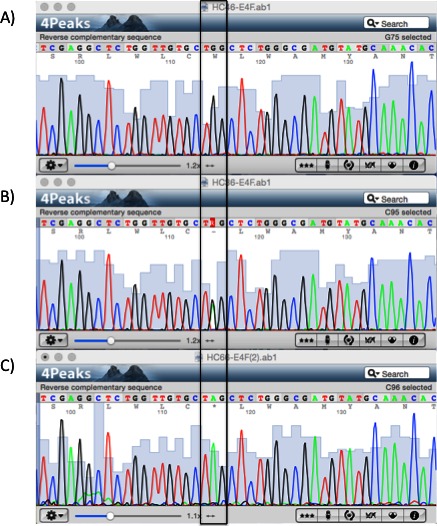
Sequencing results of the LDLRAP1 gene. Chromatograms showing part of exon 4 for the (A) wild‐type, (B) heterozygous, and (C) homozygous p.Q136* variant (boxed).
Figure 2.
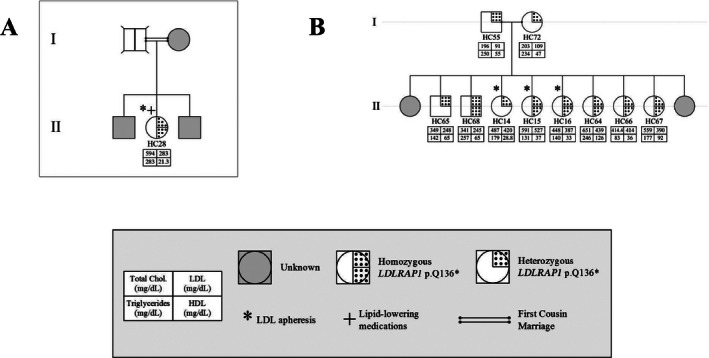
Familial cases of LDLRAP1. Pedigrees of two families A and B with only the p.Q136* variant and the associated phenotypes for each member.
Figure 3.
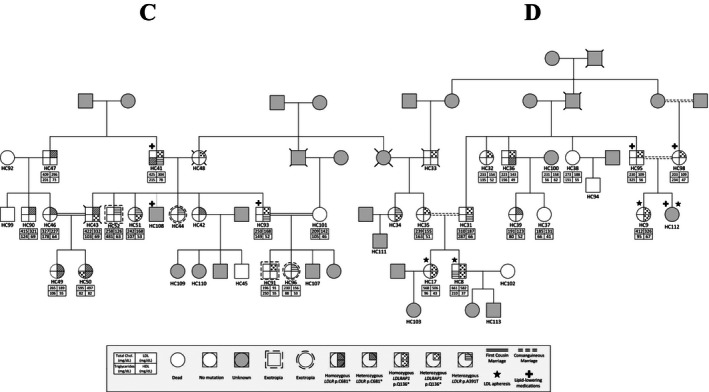
Co‐occurrence of LDLR and LDLRAP1 mutations in familial cases. Pedigrees of two families C and D distantly related with the different genotypes in LDLR (p.C681*, p.A391T) and LDLRAP1 (p.Q136*) and their corresponding phenotypes.
Table 1.
Lipid levels and general characteristics of families enrolled in the study
| Subjects description | |
| Families | 4 |
| Total subjects | 44 |
| With normal LDL | 18 |
| With mild FH | 9 |
| With severe FH | 17 |
| Characteristics | |
| Age (years) | |
| Mean | 38.4 |
| Median | 39.5 |
| Range | 7–70 |
| Gender | |
| Male | 16 (36%) |
| Female | 28 (64%) |
| External manifestations | |
| Xanthomas | 12 (27%) |
| Xanthelasmas | 5 (11%) |
| Lipid levels | |
| TC (mg/dL) | |
| Average | 360 |
| Range | 185–661 |
| SD | 150 |
| LDL (mg/dL) | |
| Average | 270.5 |
| Range | 91–582 |
| SD | 144.48 |
| HDL (mg/dL) | |
| Average | 56.3 |
| Range | 22–126 |
| SD | 19.3 |
| TG (mg/dL) | |
| Average | 162 |
| Range | 66–325 |
| SD | 67 |
LDL, low‐density lipoprotein; FH, familial hypercholesterolemia; TC, total cholesterol; HDL, high‐density lipoprotein; TG, triglycedride; SD, standard deviation.
Table 1 shows the characteristics of these 44 subjects. The pedigrees are shown in Figures 2 (families A and B) and 3 (families C and D, distantly related).
Mutations in FH genes in Lebanon
All four families share the same LDLRAP1 variant, c.C406T that leads to a truncated protein at position 136 (p.Q136*). Eighteen subjects were heterozygous for this variant and only 10 of them had normal LDL‐C levels (Table 2). The average LDL‐C for all heterozygotes was 218 ± 124 mg/dL. Ten patients were homozygous for LDLRAP1 p.Q136* and all had elevated LDL‐C levels (432 ± 101 mg/dL). In families C and D, mutations in LDLR and LDRAP1 co‐occurred in the same patient in various combinations. Table 3 summarizes the average LDL‐C levels and external manifestations of disease for the different mutation combinations in the two genes.
Table 2.
List of all different genetic variants causing FH in Lebanon with their respective inheritance pattern
| Gene | Variant | Amino acid name | ExAc AF (%) | Number of families | Number of carriers, total (normal LDL‐C) | Heterozygous | Homozygous | Other populations reported | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of carriers (with normal LDL‐C) | Average ± SD LDL‐C (mg/dL) | Number of carriers (with normal LDL‐C) | Average ± SD LDL‐C (mg/dL) | |||||||
| LDLR | c.C2043A | p.C681* | 8.26E‐06 | 20 | 47 (4) | 24 (3) | 257 ± 111 | 23 (1) | 490 ± 145 | Israel, Brazil, UK, USA |
| LDLR | C.G1171A | p.A391T | 0.0467 | 3 | 9 (4) | 9 (4) | 197 ± 61 | 0 | NA | South Africa, Russia, Canada, Morocco, Denmark |
| LDLR | c.T1352C | p.I451T | 0 | 1 | 4 (1) | 2 (1) | 161 ± 28 | 2 (0) | 596 ± 74 | Greece, Netherlands |
| LDLR | C.C2177T | p.T726I | 0.006261 | 1 | 2 (0 | 2 (0) | 194 | 0 (0) | NA | USA, UK, Netherlands, Germany, France |
| LDLR | 980_981insA | His327fsX5 | 0 | 2 | 3 (0) | 0 | NA | 2 (0) | 589 ± 26 | Denmark |
| LDLRAP1 | c.C406T | p.Q136* | 0 | 4 | 28 (10) | 18 (10) | 218 ± 124 | 10 (0) | 432 ± 101 | Turkey |
FH, familial hypercholesterolemia; LDL‐C, low‐density lipoprotein cholesterol; SD, standard deviation.
Table 3.
Gentoype/phenotype of all patients with LDLR and/or LDLRAP1 variants
| Genotype | N | % | LDL (mg/dL) (average ± SD) | XO | XA | LDL apheresis |
|---|---|---|---|---|---|---|
| No mutation | 6 | 13.6 | 150 ± 22 | 1/6 | 0/6 | 0/6 |
| LDLRAP hetero Q136* | 11 | 25.0 | 184 ± 116 | 0/11 | 0/11 | 1/11 |
| LDLRAP homo Q136* | 9 | 20.5 | 391 ± 95 | 7/9 | 3/9 | 5/9 |
| LDLR hetero A391T | 3 | 6.8 | 156 | 0/3 | 0/3 | 0/3 |
| LDLR hetero C681* | 5 | 11.4 | 242 ± 88 | 1/5 | 1/5 | 0/5 |
| LDLR homo C681* | 1 | 2.3 | 189 | 0/1 | 0/1 | 0/1 |
| LDLRAP hetero Q136* + LDLR hetero C681* | 2 | 4.5 | 238 ± 134 | 1/2 | 0/2 | 0/2 |
| LDLRAP hetero Q136* + LDLR homo C681* | 1 | 2.3 | 497 | 1/1 | 1/1 | 0/1 |
| LDLRAP hetero Q136* + LDLR hetero A391T | 4 | 9.1 | 174 ± 11 | 0/4 | 0/4 | 0/4 |
| LDLRAP homo Q136* + LDLR hetero A391T | 1 | 2.3 | 582 | 1/1 | 0/1 | 1/1 |
| LDLR hetero C681* + LDLR hetero A391T + LDLRAP hetero Q136* | 1 | 2.3 | 304 | 0/1 | 0/1 | 0/1 |
LDLR, low‐density lipoprotein cholesterol receptor; LDLRAP1, LDLR‐associated protein; LDL, low‐density lipoprotein; XO, xanthomas; XA, xanthelasmas; SD, standard deviation.
Discussion
FH is one of the best described genetic disorders in Lebanon with a high incidence in a small (four millions) and highly consanguineous population. By studying the entire registered FH population in Lebanon, we reported the prevalence of the LDLR p.C681* allele variant among affected individuals. In this report we close the genetic gap by unraveling the prevalence of yet another founder mutation in LDLRAP1 that combined with the LDLR variants (p.C681*, p.H327fsX5, p.A391T, and p.I451T) accounts to 100% of the population prevalence of FH in Lebanon.
Consanguinity, inbreeding, and founder mutations in FH
The high prevalence of consanguineous marriages in the Middle‐East and North African region contributes to a high prevalence of genetic disease, neonate deaths and miscarriages. Both dominant forms due to inbreeding and recessive forms of disease are increased. In Lebanon, the rate of these marriages was estimated to be around 35% based on a cross‐sectional study (Barbour and Salameh 2009). It has been long debated that FH in Lebanon has a founder component linked directly to the high prevalence of consanguineous marriages. The characterization of the Lebanese allele in the LDLR gene (p.C681*) in Lebanese expatriates was corroborated recently by our group in showing that it accounts for 80% of the studied families in Lebanon (Table 3). Four percent of the families carried other LDLR variants, and the remaining 16% carried the p.Q136* LDLRAP1 alone or in conjunction with the p.C681*. The high prevalence of consanguinity and inbreeding results in a higher prevalence of homozygous/severe disease as well as in compound heterozygosity of various disease alleles as seen in families C and D.
Five mutations in LDLR and one mutation in LDLRAP1 explain the FH phenotype in 100% of Lebanese patients in the National FH Database (Table 2). All mutations have been reported in other populations, although primarily in patients of Lebanese ancestry. The LDLR p.C681* is present in 20 families, either as the only mutation or cosegregating with other FH‐causing mutations, such as in families C and D (Fig. 2).
Founder mutations for FH have been described in many populations, and the most studied one with its relative small population is Sardinia in Italy whereby two LDLRAP1 variants (p.W22X and Fs144>X170) account for all the cases, and the estimated frequency of the heterozygous carriers is ~1:143 (Filigheddu et al. 2009). The LDLRAP1 p.Q136* variant was previously described in a Lebanese expatriate family living in Sardinia (Garcia et al. 2001), and in two Turkish families with severely delayed LDL catabolism (Schmidt et al. 1998; Tietge et al. 2003; Soufi et al. 2013). The results of our current study suggest that the variant likely has a founder effect in Lebanon and is the second “Lebanese allele” for FH. It is also possible that the variant migrated between modern day Turkey and Lebanon due to intermarriages during the 600‐year Turkish Empire rule of Lebanon (Ottoman Empire 1299–1922 AC).
Variable expressivity of FH mutations
Although founder mutations are usually associated with a viable phenotype that could be described as mild allowing the transmission of the genotypes across generations, in most cases of FH whether dominant or recessive, the clinical features appear very early on in the patients as is the case of the Lebanese LDLR allele (Khachadurian 1964; Fahed et al. 2011). Management of such cases could thus be ameliorated based on family history. The big challenge remains, however, the variable expressivity of the phenotype despite sharing the same genotype.
The gaps in establishing phenotype–genotype correlations might be overcome by whole exome sequencing of all affected and nonaffected familial cases. Although no single variant modifier allele has been linked so far to amelioration or exacerbation of the clinical phenotype when concomitant with a FH genotype, some variants have been previously reported in carriers of the dominant hypercholesterolemia mutations with low levels of LDL‐C (Brusgaard et al. 2010; Huijgen et al. 2012). These variants affect mainly PCSK9, APOB, and ANGPTL3 (data not shown); total loss of the latter causes familial hypolipidemia (Musunuru et al. 2010). In our cohort of patients we failed to detect any potential causative variant in the three genes except for the PCSK9 variant (p.R46L) which was previously associated with low LDL‐C levels in normal and hypercholesterolemia patients (Humphries et al. 2009), and was found in only one patient (HC28 in Fig. 2A). This patient has a very severe phenotype and thus the p.R46L variant has no effect on a case where the adaptor protein is presumably not produced, confirming thus, the hypothesis that the action of PCSK9 is dependent on its interaction directly or indirectly via the LDLR to LDLRAP1 (Soutar and Naoumova 2007; Fahed and Nemer 2011; Urban et al. 2013). This contrasts the results obtained for the p.L21dup PCSK9 allele found in patients with the p.C681* variant and conferring low LDL‐C levels (Abifadel et al. 2009).
Failure of the dominant and recessive dichotomy in FH
The concomitant presence of the two forms of FH, recessive and dominant, within the same family represents a major challenge for treatment and follow‐up because of the complexity of the phenotype. Only two cases such as the ones we are hereby reporting are published. The first reported a double heterozygous case for both LDLR and LDLRAP1 suggesting that the additional LDLRAP1 variant confers more severity to the phenotype observed in the LDLR variant alone (Tada et al. 2011). The second involves a Turkish family with the same homozygous p.Q136* LDLRAP1 variant as in this report, in combination with the p.Q254P LDLR variant (Soufi et al. 2013). The authors noted that the phenotype observed in the patients with combined heterozygous LDLR variant and LDLRAP1 homozygous variant is more severe than the one observed in the homozygous LDLR variant alone. In our case, we could not identify a universal pattern but we noticed that the combination of homozygous variant of one gene with the heterozygous variant of the other gene results in a drastic increase of LDL‐C levels as is the case of patient HC50 in family C and patient HC8 in family D (Fig. 3). Nevertheless, the presence of three heterozygous variants does not equally increase the LDL‐C levels as in the case of HC41 (Fig. 3).
While the gene LDLRAP1 is presumed to act in a recessive fashion, we report heterozygous carriers of p.Q136* with a severe FH phenotype equivalent to the homozygous carriers (Figs. 2, 3). This goes in parallel with our previous findings on the heterogeneous phenotypes observed in patients with the LDLR p.C681* variant (Fahed et al. 2012). While gene dosage of LDLR and LDRAP1 is playing a role in dictating the serum LDL‐C levels, it is not possible to obtain enough statistical power with current numbers to better understand these associations. Other players such as environmental, genetic modifiers, and treatment are also likely confounding the relationship with LDL‐C levels. Finally, the polygenic nature of the disease is to be taken into consideration whereby in some cohorts up to 60% of the cases are still with no genetic clues (Talmud et al. 2013; Futema et al. 2015) (Fig. 4).
Figure 4.
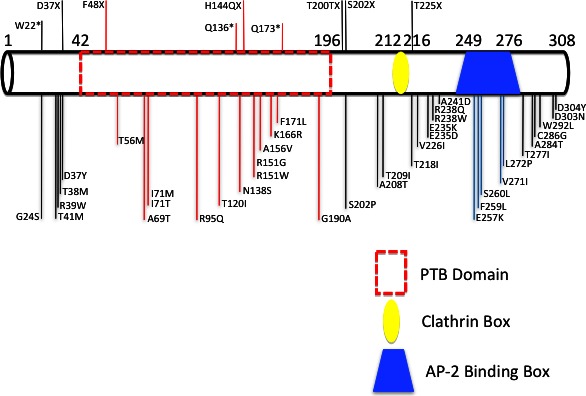
Diagram of all LDLRAP1 mutations ever reported and their location on the protein.
Overall, a better understanding of the different genetic variants in the physiological context is required to better define the role of each of the players in cholesterol synthesis and uptake process in a particular patient. Whole‐genome sequencing of large cohorts of FH are needed to establish statistical power for better understanding of LDL‐C level modifiers. Meanwhile, cascade genetic screening allows identification of cases early and serum LDL‐C level remain the best predictor of cardiovascular disease risk (Fahed et al. 2014).
Conflict of Interest
None declared.
Acknowledgments
We thank the families for their contribution to this research. We also thank members of the Congenital Heart Disease Genetic Program (CHDGP) for critical reading of the manuscript. This work is supported by a grant from the National Council for Scientific Research of Lebanon (CNRS‐L).
References
- Abifadel, M. , Varret M., Rabes J. P., Allard D., Ouguerram K., Devillers M., et al. 2003. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34:154–156. [DOI] [PubMed] [Google Scholar]
- Abifadel, M. , Rabes J. P., Jambart S., Halaby G., Gannage‐Yared M. H., Sarkis A., et al. 2009. The molecular basis of familial hypercholesterolemia in Lebanon: spectrum of LDLR mutations and role of PCSK9 as a modifier gene. Hum. Mutat. 30:E682–E691. [DOI] [PubMed] [Google Scholar]
- Barbour, B. , and Salameh P.. 2009. Consanguinity in Lebanon: prevalence, distribution and determinants. J. Biosoc. Sci. 41:505–517. [DOI] [PubMed] [Google Scholar]
- Benn, M. , Watts G. F., Tybjaerg‐Hansen A., and Nordestgaard B. G.. 2012. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol‐lowering medication. J. Clin. Endocrinol. Metab. 97:3956–3964. [DOI] [PubMed] [Google Scholar]
- Brown, M. S. , and Goldstein J. L.. 1986. A receptor‐mediated pathway for cholesterol homeostasis. Science 232:34–47. [DOI] [PubMed] [Google Scholar]
- Brusgaard, K. , Kjaersgaard L., Hansen A. B., and Husby S.. 2010. New mutations in APOB100 involved in familial hypobetalipoproteinemia. J. Clin. Lipidol. 4:181–184. [DOI] [PubMed] [Google Scholar]
- Cohen, J. C. , Kimmel M., Polanski A., and Hobbs H. H.. 2003. Molecular mechanisms of autosomal recessive hypercholesterolemia. Curr. Opin. Lipidol. 14:121–127. [DOI] [PubMed] [Google Scholar]
- Fahed, A. C. , and Nemer G. M.. 2011. Familial hypercholesterolemia: the lipids or the genes? Nutr. Metab. (Lond.) 8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahed, A. C. , Safa R. M., Haddad F. F., Bitar F. F., Andary R. R., Arabi M. T., et al. 2011. Homozygous familial hypercholesterolemia in Lebanon: a genotype/phenotype correlation. Mol. Genet. Metab. 102:181–188. [DOI] [PubMed] [Google Scholar]
- Fahed, A. C. , Bitar F. F., Khalaf R. I., Moubarak E. M., Azar S. T., and Nemer G. M.. 2012. The Lebanese allele at the LDLR in normocholesterolemic people merits reconsideration of genotype phenotype correlations in familial hypercholesterolemia. Endocrine 42:445–448. [DOI] [PubMed] [Google Scholar]
- Fahed, A. C. , Habib R. H., Nemer G. M., Azar S. T., Andary R. R., Arabi M. T., et al. 2014. Low‐density lipoprotein levels and not mutation status predict intima‐media thickness in familial hypercholesterolemia. Ann. Vasc. Surg. 28:421–426. [DOI] [PubMed] [Google Scholar]
- Filigheddu, F. , Quagliarini F., Campagna F., Secci T., Degortes S., Zaninello R., et al. 2009. Prevalence and clinical features of heterozygous carriers of autosomal recessive hypercholesterolemia in Sardinia. Atherosclerosis 207:162–167. [DOI] [PubMed] [Google Scholar]
- Futema, M. , Shah S., Cooper J. A., Li K., Whittall R. A., Sharifi M., et al. 2015. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries. Clin. Chem. 61:231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, C. K. , Wilund K., Arca M., Zuliani G., Fellin R., Maioli M., et al. 2001. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 292:1394–1398. [DOI] [PubMed] [Google Scholar]
- Goldberg, A. C. , Hopkins P. N., Toth P. P., Ballantyne C. M., Rader D. J., Robinson J. G., et al. 2011. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Clin. Lipidol. 5:133–140. [DOI] [PubMed] [Google Scholar]
- Huijgen, R. , Sjouke B., Vis K., de Randamie J. S., Defesche J. C., Kastelein J. J., et al. 2012. Genetic variation in APOB, PCSK9, and ANGPTL3 in carriers of pathogenic autosomal dominant hypercholesterolemic mutations with unexpected low LDL‐Cl Levels. Hum. Mutat. 33:448–455. [DOI] [PubMed] [Google Scholar]
- Humphries, S. E. , Neely R. D., Whittall R. A., Troutt J. S., Konrad R. J., Scartezini M., et al. 2009. Healthy individuals carrying the PCSK9 p. R46L variant and familial hypercholesterolemia patients carrying PCSK9 p.D374Y exhibit lower plasma concentrations of PCSK9. Clin. Chem. 55:2153–2161. [DOI] [PubMed] [Google Scholar]
- Khachadurian, A. K. 1964. The inheritance of essential familial hypercholesterolemia. Am. J. Med. 37:402–407. [DOI] [PubMed] [Google Scholar]
- Knowles, J. W. , Stone N. J., and Ballantyne C. M.. 2015. Familial hypercholesterolemia and the 2013 American College of Cardiology/American Heart Association Guidelines: myths, oversimplification, and misinterpretation versus facts. Am. J. Cardiol. 116:481–484. [DOI] [PubMed] [Google Scholar]
- Kolovou, G. D. , Kostakou P. M., and Anagnostopoulou K. K.. 2011. Familial hypercholesterolemia and triglyceride metabolism. Int. J. Cardiol. 147:349–358. [DOI] [PubMed] [Google Scholar]
- Lahtinen, A. M. , Havulinna A. S., Jula A., Salomaa V., and Kontula K.. 2015. Prevalence and clinical correlates of familial hypercholesterolemia founder mutations in the general population. Atherosclerosis 238:64–69. [DOI] [PubMed] [Google Scholar]
- Musunuru, K. , Pirruccello J. P., Do R., Peloso G. M., Guiducci C., Sougnez C., et al. 2010. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N. Engl. J. Med. 363:2220–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemer, G. , Fadlalah F., Usta J., Nemer M., Dbaibo G., Obeid M., et al. 2006. A novel mutation in the GATA4 gene in patients with tetralogy of Fallot. Hum. Mutat. 27:293–294. [DOI] [PubMed] [Google Scholar]
- Nissen, H. , Lestavel S., Hansen T. S., Luc G., Bruckert E., and Clavey V.. 1998. Mutation screening of the LDLR gene and ApoB gene in patients with a phenotype of familial hypercholesterolemia and normal values in a functional LDL receptor/apolipoprotein B assay. Clin. Genet. 54:79–82. [DOI] [PubMed] [Google Scholar]
- Pisciotta, L. , Priore Oliva C., Pes G. M., Di Scala L., Bellocchio A., Fresa R., et al. 2006. Autosomal recessive hypercholesterolemia (ARH) and homozygous familial hypercholesterolemia (FH): a phenotypic comparison. Atherosclerosis 188:398–405. [DOI] [PubMed] [Google Scholar]
- Pullinger, C. R. , Kane J. P., and Malloy M. J.. 2003. Primary hypercholesterolemia: genetic causes and treatment of five monogenic disorders. Expert Rev. Cardiovasc. Ther. 1:107–119. [DOI] [PubMed] [Google Scholar]
- Quagliarini, F. , Vallve J. C., Campagna F., Alvaro A., Fuentes‐Jimenez F. J., Sirinian M. I., et al. 2007. Autosomal recessive hypercholesterolemia in Spanish kindred due to a large deletion in the ARH gene. Mol. Genet. Metab. 92:243–248. [DOI] [PubMed] [Google Scholar]
- Rabes, J. P. , Varret M., Devillers M., Aegerter P., Villeger L., Krempf M., et al. 2000. R3531C mutation in the apolipoprotein B gene is not sufficient to cause hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 20:E76–E82. [DOI] [PubMed] [Google Scholar]
- Robles‐Osorio, L. , Huerta‐Zepeda A., Ordonez M. L., Canizales‐Quinteros S., Diaz‐Villasenor A., Gutierrez‐Aguilar R., et al. 2006. Genetic heterogeneity of autosomal dominant hypercholesterolemia in Mexico. Arch. Med. Res. 37:102–108. [DOI] [PubMed] [Google Scholar]
- Schmidt, H. H. , Stuhrmann M., Shamburek R., Schewe C. K., Ebhardt M., Zech L. A., et al. 1998. Delayed low density lipoprotein (LDL) catabolism despite a functional intact LDL‐apolipoprotein B particle and LDL‐receptor in a subject with clinical homozygous familial hypercholesterolemia. J. Clin. Endocrinol. Metab. 83:2167–2174. [DOI] [PubMed] [Google Scholar]
- Singh, S. , and Bittner V.. 2015. Familial hypercholesterolemia – epidemiology, diagnosis, and screening. Curr. Atheroscler. Rep. 17:482. [DOI] [PubMed] [Google Scholar]
- Soria, L. F. , Ludwig E. H., Clarke H. R., Vega G. L., Grundy S. M., and McCarthy B. J.. 1989. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B‐100. Proc. Natl. Acad. Sci. USA 86:587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi, M. , Rust S., Walter M., and Schaefer J. R.. 2013. A combined LDL receptor/LDL receptor adaptor protein 1 mutation as the cause for severe familial hypercholesterolemia. Gene 521:200–203. [DOI] [PubMed] [Google Scholar]
- Soutar, A. K. 2010. Rare genetic causes of autosomal dominant or recessive hypercholesterolaemia. IUBMB Life 62:125–131. [DOI] [PubMed] [Google Scholar]
- Soutar, A. K. , and Naoumova R. P.. 2007. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 4:214–225. [DOI] [PubMed] [Google Scholar]
- Tada, H. , Kawashiri M. A., Ohtani R., Noguchi T., Nakanishi C., Konno T., et al. 2011. A novel type of familial hypercholesterolemia: double heterozygous mutations in LDL receptor and LDL receptor adaptor protein 1 gene. Atherosclerosis 219:663–666. [DOI] [PubMed] [Google Scholar]
- Talmud, P. J. , Shah S., Whittall R., Futema M., Howard P., Cooper J. A., et al. 2013. Use of low‐density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case‐control study. Lancet 381:1293–1301. [DOI] [PubMed] [Google Scholar]
- Tietge, U. J. , Genschel J., and Schmidt H. H.. 2003. A Q136Stop mutation in the ARH gene causing autosomal recessive hypercholesterolaemia with severely delayed LDL catabolism. J. Intern. Med. 253:582–583. [DOI] [PubMed] [Google Scholar]
- Urban, D. , Poss J., Bohm M., and Laufs U.. 2013. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J. Am. Coll. Cardiol. 62:1401–1408. [DOI] [PubMed] [Google Scholar]
- Wilund, K. R. , Yi M., Campagna F., Arca M., Zuliani G., Fellin R., et al. 2002. Molecular mechanisms of autosomal recessive hypercholesterolemia. Hum. Mol. Genet. 11:3019–3030. [DOI] [PubMed] [Google Scholar]
- Yehya, A. , Souki R., Bitar F., and Nemer G.. 2006. Differential duplication of an intronic region in the NFATC1 gene in patients with congenital heart disease. Genome 49:1092–1098. [DOI] [PubMed] [Google Scholar]