

Abstract
Background
Autosomal recessive distal renal tubular acidosis (dRTA) is a rare disease characterized by a hyperchloremic metabolic acidosis with normal anion gap, hypokalemia, hypercalciuria, hypocitraturia, nephrocalcinosis, and conserved glomerular filtration rate. In some cases, neurosensorial deafness is associated. dRTA is developed during the first months of life and the main manifestations are failure to thrive, vomiting, dehydration, and anorexia.
Methods
Nine unrelated families were studied: seven children, a teenager, and an adult with dRTA. Hearing was preserved in four children. Coding regions of the genes responsible for recessive dRTA were analysed by Sanger sequencing.
Results
Molecular defects were found in the genes ATP6V1B1 and ATP6V0A4. We identified three homozygous variants in ATP6V1B: a frameshift mutation (p.Ile386Hisfs*56), a nucleotide substitution in exon 10 (p.Pro346Arg), and a new splicing mutation in intron 5. Three patients were homozygous for one novel (p.Arg743Trp) and one known (p.Asp411Tyr) missense mutations in the ATP6V0A4 gene. Three patients were compound heterozygous: one proband displayed two novel mutations, the frameshift mutation p.Val52Metfs*25, and a large deletion of exons 18–21; two probands showed the missense mutation p.Asp411Tyr and as a second mutation, p.Arg194Ter and c.1691+2dup, respectively.
Conclusion
ATP6V0A4 and ATP6V1B1 genes were involved in recessive dRTA of Mexican families. All ATP6V1B1 mutations detected were homozygous and all patients developed sensorineural hearing loss (SNHL) early in infancy. ATP6V0A4 mutations were found in one infant and three children without SNHL, and in one teenager and one adult with SNHL confirming the phenotypic variability in this trait. The mutation p.Asp411Tyr detected in four Mexican families was due to a founder effect. Screening of these mutations could provide a rapid and valuable tool for diagnosis of dRTA in this population.
Keywords: Hearing loss, hypokalemia, nephrocalcinosis, renal tubular acidosis
Introduction
Hereditary distal renal tubular acidosis (dRTA) results from mutations in genes encoding for three proteins expressed in α‐intercalated cells of the collecting duct: the a4 and B1 subunits of the V‐ATPase and the anion exchanger Cl−/HCO3 − (kAE1). Impairment of apical proton secretion or basolateral bicarbonate reabsorption, produced by abnormal function of one of these proteins, is responsible for decreased ammonium (NH4 +) excretion and defect in urine acidification, leading to simultaneous metabolic acidosis, hypokalemia, hypercalciuria, hypocitraturia, and nephrocalcinosis (Escobar Pérez et al. 2013; Gil‐Peña et al. 2014).
ATP6V1B1 and ATP6V0A4 genes encode the B1 and a4 subunits of the V‐ATPase, respectively. The V‐ATPase is expressed in the acid secretory α‐intercalated cells of the cortical and medullary collecting duct in the kidney and in the epithelial cells of the endolymphatic sac in the cochlea (Dou et al. 2004). Mutations in these genes impair the V‐ATPase proton‐secreting function and produce the autosomal recessive form of dRTA, which can be associated with sensorineural hearing loss (SNHL) (Smith et al. 2000; Stover et al. 2002; Vargas‐Poussou et al. 2006). ATP6V1B1 mutations are mostly associated with onset SNHL during infancy whereas ATP6V0A4 mutations are associated with variable hearing phenotypes ranging from early to late onset SNHL (between the ages of 10 and 40 years) (Karet et al. 1999a; Stover et al. 2002; Vargas‐Poussou et al. 2006; Gao et al. 2014).
Materials and Methods
Six kids were diagnosed with dRTA at the Hospital General del Centro Medico Nacional La Raza IMSS, a teenager at the Hospital Infantil de Mexico Federico Gomez, Mexico city and a child at the Hospital de Especialidades No. 25, Centro Medico Nacional del Noreste, Monterrey (Guerra‐Hernandez et al. 2014). The adult patient was contacted by the web site www.acidosistubular.unam.mx. Clinical diagnosis was supported by the presence of hyperchloremic metabolic acidosis with normal anion gap, hypercalciuria, hypokalemia, nephrocalcinosis, polyuria, and failure to thrive. Hearing was assessed by pure‐tone audiometry.
Informed consent was obtained for blood collection and genetic analysis from patients and children's parents. Patient clinical evolution was followed up from 1 to 3 years. Experiments were performed according to the Declaration of Helsinki and were approved by the hospital′s ethics committee.
Peripheral blood samples were collected on EDTA tubes. Pure DNA was obtained using the QIAamp DNA blood Midi kit (Qiagen) according to the manufacturer′s instructions. The coding exons and intron–exon junctions were amplified with specific primers as previously described (Vargas‐Poussou et al. 2006). Direct sequencing was performed using the dideoxy chain termination method on an automated Perkin Elmer/Applied Biosystems (Foster City, CA).
DNA mutations were identified using Sequencher software by comparison to ATP6V0A4 and ATP6V1B1 genes reference sequences: NM_130841 and NM_001692. Each mutation was confirmed by sequencing a second independent PCR product. Missense and splicing mutations were interpreted with Alamut V.2.5.1 software (Interactive Biosoftware, Rouen, France; http://www.interactivebiosoftware.com). Complementary analyses were performed with SIFT ( http://sift.jcvi.org/), PolyPhen‐2 ( http://genetics.bwh.harvard.edu/pph2/index.shtml), Mutpred ( http://mutpred.mutdb.org/about.html), SNPs&Go ( http://snps-and-go.biocomp.unibo.it/snps-and-go/info.htm), and mutation taster ( http://www.mutationtaster.org/).
Quantitative multiplex PCR of short fluorescent fragments
We adapted the Quantitative multiplex PCR of short fluorescent fragments (QMPSF) method (Houdayer et al. 2004) to detect large deletions or duplications at the ATP6V0A4 gene. QMPSF consists of a fluorescent multiplex PCR that permits simultaneous amplification of multiple short exonic fragments under semiquantitative conditions. In each QMPSF, a fragment from the hydroxymethylbilane synthase (HMBS) gene was amplified as an internal control in each one of three multiplex reactions. After the PCR, the 6FAM‐labeled amplicons are separated by capillary electrophoresis on an ABI Prism 3730XL DNA Analyzer Sequencer (Applied Biosystems). Data were analyzed using GeneMarker Software version 1.85 (Applied Biosystems). For each patient, the mean value of each amplicon was obtained by comparing the peaks between the patient and a reference sample. If this value was below 0.7, the respective exon was defined as deleted; a value between 0.7 and 1.3 was defined as normal. Primers used are shown in Table S1.
Haplotype analysis
Haplotype analysis was carried out in families harboring the recurrent mutation p.Asp411Tyr for the ATP6V0A4 gene, to determine whether these families were descended from a common ancestor. Haplotypes were defined by genotyping by direct sequencing three common intragenic single‐base pair polymorphisms (SNPs): rs10258719, rs1026435, and rs3807154, located in exons 2, 15, and 16, respectively.
Results
Clinical findings
The main clinical manifestations were dehydration episodes, failure to thrive, malnutrition, and vomiting (Table 1). dRTA is prone to constipation and inability to concentrate the urine due to renal water and potassium losses (Escobar Pérez et al. 2013; Gil‐Peña et al. 2014). Previously, we published the biochemical and clinical findings of five of these patients (Guerra‐Hernandez et al. 2014). Briefly, patients had a clinical history of hyperchloremic metabolic acidosis (venous blood gases with pH 7.2, pCO2 26 mmHg and bicarbonate <14 mEq/L), hypokalemia (potassium 2.2 mEq/L), hypercalciuria, hypocitraturia, and nephrocalcinosis. dRTA is characterized by the loss of the ability to acidify urine by a defect in acid excretion (mainly ammonium) by the collecting tubule. Even when it is not necessary to perform an acidification test for diagnosis of dRTA, impairment of urine acidification was confirmed with the maximum urinary pCO2 test using acetazolamide and sodium bicarbonate (Guerra‐Hernández et al. 2015). From the nine patients, only four had bilateral and one unilateral SNHL (Table 1), varying from mild (40 dB) to severe (80 dB).
Table 1.
Clinical features at diagnosis and current conditions in Mexican patients with recessive dRTA
| Patient | Age at diagnosis (months) | BW (kg) length (cm) at birth | Clinical features | Sensorineural hearing loss | Nephrocalcinosis | Current age, years (y), months (m) | Current weight, z score | Current height, z score |
|---|---|---|---|---|---|---|---|---|
| I – Female | 12 | 3.1, 49 | Vomiting, hypokalemia, dehydration pneumonia, malnutrition | No | Yes | 5 y 8 m | −1.99 | −2.15 |
| II – Male | 4 | 3.3, 51 | Lack of appetite,hypokalemia, vomiting, dehydration, urinary infections | Yes bilateral | Yes | 27 y | 1.61 | 0.58 |
| III – Female | 2 | 3.15, 50 | Anemia, dehydration, failure to thrive, hypokalemia | Yes unilateral | Yes | 13 y | 0.61 | −1.45 |
| IV – Male | 12 | 3.7, 50 | Dehydration, hyperammonemia, hypokalemia, hyperchloremia | No | Yes | 4 y 7 m | −1.0 | −1.1 |
| V – Female | 3 | 3.2, 51 | Vomiting, dehydration, failure to thrive, hypokalemia, diarrhea | No | Yes | 4 y 9 m | 0.68 | −0.07 |
| VI – Male | 3 | 2.4, 47 | Dehydration, failure to thrive, hypokalemia | No | Yes | 1 y 3 m | −1.6 | −1.7 |
| VII – Male | 12 | 2.9, 49 | Vomiting, dehydration, failure to thrive, hypokalemia | Yes bilateral | Yes | 9 y | 1.11 | −1.17 |
| VIII – Male | 9 | 3.0, 53 | Dehydration, failure to thrive, diarrhea, hypokalemia | Yes bilateral | Yes | 5 y 3 m | −2.22 | −3.19 |
| IX – Male | 41 | 2.95, 50 | Dehydration, failure to thrive, muscle paralysis, delayed motor skills, hypokalemia | Yes bilateral | Yes | 4 y 3 m | −0.13 | −0.67 |
Mutations in the ATP6V0A4 gene
We identified six different mutations in the ATP6V0A4 gene in six probands including three novel mutations. The novel mutations comprised a missense mutation (p.Arg743Trp) (patient VI), one small frameshift deletion (p. Val52Metfs*25), and a large deletion of exons 18–21 (patient I). The compound heterozygote (I) with deletion of four nucleotides (154_157del) in exon 3, produced a premature stop codon at Val52Metfs*25. Additionally, direct analysis of exon 3 showed that her mother was heterozygous for this mutation.
Cases III and IV were homozygous for p.Asp411Tyr missense mutation; accordingly, their parents were heterozygous for this mutation.
The adult patient (II) was compound heterozygous for one previously reported nonsense mutation (p.Arg194Ter) and the missense p.Asp411Tyr mutation detected in three other patients.
SNHL was developed during the second decade of life in 2 patients with mutations in ATP6V0A4 (Table 1).
Mutations in the ATP6V1B1 gene
Analysis of the nucleotide sequence of the coding region of the ATP6V1B1 gene identified mutations in three patients, all of them with SNHL (Table 1).
A homozygous duplication (case VIII) causes a shift in the reading frame from isoleucine 386 introducing a premature stop codon (Stover et al. 2002); his mother was heterozygous for this mutation. One patient (case IX) harbor the homozygous missense mutation p.Pro346Arg previously reported (Karet et al. 1999a) and his parents were heterozygous for this mutation. Case VII was homozygous for a novel splicing mutation (445+1G>C) that likely promotes the exon 5 skipping (Table 2). Although his mother was heterozygous for this intron splicing, his father was not, suggesting the loss of one allele in the father and son. Unfortunately, QMPSF for ATP6V1B1 gene was not available to analyze this hypothesis.
Table 2.
Mutations detected in Mexican patients with recessive dRTA
| Patient | Gene | Status | Nucleotidea | Protein | Exon/Intron | Reference | Nucleotide* | Protein | Exon/Intron | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| I | ATP6V0A4 | Compound heterozygous | c.154_157del | p. Val52Metfs*25 | 3 | This study | c.2011‐?_2523+?delb | p.? | 18–21 | This study |
| II | ATP6V0A4 | Compound heterozygous | c.580C>T | p.Arg194Ter | 7 | Stover et al. (2002) | c.1231G>T | p.Asp411Tyr | 12 | Pereira et al. 2015 |
| III | ATP6V0A4 | Homozygous | c.1231G>T | p.Asp411Tyr | 12 | Barros‐Pereira et al. (2015) | c.1231G>T | p.Asp411Tyr | 12 | Pereira et al. 2015 |
| IV | ATP6V0A4 | Homozygous | c.1231G>T | p.Asp411Tyr | 12 | Barros‐Pereira et al. (2015) | c.1231G>T | p.Asp411Tyr | 12 | Pereira et al. 2015 |
| V | ATP6V0A4 | Compound heterozygous | c.1231G>T | p.Asp411Tyr | 12 | Barros‐Pereira et al. (2015) | c.1691+2dup | p.? | 15 | Stover et al. 2002 |
| VI | ATP6V0A4 | Homozygous | c.2227C>T | p.Arg743Trp | 19 | This study | c.2227C>T | p.Arg743Trp | 19 | This study |
| VII | ATP6V1B1 | Homozygous | c.445+1G>Cc | p.? | 5 | This study | c.445+1G>Cc | p.? | 5 | This study |
| VIII | ATP6V1B1 | Homozygous | c.1155dup | p.Ile386Hisfs*56 | 12 | Stover et al. (2002) | c.1155dup | p.Ile386Hisfs*56 | 12 | Stover et al. 2002 |
| IX | ATP6V1B1 | Homozygous | c.1037C>G | p.Pro346Arg | 10 | Karet et al. (1999a, 1999b) | c.1037C>G | p.Pro346Arg | 10 | Karet et al. 1999a, 1999b |
Nucleotides numbered according to the sequence in GenBank NM_130841 for ATP6V0A4 and NM_001692 for ATP6V1B1. The A of the ATG of the Methionine initiation codon is defined as nucleotide 1. Mutations are described following version 2.0 HGVS recommendations ( http://hgvs.org/mutnomen/).
Deletion of exons 18 to 21.
Splice site score is abolished.
?, is the nomenclature used for splicing mutation when the consequence on protein is unknown.
Mutations in the two genes are summarized in Table 2 and the corresponding DNA sequences in Figure 1.
Figure 1.

Chromatograms of mutations in the ATP6V0A44 and ATP6V1B1 genes detected by direct sequencing and QMPSF. For the QMPSF, each peak represents one analyzed exon and the HMBS internal control. Control samples are shown in red and patients' samples in blue. Proband has QMPSF half doses for exons 18 to 21.
Discussion
We studied nine probands from independent families who presented clinical features of dRTA. All probands were from nonconsanguineous families. Loss‐of‐function mutations were identified in the two alleles in probands of all families: six probands had mutations in the ATP6V0A4 gene and three in the ATP6V1B1 gene.
Mutations in the ATP6V0A4 gene include 1 novel large deletion, 1 novel frameshift, 1 nonsense, 2 missense, and 1 splicing mutations (Table 2). Most of them, excepting the 2 missense, could result in unstable mRNA or truncated proteins and could be classified as pathogenic variants according to ACMG recommendations (Richards et al. 2015). The two missense mutations, the recently described p.Asp411Tyr and the novel p.Arg743Trp, could be classified as likely pathogenic. Indeed, they affect highly conserved amino acids and are predicted as pathogenic by all the in silico tools; in addition, they have a low frequency in ExAC database. A detailed classification of these variants is given in Table S2. Concerning the large deletion of exons 18–21, to the best of our knowledge, this is the second description of a large rearrangement implicating this gene. Miura et al. described a deletion of exon 15 and a deletion of exons 1–8 (Miura et al. 2013).
ATP6V0A4 mutations were found in one infant and three children without SNHL, and in one teenager and one adult with SNHL confirming the phenotypic variability in this trait.
SNP haplotype analysis suggests that mutation p.Asp411Tyr, detected in four Mexican families, is a founder effect. Indeed, patients carrying this mutation shared the same haplotype (CTC) at the disease locus (Fig. 2). Interestingly, this mutation was recently found in one family (a boy and a girl) from Brazil (Pereira et al. 2015).
Figure 2.
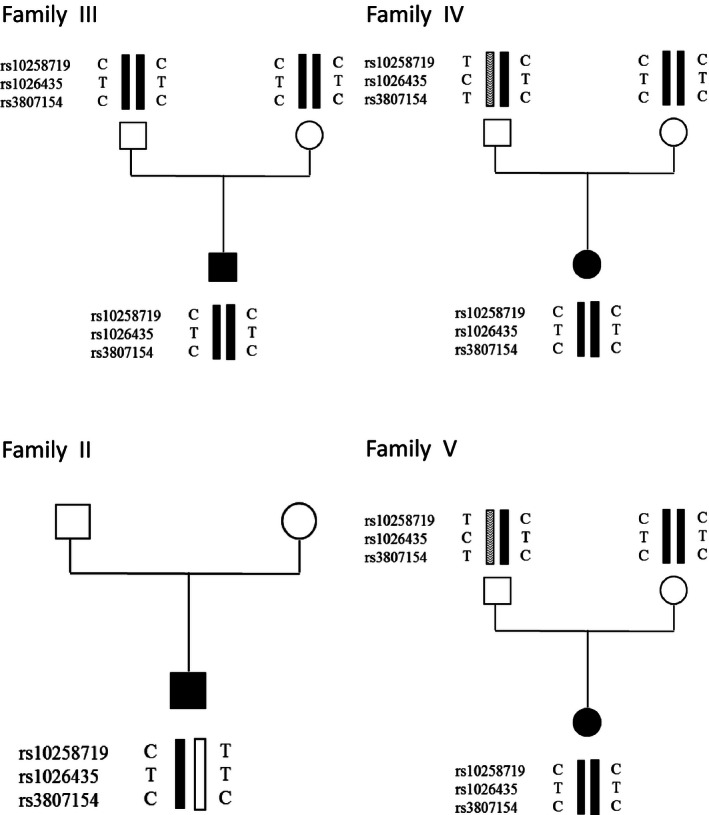
Haplotypes of four Mexican families carrying the p.Asp411Tyr mutation: families III and IV have no history of consanguinity but the mutation was homozygous, as well as the haplotypes. In probands of families II and V the mutation was heterozygous and associated with a second mutation. For patient II, DNA from parents was not available, but he harbors the CTC haplotype in one allele. For patient V, the CTC allele and p.Asp411Tyr mutation were inherited from her mother.
All ATP6V1B1 mutations detected were homozygous and all patients developed SNHL early in infancy. Mutations comprise one frameshift that provoked a premature stop codon, one missense mutation, and a novel mutation in intron 5 (a substitution in the first base of splice donor site). The frameshift and splice site mutation could be classified as pathogenic and the known missense mutation as likely pathogenic (Table S2).
ATP6V0A4 and ATP6V1B1 genes have been associated with autosomal recessive dRTA of families from Turkey (Karet et al. 1999b), Tunisia (Elhayek et al. 2013) and North Africa (Vargas‐Poussou et al. 2006), mostly from consanguineous marriages, and also from Algeria (Vargas‐Poussou et al. 2006), France (Vargas‐Poussou et al. 2006), Saudi Arabia (Karet et al. 1999b), China (Gao et al. 2014), Greece (Feldman et al. 2006), Italy (Andreucci et al. 2009), Iran (Zeinali et al. 2014), India (Naveen et al. 2015) Pakistan (Vargas‐Poussou et al. 2006), Spain (Gil‐Peña et al. 2007), Serbia (Mohebbi et al. 2013), and Brasil (Pereira et al. 2015).
In conclusion, ATP6V0A4 and ATP6V1B1 genes are involved in recessive dRTA of Mexican families. This study constitutes the first genetic analysis of Mexican families with autosomal recessive dRTA. These data show that analysis of these genes is a good predictor for future screenings and molecular diagnostic of dRTA in this population.
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
Table S1. Primers used for QMPSF
Table S2. Classification of the variants detected in Mexican families with distal renal tubular acidosis
Acknowledgments
We thank patients and families for their valuable participation. This work was supported by grant IN214316 Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT; DGAPA,UNAM), Conacyt Grant 166913 and 253586, Fondos Federales Hospital Infantil de Mexico, HIM/2012/036 and Fundación para la Acidosis Tubular Renal Infantil Mexicana AC www.acidosistubular.unam.mx.
References
- Andreucci, E. , Bianchi B., Carboni I., Lavoratti G., Mortilla M., Fonda C., et al. 2009. Inner ear abnormalities in four patients with dRTA and SNHL: clinical and genetic heterogeneity. Pediatr. Nephrol. 24:2147–2153. [DOI] [PubMed] [Google Scholar]
- Dou, H. , Xu J., Wang Z., Smith A. N., Soleimani M., Karet F. E., et al. 2004. Co‐expression of pendrin, vacuolar H+‐ATPase alpha4‐subunit and carbonic anhydrase II in epitelial cells of the murine endolymphatic sac. J. Histochem. Cytochem. 52:1377–1384. [DOI] [PubMed] [Google Scholar]
- Elhayek, D. , Perez de Nanclares G., Chouchane S., Hamami S., Mlika A., Troudi M., et al. 2013. Molecular diagnosis of distal renal tubular acidosis in Tunisian patients: proposed algorithm for Northern Africa populations for the ATP6V1B1, ATP6V0A4 and SCL4A1 genes. BMC Med. Genet. 14:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar Pérez, L. I. , Mejía N., Gil H., and Santos F.. 2013. Distal renal tubular acidosis: a hereditary disease with an inadequate urinary H⁺ excretion. Nefrología 33:289–296. [DOI] [PubMed] [Google Scholar]
- Feldman, M. , Prikis M., Athanasiou Y., Elia A., Pierides A., and Deltas C. C.. 2006. Molecular investigation and long‐term clinical progress in Greek Cypriot families with recessive distal renal tubular acidosis and sensorineural deafness due to mutations in the ATP6V1B1 gene. Clin. Genet. 69:135–144. [DOI] [PubMed] [Google Scholar]
- Gao, Y. , Xu Y., Li Q., Lang Y., Dong Q., and Shao L.. 2014. Mutation analysis and audiologic assessment in six Chinese children with primary distal renal tubular acidosis. Ren. Fail. 36:1226–1232. [DOI] [PubMed] [Google Scholar]
- Gil, H. , Santos F., Garcia E., Alvarez M. V., Ordoñez F. A., Malaga S., et al. 2007. Distal RTA with nerve deafness: clinical spectrum and mutational analysis in five children. Pediatr. Nephrol. 22:825–828. [DOI] [PubMed] [Google Scholar]
- Gil‐Peña, H. , Mejía N., and Santos F.. 2014. Renal tubular acidosis. J. Pediatr. 164:691–698. [DOI] [PubMed] [Google Scholar]
- Guerra‐Hernández Norma, E. , Ordaz‐López Karen V., Laura E. P., García Nieto Víctor M., and Circe G. T.. 2015. Screening of distal renal tubular acidosis by urinary acidification tests in mexican children. Rev. Invest. Clin. 67:191–198. [PubMed] [Google Scholar]
- Guerra‐Hernandez, N. , Matos‐Martinez M., Ordaz‐Lopez V., Camargo‐Muñiz M. D., Medeiros M., and Escobar‐Perez L.. 2014. Clinical and biochemical findings in mexican patients with distal renal tubular acidosis. Rev. Invest. Clin. 66:386–392. [PubMed] [Google Scholar]
- Houdayer, C. , Gauthier‐Villars M., Laugé A., Pagès‐Berhouet S., Dehainault C., Caux‐Moncoutier V., et al. 2004. Comprehensive screening for constitutional RB1 mutations by DHPLC and QMPSF. Hum Mutat. 23:193–202. [DOI] [PubMed] [Google Scholar]
- Karet, F. E. , Fiberg K. E., Nelson R. D., Nayir A., Mocan H., Sanjad S. A., et al. 1999a. Mutations in the gene encoding B1 subunti of H+‐ATPase cause renal tubular acidosis with sensorineural deafness. Nat. Genet. 21:84–90. [DOI] [PubMed] [Google Scholar]
- Karet, F. E. , Fingberg K. E., Nayir A., Bakkaloglu A., Ozen S., Hulton S. A., et al. 1999b. Localization of a gene for autosomal recessive distal renal tubular acidosis with normal hearing (rdRTA2) to 7q33‐34. Am. J. Hum. Genet. 65:1656–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura, K. , Sekine T., Takahashi K., et al. 2013. Mutational analysis of the ATP6V1B1 and ATP6V0A4 genes in patients with primary distal renal tubular acidosis. Nephrol. Dial. Transplant. 28:2123–2130. [DOI] [PubMed] [Google Scholar]
- Mohebbi, N. , Vargas‐Poussou R., Hegemann S. C., Schuknetch B., Kistler A. D., Wüthrich R. P., et al. 2013. Homozygous and compound heterozygous mutations in the ATP6V1B1 gene in patients with renal tubular acidosis and sensorineural hearing loss. Clin. Genet. 83:274–278. [DOI] [PubMed] [Google Scholar]
- Naveen, P. S. , Srikanth L., Venkatesh K., Sarma P. V., Sridhar N., Krishnakishore C., et al. 2015. Distal renal tubular acidosis with nerve deafness secondary to ATP6B1 gene mutation. Saudi J. Kidney Dis. Transpl. 26:119–121. [DOI] [PubMed] [Google Scholar]
- Pereira, P. C. , Melo F. M., De Marco L. A., Oliveira E. A., Miranda D. M., and Simões E Silva A. C.. 2015. Whole‐exome sequencing as a diagnostic tool for distal renal tubular acidosis. J Pediatr (Rio J) 91:581–589. [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz N., Bale S., Bick D., Das S., Gastier‐Foster J., et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, A. N. , Skaug J., Choate K. A., Nayir A., Bakkaloglu A., Ozen S., et al. 2000. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116‐kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat. Genet. 26:71–75. [DOI] [PubMed] [Google Scholar]
- Stover, E. H. , Borthwick K. J., Bavalia C., Eady N., Fritz D. M., Rungroj N., et al. 2002. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J. Med. Genet. 39:796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas‐Poussou, R. , Houillier P., Le Pottier N., Strompf L., Loirat C., Baudouin V., et al. 2006. Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J. Am. Soc. Nephrol. 17:1437–1443. [DOI] [PubMed] [Google Scholar]
- Zeinali, F. , Mohseni M., Fadaee M., Fattahi Z., Najmabadi H., Otukesh H., et al. 2014. Investigation of ATP6V1B1 and ATP6V0A4 genes causing hereditary hearing loss associated with distal renal tubular acidosis in Iranian families. J. Laryngol. Otol. 128:1056–1059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used for QMPSF
Table S2. Classification of the variants detected in Mexican families with distal renal tubular acidosis