

Abstract
To sense and defend against oxidative stress, cells depend on signal transduction cascades involving redox‐sensitive proteins. We previously identified SUMO (small ubiquitin‐related modifier) enzymes as downstream effectors of reactive oxygen species (ROS). Hydrogen peroxide transiently inactivates SUMO E1 and E2 enzymes by inducing a disulfide bond between their catalytic cysteines. How important their oxidation is in light of many other redox‐regulated proteins has however been unclear. To selectively disrupt this redox switch, we identified a catalytically fully active SUMO E2 enzyme variant (Ubc9 D100A) with strongly reduced propensity to maintain a disulfide with the E1 enzyme in vitro and in cells. Replacement of Ubc9 by this variant impairs cell survival both under acute and mild chronic oxidative stresses. Intriguingly, Ubc9 D100A cells fail to maintain activity of the ATM–Chk2 DNA damage response pathway that is induced by hydrogen peroxide. In line with this, these cells are also more sensitive to the ROS‐producing chemotherapeutic drugs etoposide/Vp16 and Ara‐C. These findings reveal that SUMO E1~E2 oxidation is an essential redox switch in oxidative stress.
Keywords: ATM, oxidative stress, redox regulation, SUMO, Ubc9
Subject Categories: DNA Replication, Repair & Recombination; Metabolism; Post-translational Modifications, Proteolysis & Proteomics
Introduction
At elevated concentrations, reactive oxygen species (ROS) can randomly damage many intracellular components (Winyard et al, 2005; Imlay, 2008); at lower levels, ROS such as hydrogen peroxide or nitric oxide are essential and specific players in physiological signal transduction processes (Rhee, 2006; D'Autreaux & Toledano, 2007; Finkel, 2011). ROS concentrations can temporally or chronically increase in cells and organisms due to exogenous sources (e.g., ionizing radiation, xenobiotics, chemotherapeutics) or (patho)physiological changes that trigger endogenous ROS production (e.g., activation of NADPH oxidases in signal transduction, mitochondrial malfunction). Increased ROS levels induce signal transduction cascades that depend on proteins able to sense ROS (Veal et al, 2007; Brandes et al, 2009). An emerging concept in the field is that these proteins act as thiol switches, which are defined as proteins that are specifically and reversibly modified by oxidation (Groitl & Jakob, 2014). Protein thiol switches are involved in numerous pathways, including signal transduction (e.g., the phosphotyrosine phosphatase PTP1B (Tonks, 2005)) and transcription regulation (e.g., the bacterial transcription factor OxyR (Zheng et al, 1998), the yeast transcription factor YAP1 (Delaunay et al, 2002; Wood et al, 2004) or the mammalian transcription factor STAT3 (Sobotta et al, 2015)). Another example is the ataxia‐telangiectasia mutated (ATM) protein kinase (Guo et al, 2010), a key player in DNA double‐strand break (DBS) repair pathways (Paull, 2015). In 2006, we added SUMO pathway enzymes to the growing list of thiol switches (Bossis & Melchior, 2006).
Attachment of the ubiquitin‐like protein (Ubl) SUMO (small ubiquitin‐related modifier) serves to regulate hundreds of proteins in all eukaryotic cells and is essential for viability in most organisms including mammals (Gareau & Lima, 2010; Flotho & Melchior, 2013). SUMO targets include numerous transcription factors and co‐regulators, proteins involved in cell cycle control and signal transduction, and proteins involved in chromatin organization and repair. Attachment of SUMO to its targets requires an enzymatic cascade: The E1‐activating enzyme Aos1/Uba2 (also known as SAE1/SAE2) utilizes ATP to form a thioester bond between its catalytic cysteine (Uba2 C173) and the C‐terminal carboxy group of SUMO. From there, SUMO is transferred to the catalytic cysteine (C93) of the single E2‐conjugating enzyme Ubc9. Finally, an isopeptide bond is formed between SUMO and the ε‐amino group of a lysine side chain in the target, usually with the help of an E3 ligase. SUMO‐specific isopeptidases, all of which are cysteine proteases, reverse SUMOylation (Gillies & Hochstrasser, 2012; Nayak & Muller, 2014).
Oxidative stress affects SUMOylation via multiple direct and indirect mechanisms (Fig 1A): First, we found that H2O2 can inhibit SUMOylation reversibly by inducing a disulfide bond between the catalytic cysteines of the SUMO E1 and SUMO E2 enzymes (Bossis & Melchior, 2006). Intriguingly, chemotherapeutic drugs that are used to treat acute myeloid leukemia (AML) also induce E1–E2 disulfide bond formation and concomitant loss of SUMO conjugates (Bossis et al, 2014). Second, H2O2 can activate deSUMOylation by reversibly oxidizing a cysteine motif in the isopeptidase Senp3; this protects Senp3 from proteasomal degradation and leads to its relocalization from nucleoli to the nucleoplasm (Huang et al, 2009; Yan et al, 2010). Third, ROS can inactivate SUMO isopeptidases—either reversibly by disulfide bond‐induced dimerization or irreversibly by overoxidation of their catalytic cysteine (Xu et al, 2008). The latter may explain the observed increase in SUMO conjugates in mammalian cells at lethal doses of hydrogen peroxide (Saitoh & Hinchey, 2000). Fourth, ROS can influence SUMO E3 ligase activities. For example, the interaction of the E3 ligase PIAS4 with its target NEMO is enhanced upon H2O2 treatment (Mabb et al, 2006). Whether this involves oxidation of the E3 ligase or the target remains to be seen. Moreover, ROS can activate the E3 ligase PIAS1 indirectly by hydrogen peroxide‐induced phosphorylation. This in turn seems to increase SUMOylation of various transcription factors (Leitao et al, 2011). Finally, since H2O2 is known to influence the activity of numerous kinases and phosphatases, it may cause altered SUMO target phosphorylations, which in turn influence their SUMOylation. Among several specific targets that were found to change SUMOylation in response to oxidative stress are, e.g., HIPK2 (de la Vega et al, 2012) or TP53INP1 (Peuget et al, 2014).
Figure 1. While ROS susceptibility of SUMO targets vary between cells, SUMO E1~E2 redox regulation is conserved.
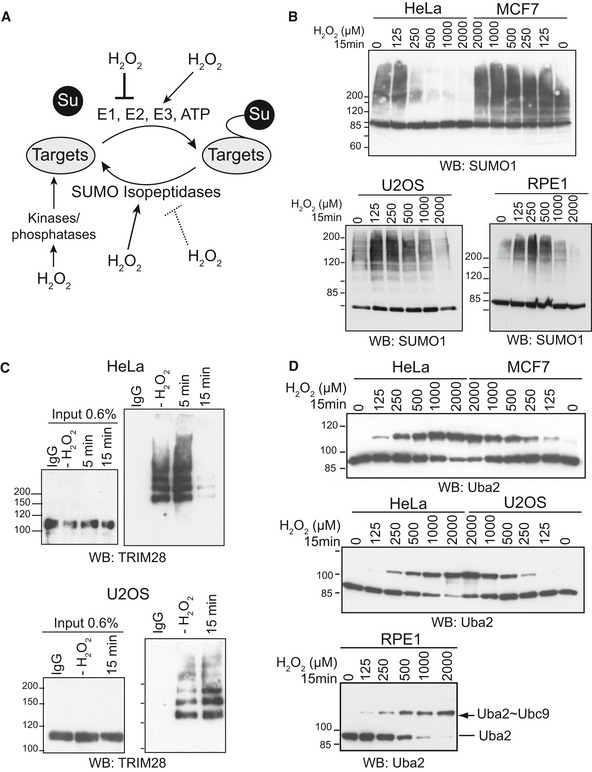
- Multiple pathways alter SUMOylation in response to oxidative stress.
- Consequences of ROS on the SUMO proteome vary among cell lines. HeLa, MCF7, U2OS or RPE1 cells were treated with increasing H2O2 concentration for 15 min and lysed in Laemmli buffer supplemented with 20 mM N‐ethylmaleimide (NEM). Samples were analyzed by immunoblotting with SUMO1 antibodies.
- H2O2 induces loss of TRIM28 SUMOylation in HeLa cells but not in U2OS cells. HeLa or U2OS cells were mock treated or treated with 500 μM H2O2 for 5 or 15 min. Cells were lysed with 1% SDS in PBS buffer and 20 mM NEM. Upon immunoprecipitation of the SUMO1 proteome, samples were analyzed by immunoblotting with TRIM28 antibodies.
- Induction of the Uba2˜Ubc9 disulfide is a conserved mechanism. HeLa, MCF7, U2OS or RPE1 cells were treated with increasing concentration of H2O2 for 15 min, lysed in non‐reducing buffer supplemented with 20 mM NEM, and analyzed by immunoblotting with Uba2 antibodies.
To gain deeper insights into the interplay between SUMOylation and oxidative stress response, we decided to specifically interfere with SUMO E1~E2 disulfide bond formation. As described below, our findings reveal that the reversible SUMO E1~E2 oxidation is an essential thiol switch required for cell survival in oxidative stress that is accompanied by DNA damage.
Results
SUMO E1~E2 disulfide formation is conserved between various cell lines
Reactive oxygen has been reported to either increase or decrease SUMO conjugate levels in mammalian cells (Saitoh & Hinchey, 2000; Manza et al, 2004; Zhou et al, 2004; Bossis & Melchior, 2006; Leitao et al, 2011). To directly compare different cell lines for global changes in the SUMO proteome upon exposure to hydrogen peroxide, we analyzed the cancer cell lines HeLa, U2OS and MCF7 and the immortal normal cell line hTERT‐RPE1 for their response to increasing doses of hydrogen peroxide by immunoblotting with anti‐SUMO1 or anti‐SUMO2 antibodies. The differences were indeed striking: Whereas HeLa cells lost detectable SUMO conjugates within 15 min of exposure to 250 μM H2O2 (Fig 1B, upper panel and Appendix Fig S1), U2OS or hTERT‐RPE1 cells required much higher doses (between 1 and 2 mM, Fig 1B, lower panel); MCF7 cells showed no deSUMOylation at the tested concentrations (Fig 1B, upper panel). Of note, analysis of global SUMO patterns by immunoblotting reveals only the most abundant SUMO targets. However, analysis of the specific SUMO target TRIM28 (Ivanov et al, 2007) led to similar differences between cell lines: While 15‐min exposure to 250 μM hydrogen peroxide led to loss of SUMOylated TRIM28 in HeLa cells, it increased TRIM28 SUMOylation in U2OS cells (Fig 1C). The underlying reasons for these differences are presently unclear. However, as described in the introduction, numerous mechanisms have been identified that contribute to ROS‐induced changes in the SUMO proteome (Fig 1A). If one assumes that the relative activity of stimulatory and inhibitory pathways varies between cell types, the degree of steady‐state SUMOylation may differ dramatically between cell lines.
Much more revealing than the fate of individual SUMOylated proteins may thus be analysis of upstream ROS‐sensing mechanisms. We thus decided to compare the different cell lines for their ability to induce the disulfide bond between the SUMO E1 and E2 enzymes in response to ROS. For this, we treated HeLa, U2OS, MCF7 and hTERT‐RPE1 cells again with increasing H2O2 concentration for 15 min, lysed the cells in Laemmli buffer (2% SDS) without reducing agent, and tested for the presence of the Uba2~Ubc9 disulfide upon non‐reducing SDS–PAGE and immunoblotting with anti‐Uba2 antibodies. As shown in Fig 1D, all four cell lines formed the Uba2~Ubc9 disulfide with comparable dose–response curves. Thus, while the fate of individual SUMO targets may differ between cell lines, Uba2~Ubc9 disulfide formation upon H2O2 treatment is a conserved mechanism, both in cancer and in immortalized normal cell lines. Uba2~Ubc9 oxidation may thus play a general role in ROS‐dependent signaling pathways including oxidative stress response.
A random mutagenesis screen to identify H2O2‐resistant Ubc9 variants
To gain insights into the specific contribution of the SUMO E1~E2 disulfide in oxidative stress response, we decided to search for SUMO enzyme variants that are resistant to oxidative inactivation. This seemed feasible in light of the observation that E1~E2 disulfide bond formation is not a general feature of all Ubl E1–E2 pairs (Kumar et al, 2007; Doris et al, 2012). Susceptibility to oxidation should hence not be an intrinsic consequence of E1 and E2 enzyme chemistry. We decided to focus on Ubc9, a monomeric 18‐kDa protein, for which we had established a straightforward bacterial expression/purification scheme. To identify the desired Ubc9 variant, we designed a random mutagenesis screen that involves an in vitro FRET‐based SUMOylation assay (Bossis et al, 2005; Stankovic‐Valentin et al, 2009) (Fig 2A). In brief, random Ubc9 mutants were generated by PCR, expressed in E. coli and released from bacteria by simple freezing/thawing (Bossis et al, 2005). The Ubc9‐containing supernatant was incubated with recombinant SUMO E1 enzymes with or without 1 mM H2O2 for 30 min at 37°C, before YFP‐SUMO, CFP‐RanGAP1tail and ATP were added to start the SUMOylation reaction. Of note, 0.06 mM DTT were introduced at this point due to its presence in the storage buffer of the substrates. Approximately 300 individual clones were analyzed in this assay, 40% of which behaved like wt Ubc9. They were active without H2O2 and inactive in the presence of H2O2. Approximately 60% of the clones did not show activity at all, probably because of severe mutations. Importantly, lysate from one clone showed activity both with and without H2O2 treatment. Sequencing of the corresponding plasmid revealed that this clone expressed an Ubc9 variant in which tryptophan 103 was mutated to arginine (Ubc9 W103R, Fig 2A).
Figure 2. A random mutagenesis screen for the identification of H2O2‐resistant Ubc9.
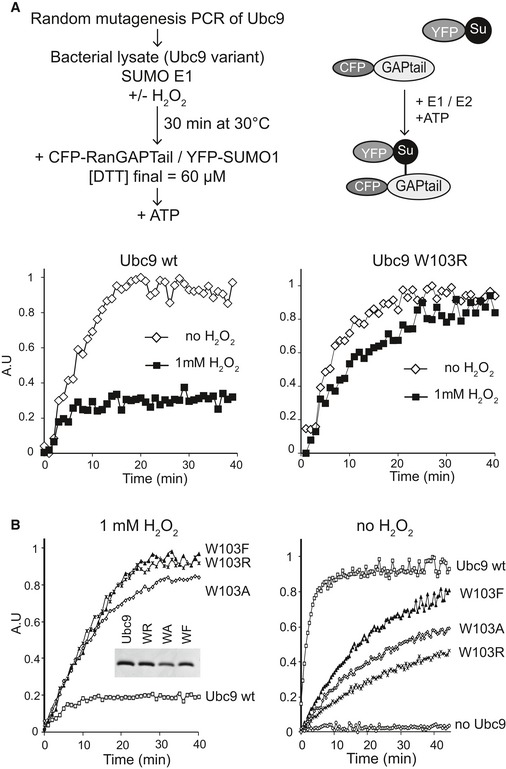
- Upper panel: Strategy for the identification of oxidation‐resistant Ubc9 variants. Ubc9 was mutagenized by PCR under conditions that introduced 1–3 random mutations. Ubc9 variants were expressed in bacteria and the bacterial lysates of individual clones tested for E2 activity in the presence or absence of 1 mM H2O2. For this purpose, we used a FRET‐based in vitro SUMOylation assay with recombinant SUMO E1, YFP‐SUMO and CFP‐RanGAPtail and ATP. Lower panel: Ubc9 W103R is H2O2 resistant. Bacterial lysates containing Ubc9 wt (left panel) or Ubc9 W103R (right panel) were tested as described.
- Ubc9 W103 mutants are H2O2 resistant but not fully active. Left panel: Recombinant Ubc9 W103R, W103A and W103F were purified (embedded panel). Resistance against oxidation was tested under conditions of limiting E1 enzyme: 21 nM Aos1/Uba2 and 73 nM Ubc9 were incubated with H2O2 prior to the addition of 160 nM each of YFP‐SUMO1 and CFP‐RanGAPtail. Right panel: To compare specific activities of wt and mutants in the absence of H2O2, SUMOylation assays were carried out using limiting Ubc9 concentration. Reactions contained 35 nM Aos1/Uba2, 11 nM Ubc9, 85 nM each of YFP‐SUMO1 and CFP‐RanGAPtail and 1 mM ATP.
To confirm H2O2 resistance of Ubc9 W103R, and to test whether less drastic mutations of Trp103 also showed resistance, we generated Ubc9 W103R, Ubc9 W103A and Ubc9 W103F and compared their activity to wt Ubc9 in the presence of H2O2. Indeed, each of the three variants remained active in the assay (Fig 2B, left panel), indicating that W103 is critical for stable oxidation. We then compared specific activities of wt and Ubc9 W103 mutants in SUMOylation assays with limiting concentration of Ubc9 (35 nM SUMO E1, 11 nM Ubc9). While Ubc9 W103R was severely impaired, Ubc9 W103F was only 2.5‐fold reduced in activity, indicating that ROS susceptibility and catalytic activity can indeed be separated (Fig 2B, right panel).
Ubc9 D100A is a suitable variant to study the relevance of SUMO E1–E2 oxidation
While Ubc9 W103F was a promising mutant, its twofold reduction in specific activity compared to wt Ubc9 may cause problems in subsequent cell‐based assays. Inspection of Ubc9's crystal structure (Fig 3A) suggested that mutating the conserved tryptophane residue may influence the orientation of a small Ubc9‐specific loop that is formed by insertion of two amino acids between W103 and the catalytic cysteine. To test the idea that the loop residues D100 and K101 contribute to formation or stability of the Uba2~Ubc9 disulfide, we generated and tested the four Ubc9 variants D100A, K101A, K101Q (mimicking yeast Ubc9) and DK100AA (Appendix Fig S2A, Fig 3A). Whereas Ubc9 K101A and K101Q were inactivated like wt Ubc9, the double mutant Ubc9 DK100AA and the single‐point mutant Ubc9 D100A remained catalytically active upon pretreatment with H2O2 (Fig 3B and Appendix Fig S2B). Importantly, without H2O2 treatment, Ubc9 D100A was as active as wt Ubc9 (Fig 3C). Thus, Ubc9 D100A seemed to be much better suited than Ubc9 W103F to study consequences of impaired redox regulation.
Figure 3. Ubc9 D100A remains active in the presence of H2O2 .
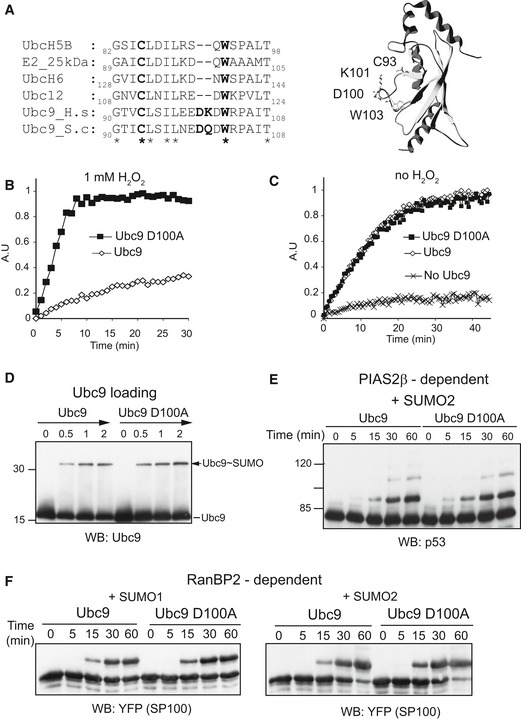
- Ubc9 possesses a specific insertion of two amino acids between the catalytic cysteine and tryptophane W103. Sequences of human UbcH5B (GI: 1145689), human E2‐25k (GI: 1381164), human UbcH6 (GI: 1064914), human Ubc12 (GI: 4507791), human Ubc9 (GI: 4507785) and S. cerevisiae Ubc9 (GI: 1431070) were aligned using Clustalw with default parameters. The catalytic cysteine is shown in bold. Identical residues are indicated by an asterisk under the aligned sequences. The model of Ubc9 (PDB: 1A3S) was generated using Deepview and rendered using POV‐Ray.
- In FRET‐based assay, Ubc9 D100A is active upon H2O2 treatment. Ubc9 D100A was purified and compared for activity in the presence of H2O2 as described in Fig 2B.
- Ubc9 wt and D100A are equally active in CFP‐RanGAPtail SUMOylation. Assays were with 35 nM Aos1/Uba2 and 15 nM Ubc9.
- Ubc9 D100A is fully competent to form a thioester with SUMO. Single turnover reactions were performed using 630 nM E1, 3.3 μM SUMO1 and 600 nM Ubc9. Reactions were stopped at different time points by addition of non‐reducing buffer.
- Ubc9 wt and D100A are equally active in PIAS2β‐dependent SUMOylation. Assays were with 300 nM GST‐p53, 170 nM E1, 120 nM Ubc9, 5 μM SUMO2 and 49 nM GST‐Pias2β.
- Ubc9 wt or D100A are equally active in RanBP2–dependent SUMOylation. Assays were with 650 nM YFP‐SP100, 110 nM of E1, 25 Ubc9, 40 nM RanBP2ΔFG and 3.5 μM SUMO1 (left panel) or SUMO2 (right panel).
To ensure that Ubc9 D100A indeed functions as efficiently as wt Ubc9 in SUMOylation, we subjected it to a wide range of additional assays: The first E2‐dependent event in SUMOylation is the charging of Ubc9 with a thioester‐linked SUMO. This can be tested in single turnover reactions: First, the E1‐SUMO thioester was generated by the incubation of the SUMO E1 enzyme with SUMO and ATP. Second, ATP was depleted, Ubc9 was added and the transfer of SUMO onto Ubc9 was analyzed by immunoblotting. As shown in Fig 3D, and in accordance with previous work showing a small increase in trans‐thioesterification activity (Tatham et al, 2003), Ubc9 D100A functioned efficiently in single turnover reactions. To test it in E3 ligase‐dependent reactions, we investigated GST‐p53 SUMOylation in dependence of the E3 ligase Pias2β (Fig 3E) and SUMOylation of YFP‐Sp100 in dependence of the SUMO E3 ligase RanBP2 (Fig 3F). In both assays, reaction rates were indistinguishable for wt and D100A Ubc9. These in vitro experiments indicated that Ubc9 D100A is fully functional. As a final control, we tested functionality of the Ubc9 D100A variant in Saccharomyces cerevisiae. Since yeast SUMO E1 and E2 enzymes are not readily oxidized in vitro (our unpublished observation) and SUMOylation in yeast is not inhibited by hydrogen peroxide (Zhou et al, 2004), an oxidative stress‐specific phenotype for D100A Ubc9‐expressing yeast was not expected. However, since the SUMO pathway is essential in baker's yeast (Seufert et al, 1995; Johnson et al, 1997), major defects in Ubc9's activity would be expected to impair cell doubling. Using a shuffle plasmid strategy (Pereira et al, 2001), we replaced endogenous UBC9 by either wt or mutant S.c. UBC9. Yeasts cells expressing wt or UBC9 D100A showed the same Ubc9 expression levels, steady‐state SUMOylation ability and growth rate at different temperatures (Appendix Fig S3). Together, these findings indicate that introducing D100A in Ubc9 does not affect critical functions of this essential protein in yeast. In conclusion, Ubc9 D100A seemed a perfect tool to study the relevance of SUMO E1~E2 oxidation in mammalian cells.
Ubc9 D100A renders the SUMO E1~E2 disulfide highly sensitive to reductants
Prior to moving into cells, we wanted to gain insights into why Ubc9 D100A showed activity in our primary assay. Due to the specific assay condition, we envisioned two possibilities: Either the mutant was resistant to disulfide bond formation with the SUMO E1 enzyme, or the Uba2~Ubc9 D100A disulfide was much more sensitive to reduction by the low amount of DTT (60 μM) that was added in the second step of the assay (Fig 2A). To distinguish between these two possibilities, we repeated the assays in the presence of DTT concentration that ranged from 6 to 200 μM. In contrast to wt Ubc9 that required more than 200 μM DTT to restore activity, as little as 6 μM DTT sufficed to partially activate Ubc9 D100A. With 56 μM DTT, Ubc9 D100A was fully active (Fig 4A). This suggested that Ubc9 D100A was indeed oxidized, but that this oxidation is unstable in the presence of low concentration of reductants.
Figure 4. Ubc9 D100A sensitizes the E1~E2 disulfide to reduction.
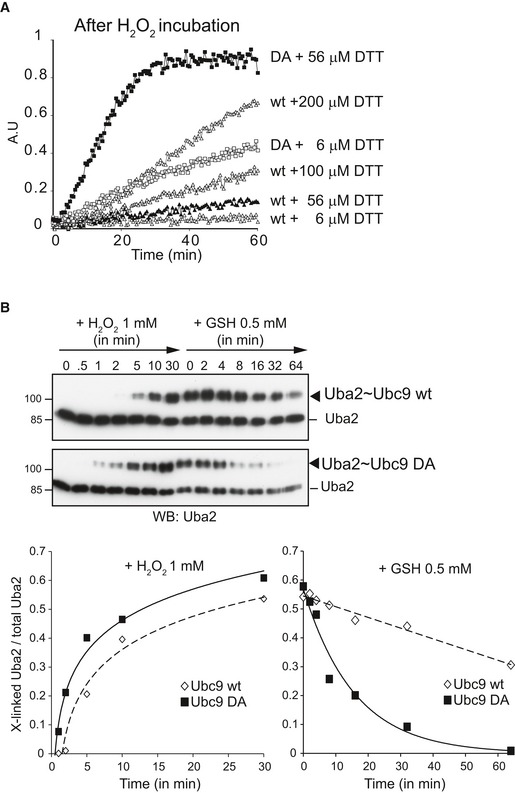
- Ubc9 D100A enzymatic activity can be recovered by limited amount of DTT. 21 nM Aos1/Uba2 and 75 nM Ubc9 were incubated with H2O2 for 30 min prior to addition of 65 nM each YFP‐SUMO1 and CFP‐RanGAPtail diluted in DTT‐free buffer. The final DTT concentration in the reaction was 6 μM. Prior to addition of ATP, extra amount of DTT was added at the indicated final concentration.
- Both Uba2˜Ubc9 disulfide formation and reduction are accelerated with Ubc9 D100A. 100 nM E1 and 1 μM E2 were incubated in the presence of 1 mM H2O2. One hour after H2O2 addition, 500 μM reduced glutathione was added. Samples were taken at the indicated time points and the Uba2˜Ubc9 disulfide was monitored by immunoblotting against Uba2. Lower panel: Quantification of the ratio between cross‐linked Uba2 vs. total Uba2.
Up to this point, we had used activity‐based assays as an indirect readout for disulfide bond formation. To get direct evidence for our interpretation, we next compared rates of disulfide formation and cleavage in non‐reducing SDS–PAGE. As shown in Fig 4B, treatment of SUMO E1 and Ubc9 with 1 mM H2O2 leads to fast appearance of the E1~E2 disulfide. Surprisingly, the rate of Ubc9 D100A disulfide formation was even faster than for wt Ubc9 (Fig 4B). Importantly, however, the rate of reduction in the disulfide by 0.5 mM glutathione, one of the most important and versatile ROS scavengers in cells, differed dramatically between disulfides formed with wt or mutant Ubc9 (Fig 4B): Whereas the Uba2~Ubc9 wt disulfide was quite resistant to reduction (Fig 4B, upper panel), the Uba2~Ubc9 D100A disulfide was rapidly resolved (Fig 4B, lower panel). In conclusion, mutating the conserved Ubc9 residue D100, a surface‐exposed residue that is unlikely to affect Ubc9's catalytic pocket and overall fold, accelerates formation but also strongly decreases the stability of the Uba2~Ubc9 disulfide.
Replacement of Ubc9 with Ubc9 D100A in mammalian cells leads to cell survival defects
Our detailed characterization of Ubc9 D100A indicated that this mutant is ideally suited to investigate physiological consequences of SUMO E1~E2 disulfide bond persistence under oxidative stress conditions. We thus decided to generate human cell lines that express untagged wt or D100A Ubc9, under conditions that would allow depleting endogenous Ubc9 by siRNA. The latter was accomplished using murine Ubc9 cDNA for transfection of human cells (mouse and human Ubc9 proteins are identical). Untagged Ubc9 was chosen, because N‐ and C‐terminal HA‐tags reduce Ubc9's specific activity (data not shown). However, multiple attempts to generate single MCF7 or HeLa cell clones that express significant levels of the oxidation‐resistant variant failed. Because such clones were readily obtained for wt Ubc9, this provided first evidence that the mutant may be toxic for the cells. To better control for Ubc9 expression levels, we turned to pIRES constructs that allow simultaneous expression of Ubc9 and GFP from one mRNA via an internal ribosome entry site (Fig 5A, upper panel). Since Ubc9 expression directly correlates with GFP expression, the GFP signal can be used for fluorescence‐based cell sorting (FACS). Upon transfection of U2OS cells and 4 weeks of selection under antibiotic pressure, we collected GFP‐positive cells by FACS. This resulted in polyclonal cell populations that expressed wt and D100A Ubc9 at a comparable expression level range, with an average expression level that was approximately 5‐times higher than endogenous Ubc9 in U2OS cells. Again, the mutant caused problems: As revealed by repeated FACS analyses, its expression levels dropped substantially within a few passages (Appendix Fig S4A and B). Together, these findings revealed that Ubc9 D100A has a dominant‐negative effect on cell proliferation or survival when expressed at elevated levels. The strategy that finally allowed stable expression of wt and variant Ubc9 over 2–3 months was to only chose cells in two rounds of FACS selection that have very low GFP expression levels (Appendix Fig S4C). Under those conditions, exogenous wt and variant Ubc9 were expressed at levels that are comparable to that of endogenous Ubc9 (Fig 5A, analysis of Ubc9 levels in stable cells with or without siRNA treatment). These cell populations in combination with siRNA depletion of the endogenous Ubc9 (from now on referred to as “Ubc9 wt” and “Ubc9 DA” cells) were used for all subsequent experiments, unless indicated otherwise. To ensure that the replacement of endogenous Ubc9 with exogenously expressed variants had no severe consequences for the SUMO proteome, we analyzed “Ubc9 wt” and “Ubc9 DA” cells by immunoblotting with SUMO1 and SUMO2 antibodies. As shown in Appendix Fig S5, we did not observe any significant differences in the pattern of SUMO conjugates without or upon treatment with 250 μM H2O2. Because immunoblotting of total lysates detects only the most abundant SUMO targets, this finding does obviously not exclude that individual low‐abundant SUMO targets differ between the two cell populations. Time‐resolved analyses of the Uba2~Ubc9 disulfide in “Ubc9 wt” and “Ubc9 DA” cells upon exposure to 250 μM H2O2 (Fig 5B and Appendix Fig S6) revealed that wt and mutant Ubc9 behaved in cells very similar to their behavior in vitro (Fig 4B): There was a striking difference in the kinetics of appearance and disappearance, indicating that the disulfide bond is more readily formed—but also much less stable—when Ubc9 is mutated. Both species could be stabilized by buthionine sulfoximine (BSO), indicating that the glutathione system is involved in resolving the disulfide bond irrespective of whether wt or mutant Ubc9 is involved (Appendix Fig S7). In conclusion, replacement of Ubc9 wt by Ubc9 D100A does not prevent initial oxidation but strongly reduces the persistence of the SUMO E1~E2 disulfide upon exposure of cells to hydrogen peroxide.
Figure 5. Ubc9 D100A compromises cell survival upon oxidative stress.

- Generation of polyclonal populations of stable cells. Top panel: Replacement strategy: U2OS cells transfected with pIRES‐hrGFP II‐based vectors were selected via addition of G418. After 4 weeks, low‐level GFP‐expressing cells were sorted by FACS and again after 8 weeks. Bottom panel: stable cells express Ubc9 wt and D100A at levels comparable to endogenous Ubc9: U2OS or U2OS stable cells were transfected with siRNA targeting human Ubc9 mRNA for 72 h or mock transfected. Cells were lysed in Laemmli buffer, and Ubc9 and α‐tubulin as loading control were detected by immunoblotting.
- Ubc9 D100A is severely impaired for disulfide bond formation with Uba2. Stable U2OS cells were transfected with siRNA against Ubc9 for 72 h, treated with 250 μM H2O2 for different time points, from 2 min to 2 h, lysed in non‐reducing buffer and analyzed by immunoblotting against Ubc9 to detect the disulfide (upper panel) or free Uba2 as loading control (see also Appendix Fig S5). Lower panel: relative intensities of Ubc9 bands were measured using imageJ. Highest intensities in both cell populations were set to 1.
- Ubc9 D100A impairs cell survival upon H2O2 treatment. Ubc9 wt or Ubc9 D100A cells were transfected with siRNA against endogenous Ubc9 for 72 h, transferred into 6‐well plates (20,000 cells/well) and treated with increasing H2O2 concentration for 1 h. Ten days later, cells were washed with PBS, fixed with 4% formaldehyde and stained with crystal violet. The area occupied by the cells was evaluated using ImageJ and the ColonyArea plugin. An arbitrary value of 1 was given to the area occupied by non‐treated cells. The graph is showing the mean of 4 independent experiments. Error bars are SEM. *P < 0.05
- Constitutive expression of Ubc9 D100A affects cell growth. 103 cells were plated per 10‐cm plate and cultivated for 10 days in 20% or 5% O2 incubators. Subsequently, cells were colored using crystal violet. The area of each clone was measured using ImageJ. Representative plates are shown in the upper panel. Lower left panel: Distribution of colony sizes, shown for one of the three biological replicates. Lower right panel: Comparison of the colonies size between “Ubc9 wt” and “Ubc9 DA” cells. The average size of “Ubc9 wt” was set to 1. Shown is the mean of 3 independent experiments, each of which was performed in technical triplicates. Error bars are SEM.
These findings allowed us to ask whether redox regulation of the SUMO E1 and E2 enzymes contributes to survival upon oxidative stress. For this, we turned to clonogenic survival assays: First, we tested consequences of acute stress by treating “Ubc9 wt” and “Ubc9 DA” cells with a single dose of the indicated H2O2 concentration for 1 h. Ten days later, we measured the area occupied by the cells as an indicator for cell survival (Fig 5C, upper panel). While high concentrations of H2O2 (250 μM) were equally detrimental for both cell populations, we observed a statistically significant reduction in “Ubc9 DA” cell survival compared to “Ubc9 wt” cells at low doses of H2O2 (62.5 μM H2O2; Fig 5C, lower panel). To confirm these findings in a condition that represents mild but chronic oxidative stress, we compared the survival of cells in standard cell culture condition (20% O2), known to create a mild oxidative stress situation (Halliwell, 2007), with cells kept at 5% O2. When cells were kept at 5% O2, colony sizes did not differ significantly. In contrast, “Ubc9 DA” colonies were significantly smaller than those formed by “Ubc9 wt” cells when cells were kept at 20% O2 (P < 0.005, Fig 5D). An example of the area distribution of “Ubc9 wt” and “Ubc9 DA” colonies is represented in Fig 5D, bottom left panel. These findings reveal that interfering with the H2O2‐induced Uba2~Ubc9 crosslink leads to a growth defect even in mild oxidative stress.
Ubc9 D100A impairs DNA damage response
Overexpression and replacement experiments indicated that redox regulation of Ubc9 contributes to cell survival even under conditions of mild chronical oxidative stress. To rule out a general defect in the glutathione system as a downstream consequence of Ubc9 D100A expression, we compared the total GSH concentration and the ratio between the reduced (GSH) and the oxidized form (GSSG) of glutathione in Ubc9 wt and D100A cells with or without H2O2 treatment. However, we did not find any significant difference between the two populations of cells (Appendix Fig S8A).
ln light of the fact that SUMO is a major player in numerous DNA repair pathways (reviewed in Sarangi & Zhao, 2015), we hypothesized that Ubc9 D100A cells may have a mild defect in one or several DNA damage repair (DDR) pathways that would lead to sporadic cell death. This would be consistent with our finding that Ubc9 D100A has long‐term detrimental effects on cell proliferation but is not acutely toxic. DNA repair pathways involve numerous proteins and a cascade of events that starts with the recognition of the damage. For example, double‐strand breaks are detected by the MRN sensor complex (Lavin, 2007) that contributes to the recruitment and the activation of sensor kinases such as ATM. These kinases will in turn phosphorylate mediator proteins such as H2AX or 53BP1. As a consequence, discrete foci containing, e.g., phosphorylated 53BP1 and phosphorylated histone H2AX, are formed. With the help of these mediators, sensor kinases activate effector kinases, such as Chk2 in the case of DSB (Polo & Jackson, 2011; Sulli et al, 2012).
Exposure of “Ubc9 wt” and “Ubc9 DA” cells to 250 μM H2O2 led to rapid and very comparable accumulation of DNA damage (Appendix Fig S8B). Because H2O2 is known to induce (among other lesions) DNA double‐strand breaks (Barzilai & Yamamoto, 2004), we followed the appearance of the above‐mentioned indicators, phosphorylated ATM, γH2AX and phosphorylated Chk2 in “Ubc9 wt” and “Ubc9 DA” cells upon exposure to 250 μM H2O2 by immunoblotting. As shown in Fig 6A–C, the two cell populations showed striking differences in the timing and maintenance of the DNA damage response: pATM could be detected in both cell populations 30 min after exposure to H2O2, persisted for 4 h after initial stress in “Ubc9 wt” cells, but rapidly declined in “Ubc9 DA” cells (after 1 h, pATM was barely detectable). Phosphorylated H2A.X appeared within 15 min in Ubc9 wt cells and peaked at 60 min, but was less abundant in Ubc9 DA cells. Similarly, phosphorylated Chk2, which could first be detected after 30 min, was significantly reduced in “Ubc9 DA” cells compared to “Ubc9 wt” cells (Fig 6C). The failure to maintain signaling correlated remarkably well with the disappearance of the SUMO E1~E2 disulfide in “Ubc9 DA” cells shown in Fig 5B, allowing to speculate that oxidation of the SUMO E1 and E2 enzymes is required to maintain ATM activity upon exposure to hydrogen peroxide.
Figure 6. Ubc9 D100A cells have a defective DNA damage response pathway.
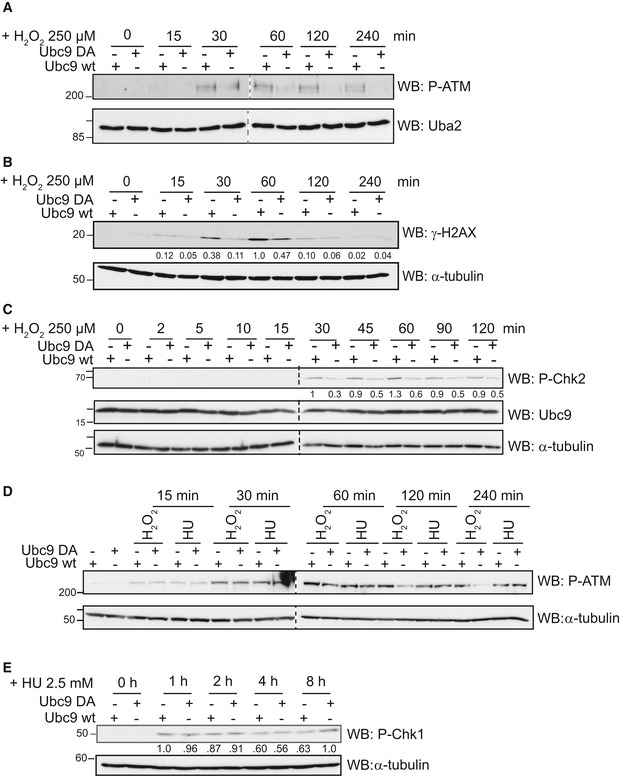
- Phosphorylation of ATM is impaired in Ubc9 D100A cells. Stable U2OS cell populations were depleted from endogenous Ubc9 by siRNA for 72 h before addition of 250 μM H2O2. At the indicated times, cells were lysed in Laemmli buffer and analyzed by immunoblotting with the indicated antibodies.
- Phosphorylation of H2AX is impaired in Ubc9 D100A. Experiment was performed as in (A).
- Phosphorylation of Chk2 is impaired in Ubc9 D100A. Experiment was performed as described in (A).
- Ubc9 D100A cells are fully competent for phosphorylation of ATM upon hydroxyurea exposure. Stable U2OS cell populations were exposed to 2.5 mM hydroxyurea or 250 μM H2O2. Cells were lysed at the indicated time points and analyzed by immunoblotting with the indicated antibodies.
- Phosphorylation of Chk1 is not impaired in Ubc9 D100A cells. Stable U2OS cells were depleted from endogenous Ubc9 by siRNA for 72 h before addition of 2.5 mM hydroxyurea. At the indicated times, cells were lysed in Laemmli buffer and analyzed by immunoblotting with the indicated antibodies.
To gain further evidence for this idea, we tested whether “Ubc9 DA” and “Ubc9 wt” cells also differ in DNA damage pathways that do not involve oxidative stress. This was not trivial, as many insults that cause DNA damage also lead to significant oxidative stress, either directly or indirectly (Caputo et al, 2012). One suitable treatment is exposure of cells to hydroxyurea, which leads to replication stress and can activate both the ATM–Chk2 (Matsuoka et al, 2000) and the ATR–Chk1 pathway (Cimprich & Cortez, 2008). Consistent with the absence of oxidative stress, exposure of cells to hydroxyurea did not promote formation of the Uba2~Ubc9 disulfide (Appendix Fig S9). Upon exposure of cells to 2.5 mM hydroxyurea or 250 μM H2O2, we followed time‐dependent appearance of pATM by immunoblotting (Fig 6D). While H2O2 led to the expected differences between “Ubc9 wt” and “Ubc9 DA” cells (compare with Fig 6A), pATM induction by HU was identical in both cell lines for up to 240 min. Finally, we compared “Ubc9 wt” or “Ubc9 DA” cells for activation of the ATR pathway upon treatment with HU: As shown in Fig 6E, levels of pChk1 were comparable in both cell lines between 1 and 8 h after addition of hydroxyurea. Together, our findings indicate that the “Ubc9 DA” cells are not generally impaired in DNA damage repair pathways, but that they have a specific defect in oxidative stress‐dependent activation (or maintenance) of the ATM–Chk2 DDR pathway.
Ubc9 D100A cells fail to maintain DNA damage repair foci
A consequence of a defect in the ATM–Chk2 DDR would be a failure to maintain or recruit DNA repair proteins at DSB sites. To test this hypothesis, we followed the accumulation of 53BP1 and γH2AX foci over several hours (Fig 7A) in the stable cell populations upon treatment with 125 μM H2O2. Three hours after H2O2 addition, the number of “Ubc9 DA” cells containing DNA damage foci was significantly lower compared to “Ubc9 wt” cells. This difference was even more pronounced 7 h after the initial treatment (< 20% compared to 45%; P < 0.05). To evaluate the role of ATM activation in foci formation, we treated cells with the ATM kinase inhibitor KU55933 one hour before exposure to H2O2. In “Ubc9 wt” cells, the number of cells with γH2AX foci was clearly decreased to a level comparable of the “Ubc9 DA” cells without inhibitor (Appendix Fig S10). This suggests a major role of ATM phosphorylation in H2O2‐dependent foci formation. Finally, to confirm that the absence of γH2AX or 53BP1 foci in most of the “Ubc9 DA” cells 7 h after exposure to H2O2 was the consequence of a defective DDR pathway rather than a very efficient repair mechanism, we quantified the number of cells with damaged DNA (Fig 7B). Indeed, comparable numbers of damaged cells (65%) were detected in comet assays for “Ubc9 wt” and “Ubc9 DA” cells. Based on these findings, we conclude that “Ubc9 DA” cells fail to form or maintain repair foci.
Figure 7. Ubc9 D100A cells fail to maintain repair foci despite DNA damage.
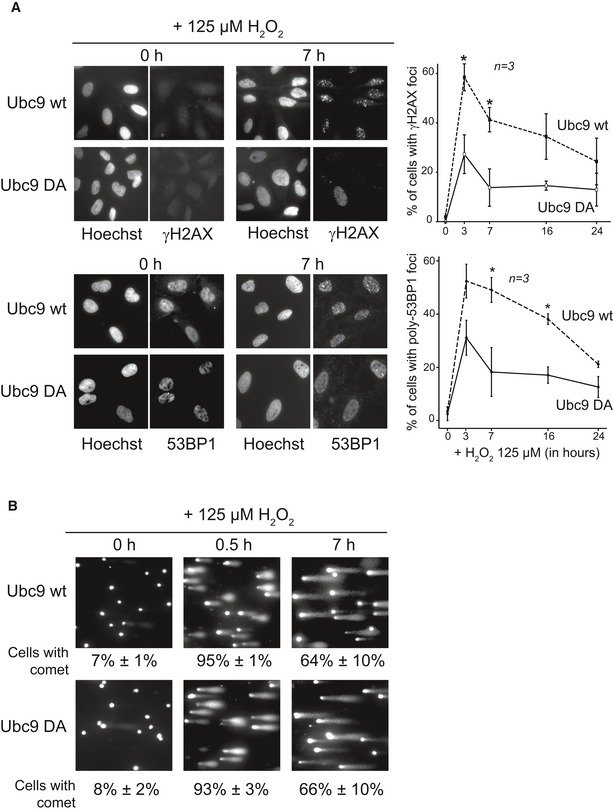
- Impaired γH2AX and 53BP1 accumulation in Ubc9 D100A cells. Stable U2OS cells were transfected with siRNA against endogenous Ubc9 for 72 h. Cells were treated with 125 μM H2O2, and immunofluorescence was performed. Left panel: representative immunofluorescence images. Right panel: quantification of cells with γH2AX and 53BP1 foci. Error bars represent SEM, n = 3 independent experiments, *P < 0.05.
- H2O2 exposure leads to prolonged DNA damage in Ubc9 wt and Ubc9 D100A cells. Cells were treated as described in (A). An alkaline comet assay was performed to evaluate the DNA damage. Representative images are shown. Error intervals represent SEM, n = 4 independent experiments.
E1~E2 disulfide formation acts downstream of ATM phosphorylation
Reduced levels of phosphorylated ATM could either be due to impaired activation or enhanced inactivation of ATM. ATM can be activated in response to DNA damage but also via DNA damage‐independent disulfide formation (Guo et al, 2010). To test whether SUMO E1~E2 disulfide formation acts upstream of these two ATM activation pathways, we treated cells with the thiol oxidizing agent diamide. Importantly, diamide is not reported to induce DNA damage. 500 μM diamide promotes SUMO E1~E2 disulfides very rapidly (Fig 8A, upper panel) and more efficiently than 500 μM H2O2. Consistent with our earlier observations with H2O2, Ubc9 D100A was more rapidly oxidized but also faster reduced than wt Ubc9. Diamide also induced rapid appearance of DTT‐sensitive ATM oligomers (Appendix Fig S11). However, diamide did not induce activation of ATM between 5 and 60 min, the times at which the E1~E2 disulfides were most abundant. Some pATM was detected at 120 min, but significantly less than with H2O2 at 60 min. In line with the low pATM signal, diamide treatment did not lead to phosphorylation of Chk2, contrary to H2O2 (Fig 8A, lower panel). From this, we conclude that SUMO E1~E2 oxidation does not lead to ATM activation in the absence of DNA damage. This leaves the idea that SUMO E1~E2 dimer formation is needed downstream of ATM activation, for example, to protect it from dephosphorylation. If so, defects in “Ubc9 DA” cells may be rescued by inhibition of phosphatases. Indeed, treatment of “Ubc9 DA” cells with okadaic acid, which has been shown to inhibit the protein phosphatase PP2A (Goodarzi et al, 2004), increases the number of cells with γH2AX foci to that of “Ubc9 wt” cells (Fig 8B).
Figure 8. The E1~E2 thiol switch maintains pATM level upon DNA damage.
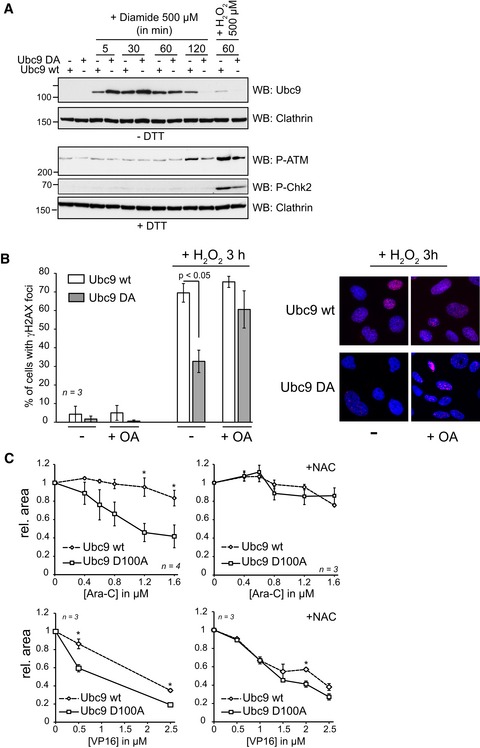
- Diamide is inducing the SUMO E1˜E2 disulfide but not ATM phosphorylation. U2OS stable cell populations were depleted from endogenous Ubc9 by siRNA for 72 h before addition of 500 μM diamide or 500 μM H2O2. Cells were subsequently washed in PBS with 20 mM NEM and lysed in Laemmli buffer. The lysates were resolved by SDS–PAGE.
- Left panel: The PP2A inhibitor okadaic acid rescues the γH2AX defect of the “Ubc9 DA” cells. “Ubc9 wt” or “Ubc9 DA” cells were treated with 0.25 μM okadaic acid for 30 min. After addition of 250 μM H2O2 in fresh medium for 3 h, immunofluorescence was performed as described in Fig 7A. Error bars represent SEM, n = 3 independent experiments. Right panel: Representative immunofluorescence images.
- Left panel: Ubc9 D100A enhances sensitivity to Ara‐C or etoposide. Cell survival assays were performed as described in Fig 5C. Cells were exposed to Ara‐C or etoposide (VP16) for 1 h and evaluated 10 days later. Right panel: N‐acetylcysteine treatment rescues the higher toxicity of Ara‐C or etoposide on “Ubc9 DA” cells. Cells were treated with 0.5 mM NAC 16 h prior to addition of etoposide or Ara‐C. Graphs represent means of three to four independent experiments, error bars represent SEM. *P < 0.05.
Figure 9. Redox regulation of SUMO enzymes is required to maintain ATM activity upon ROS‐induced DNA damage.
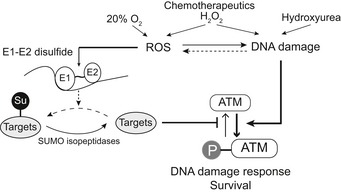
Temporal inactivation of a specific E1 and E2 pool by disulfide bond formation leads to local deSUMOylation of one or several unknown SUMO targets. This in turn is required to maintain ATM activity, which is required for DNA damage response and cell survival.
Ubc9 D100A sensitizes cells to chemotherapeutics
To gain further evidence that SUMO E1–E2 oxidation is required for DNA damage repair after oxidative stress, we turned to chemotherapeutics. Recently, Bossis et al (2014) demonstrated that chemotherapeutic drugs used against acute myeloid leukemia, a combination of the nucleoside analog cytarabine (Ara‐C), the topoisomerase II inhibitor etoposide (VP16) and the anthracycline daunorubicin (DNR), induce the SUMO E1~E2 disulfide when applied to AML cells at therapeutic concentration. While these drugs function by diverse mechanisms, ROS production seems to contribute to their efficacy (Gorrini et al, 2013). Our findings suggested that SUMO E1~E2 disulfide formation may protect cells against these therapeutics. We thus evaluated their effect on survival of “Ubc9 wt” and “Ubc9 DA” cells. For this, we treated cells with increasing concentration of Ara‐C (0–1.6 μM), VP16 (0–2.5 μM) and DNR (0–60 nM). While DNR was equally toxic for “Ubc9 wt” and “Ubc9 D100A” cells (Appendix Fig S12), cell death induced by VP16 and Ara‐C was significantly enhanced in “Ubc9 DA” cells compared to “Ubc9 wt” cells (Fig 8C, left graphs). To investigate whether this higher sensitivity is the direct consequence of ROS production induced by these drugs, we added the antioxidant N‐acetylcysteine (NAC) 16 h before VP16 or Ara‐C treatment. Indeed, addition of NAC prevented the enhanced cell death of “Ubc9 DA” cells treated with Ara‐C or VP16 (Fig 8C, right panel). In line with a contribution of the ATM pathway to these differences, pATM levels were higher in “Ubc9 wt” than in “Ubc9 DA” cells and this difference was lost by pretreatment with NAC (Appendix Fig S13). These findings revealed that Ubc9 D100A sensitizes cells to specific chemotherapeutic drugs and demonstrated that their enhanced sensitivity depends on ROS production. In conclusion, SUMO E1–E2 oxidation is required for survival after oxidative stress‐induced DNA damage and acts downstream of damage‐induced ATM phosphorylation.
Discussion
Here, we showed that the SUMO E1 and E2 enzymes are essential thiol switches whose reversible oxidation contributes to cell survival—not only upon acute oxidative stress that is caused by exposure of cells to hydrogen peroxide or selected chemotherapeutic drugs, but also in mild chronic oxidative stress that is caused by cell cultivation at 20% oxygen. Our findings provide novel insights into the regulation of SUMO enzymes, link the SUMO pathway to essential aspects of redox signaling in oxidative stress and suggest that specific interference with SUMO E1~E2 redox regulation may be an attractive strategy to enhance efficacy of chemotherapeutics.
A SUMO E2 variant that enhances E1~E2 disulfide sensitivity toward reduction
Here, we identified Ubc9 D100 as a residue whose mutation to alanine allows to selectively disrupt pathways that require reversible SUMO enzyme oxidation without interfering with Ubc9's essential role in SUMOylation. Based on multiple lines of evidence, including the observation that Ubc9 D100A impairs cell proliferation at 20%—but not at 5%—oxygen and the finding that N‐acetyl cysteine rescues “D100A cells” from increased sensitivity toward chemotherapeutics, the mutant has no other toxic defect than its negative effect on the stability of the SUMO E1~E2 disulfide. At present, we can only speculate why the Uba2~Ubc9 D100A disulfide is much more prone to reduction than wt Ubc9: Initial interaction between Ubc9 and the SUMO E1 enzyme in catalysis requires binding of Ubc9 to the ubiquitin fold domain of Uba2. During the catalytic cycle, the E1 enzyme undergoes dramatic changes, during which the catalytic cysteine of the E1 is unmasked to attack the SUMO adenylate (Olsen et al, 2010). Thioester transfer of SUMO from E1 to Ubc9 requires Ubc9's catalytic cysteine to come into immediate proximity of the E1 active site. Close proximity of these two cysteines is also essential for disulfide bond formation in oxidative stress, and we thus assume that the relative orientation of E1 and E2 during thioester transfer and during oxidation is very similar. How Ubc9's and Uba2's catalytic cysteines may face each other has been deduced from a recent crystal structure of disulfide‐linked ubiquitin E1 and the E2 enzyme UbcH4 (Olsen & Lima, 2013). Based on this, Ubc9 D100 is part of the Ubc9 surface that faces the E1, but it is distant from E1's catalytic cysteine. Since the D100A mutation leads to charge removal and to side chain shortening, we consider the following explanations: First, the D100A mutation may have a subtle influence on the exact orientation of Ubc9 on the E1. This may increase a mechanical strain on the E1~E2 disulfide, which in turn could influence its redox potential (Baldus & Grater, 2012). Second, side chain shortening and/or removal of the negative charge may facilitate access of the reducing agents DTT and glutathione. Facilitated access may in fact also explain the increased propensity for oxidation by H2O2 or diamide. While detailed insights into the in vitro and in vivo mechanism of oxidation and reduction require further investigations, the Ubc9 D100A variant turned out to be an excellent tool to study the physiological role of SUMO E1 and E2 enzymes as reversible thiol switches.
A role for reversible SUMO enzyme oxidation in ATM‐dependent DDR
Overexpression of Ubc9 D100A or replacement of endogenous Ubc9 by Ubc9 D100A is detrimental for long‐term cell proliferation, when cells are cultivated under conditions of chronic low oxidative stress (20% oxygen), exposed to acute oxidative stress or treated with two very different DNA damage‐inducing chemotherapeutics (Ara‐C or VP16). Here, we showed that this can be explained, at least in part, by a defect of “Ubc9 D100A” cells in a ROS‐dependent pathway that leads to maintenance of activated ATM.
Considering that levels of the SUMO E1~E2 disulfide are low in acute stress and undetectable in cells grown at 20% oxygen, we hypothesize that a specialized fraction of SUMO E1 and E2 enzymes rather than the freely diffusible enzyme pool is regulated by oxidation in mild oxidative stress: On the one hand, stochastic DNA damage may lead to selective oxidation of DNA‐associated SUMO enzymes. Evidence for this idea comes from studies that suggest a feedback loop, in which the DNA damage response pathway activates ROS production; this in turn seems required to maintain DDR (reviewed in Caputo et al, 2012). SUMO E1 and Ubc9 have both been observed at sites of DNA damage (Galanty et al, 2009). On the other hand, SUMO E1 and E2 may be components of a ROS‐sensing signaling complex that is required to maintain DNA damage response. Such a complex could operate in the nucleus or the cytoplasm, in line with the finding that ATM exists in both compartments (Lavin, 2008).
What could be the molecular role of a pathway that involves SUMO enzyme oxidation? A requirement for SUMO E1~E2 oxidation indicates that one or several SUMOylated proteins need to be rapidly demodified to allow persistent ATM activity. Based on our findings, these proteins influence levels of pATM, but whether directly or indirectly remains to be seen. Candidates include ATM phosphatases and their regulators, signaling kinases that may alter the activity of ATM phosphatases, but also pATM binding partners that may protect ATM from dephosphorylation. Finding the relevant proteins will be challenging, as DNA damage causes dramatic changes in the SUMO proteome (reviewed in Bekker‐Jensen & Mailand, 2010; Jackson & Durocher, 2013; Sarangi & Zhao, 2015): For example, SUMOylation and deSUMOylation are essential events in the assembly and disassembly of repair complexes (Psakhye & Jentsch, 2012; Wu et al, 2014). Our “Ubc9 D100A” cells will be a very valuable tool for the identification of proteins whose deSUMOylation specifically depends on SUMO E1~E2 oxidation.
Interference with SUMO E1~E2 oxidation—a strategy for cancer treatment?
Chemotherapeutic drugs are given in cancer therapy to induce massive DNA damage and cell death of rapidly dividing cells. While different drugs function by diverse mechanisms, a common side effect is their frequent ability to induce ROS (Gorrini et al, 2013). Bossis and co‐workers (Bossis et al, 2014) found that the chemotherapeutic drugs Ara‐C, VP16 and DNR, which are used in combination to treat AML, induce ROS‐dependent SUMO E1~E2 disulfide formation and deSUMOylation in leukemia cells. The findings presented here reveal that this is a pro‐survival pathway that counteracts ROS‐induced cell death. Specific interference with E1~E2 disulfide bond formation may thus be a suitable strategy to increase the efficacy of ROS‐producing chemotherapeutics.
Materials and Methods
DNA constructs
SUMO‐1, CFP‐RanGAPtail, YFP‐SUMO‐1, Uba2, His‐Aos1 and Ubc9 plasmids used for recombinant protein production have been described (Pichler et al, 2002; Bossis et al, 2005). Sequences of oligonucleotides used for site‐directed mutagenesis of Ubc9 are available upon request. HA‐tagged mouse Ubc9 has been described (Bossis & Melchior, 2006). For pIRES‐Ubc9 constructs, Ubc9 wt or D100A were PCR amplified and cloned into BamHI/EcoRI sites of pIRES‐hrGFPII (Agilent Technologies).
Protein purification
SUMO‐1, SUMO‐2, YFP‐SP100, PIAS1, GST‐p53, Pias2β, RanBP2 ΔFG, Aos1/Uba2, Ubc9, YFP‐SUMO‐1 and CFP‐RanGAPtail purification procedures have been described (Pichler et al, 2004; Stankovic‐Valentin et al, 2009; Werner et al, 2009).
Reagents and antibodies
Cytosine‐β‐D‐arabinofuranoside (Ara‐C, Cat. No. C1768), daunorubicin‐hydrochloride (DNR, Cat. No. 30450), etoposide (VP‐16, Cat. No. 1383), N‐ethylmaleimide (Cat. No. E3876), L‐glutathione reduced (Cat. No. G4251), diamide (Cat. No. D3648), hydroxyurea (Cat. No. H8627), hydrogene peroxide (Cat. No. H1009) andα‐tubulin antibody (Cat. No. T9026) were from Sigma. N‐acetyl‐L‐cysteine was from Merck (Cat. No. 1.12422). Rabbit anti‐Ubc9 antibody was raised against GST‐Ubc9 and affinity‐purified against Ubc9. Goat α‐Uba2 has been described (Bossis & Melchior, 2006). Okadaic acid (Cat. No. sc‐3513), anti‐GFP (Cat. No. sc‐8334), anti‐p53 (Cat. No. sc‐126) and S. cerevisiae Ubc9 (Cat. No. sc‐6721) antibodies were from Santa Cruz. Anti‐γH2AX (S139) was from Millipore (Cat. No. 05‐636). Anti‐phospho‐ATM (s1981) was from Rockland (Cat. No. 200‐301‐400, Fig 6A) or Abcam (Cat. No. ab81292). Anti‐clathrin heavy chain (Cat. No. 2410), anti‐phospho‐Chk1 (S345, Cat. No. 2341), anti‐phospho‐Chk2 (T68, Cat. No. 2661) and anti‐TRIM28 (Cat. No. 4124) were from Cell Signaling. Anti‐53BP1 was from Novus Bio (Cat. No. NB‐100‐304). Rabbit polyclonal α‐GST and rabbit α‐Smt3 were kindly provided by Dr. Ludger Hengst, Innsbruck, and Dr. Stephan Jentsch, Martinsried, respectively. Secondary antibodies were obtained from Jackson Laboratories.
Generation of random Ubc9 mutant proteins
Random mutagenesis PCR on a cDNA fragment encoding Ubc9 aa 49–153 was performed using Taq DNA polymerase, 0.1 mM dATP, 0.1 mM dCTP, 1 mM dGTP, 1 mM dTTP and 0.1 nM of MnCl2. PCR products were cloned via NcoI/BamHI into pET23a‐Ubc9 in which the corresponding wild‐type sequence was deleted. After transformation of BL21(DE3) bacteria, single colonies were grown in 96‐well plates. At OD 0.6, Ubc9 expression was induced with 1 mM IPTG for 3 h. Bacteria were resuspended in transport buffer (TB: 110 mM KOAc, 20 mM HEPES [pH 7.3], 2 mM Mg[OAc]2, 1 mM EGTA, 1 μg/ml of each leupeptin, pepstatin and aprotinin) and lysed by a single freeze/thaw cycle, which is sufficient to release Ubc9. The supernatant was used as a source of E2 in in vitro SUMOylation assays.
In vitro SUMOylation assay
SUMOylation of CFP‐RanGAPtail with YFP‐SUMO was carried out using a FRET‐based high‐throughput assay (Bossis et al, 2005). SUMOylation of YFP‐SP100 was performed at 30°C using 650 nM YFP‐SP100, 110 nM E1, 3.5 μM SUMO1 or SUMO2, 40 nM RanBP2ΔFG and 25 nM wt or mutant Ubc9 in TB buffer supplemented with protease inhibitors, 1 mM DTT, 0.05% Tween‐20 and 0.2 mg/ml ovalbumin. For GST‐p53 SUMOylation, 300 nM GST‐p53, 170 nM E1, 5 μM SUMO2, 49 nM GST‐Pias2β and 120 nM wt or D100A Ubc9 were used. SUMOylation was started by addition of 5 mM ATP, and reactions were stopped by addition of 2× SDS sample buffer.
SUMO E1 discharge
630 nM E1 and 3.3 μM SUMO1 were incubated with 6 mM of ATP at 30°C in TRB buffer (50 mM Tris pH 7.5; 10 mM NaCl; 10 mM MgCl2). After 30 min, E1 loading was stopped by addition of 30 mM EDTA and 600 nM of Ubc9 wt or mutant was added. Reactions were stopped by addition of TLB buffer (50 mM Tris pH 6.8, 2% SDS, 4 M urea, 10% glycerol).
Cell culture, transfection, siRNA
HeLa and MCF‐7 were obtained from DSMZ (Cat. No. ACC‐57, ACC‐115, respectively). U2OS cells were from ATCC (Cat. No. HTB‐96). HTERT‐RPE1 cell line was kindly provided by Dr. Oliver Gruss (University of Bonn). Mammalian cells were maintained at 37°C in 5% CO2 in Dulbecco's modified Eagles's medium (Invitrogen) with 10% fetal bovine serum (FBS, GIBCO). Transfection was with Fugene HD reagent (Promega). H2O2 treatment and lysis was performed 36 h after transfection. Silencing of human Ubc9 was performed using a pool of oligonucleotides (Ubc9‐1: 5′‐GAGGAAAGCAUGGAGGAAAUU‐3′, Ubc9‐2: 5′‐CCAUCUUAGAGGAGGACAAUU‐3′, Ubc9‐3: 5′‐GGGAAGGAGGCUUGUUUAAUU‐3′) and Lipofectamine RNAimax reagent (Invitrogen). For analysis of the SUMO E1~E2 disulfide, cells were washed in PBS supplemented with 20 mM N‐ethylmaleimide (NEM) and lysed in non‐reducing TLB buffer supplemented with 20 mM NEM. Otherwise, cells were directly lysed in 2× SDS sample buffer.
Generation of cell population stably expressing Ubc9
U2OS cells were transfected with the pIRES‐hrGFPII constructs using Fugene HD. 48 h after the transfection, G418 (Sigma‐Aldrich) was added to the medium (800 μg/ml). After 4 weeks of selection, cells were harvested and GFP‐positive cells were sorted for the first time using a FACSAria. Stable cells were maintained in the presence of G418 and sorted a second time as indicated.
Clonogenical survival assay
U2OS stable cells were transfected with siRNA against human Ubc9. Seventy‐two hours later, 2 × 104 cells per well of a 6‐well plate were seeded with DMEM + 10% FBS, and treated with H2O2 or chemotherapeutic drugs for 1 h. After 10 days, wells were washed with PBS, fixed with 4% formaldehyde for 10 min, stained with 0.05% crystal violet for 20 min, rinsed with tap water and dried overnight. Areas of the well occupied by cells were measured with ImageJ using the “colonyArea” plugin. Graphs represent the mean of at least three independent experiments.
Immunofluorescence
After pre‐extraction with 2% Triton X‐100 in PBS for 2 min, cells were fixed with 3.7% formaldehyde for 20 min, and blocked with 2% BSA in PBS. Cells were stained with rabbit α‐53BP1 or mouse α‐γH2AX; α‐rabbit‐Alexa594 and α‐mouse‐Alexa594 were used for detection. Samples were analyzed with a LSM780 confocal microscope (Zeiss) set up with Zen 2010 software, using a Plan‐Apochromat 63×/1.4 oil immersion objective.
Comet assay
Comet assay was adapted from Olive and Banath (2006). Briefly, 72 h after siRNA transfection, cells were treated with H2O2, resuspended in trypsin and washed with PBS. 5,000 cells combined with 50 μl low melting point agarose (Sigma) at 37°C were dropped off on a CometSlide (Trevigen). After gelling, slides were immersed in the lysis buffer (10 mM Tris, 100 mM EDTA, 2.5 M NaCl, 1% Triton X‐100, pH 10) overnight at 4°C in the dark. Then, slides were immersed in alkaline electrophoresis solution (300 mM NaOH, 1 mM EDTA, pH > 13) for 1 h at 4°C before electrophoresis. Electrophoresis was performed in alkaline electrophoresis solution at 20 V for 15 min. Slides were immersed twice in 0.4 M Tris pH 7.4, once in 70% EtOH and dried at 37°C. DNA was stained by propidium iodide (10 μg/ml for 20 min) and visualized with a Zeiss Axioskop 2 (CellObserver, Zeiss) and a 10× Plan‐apochromat objective.
Immunoprecipitation
To investigate TRIM28 SUMOylation, the SUMO proteome of U2OS or HeLa cells was precipitated upon denaturing cell lysis as described previously (Barysch et al, 2014) and analyzed by immunoblotting with TRIM28 antibodies.
Statistical analysis
Experimental data were analyzed using two‐tailed Student's t‐test. P‐values < 0.05 were considered statistically significant.
Author contributions
NS‐V designed and carried out most experiments and wrote the manuscript. KD designed and carried out several experiments, CK carried out and ES guided experiments with S. cerevisiae. FM guided the project, designed experiments and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
We gratefully acknowledge Katja Curth and Anja Schubert for excellent technical assistance, Iker Valle Aramburu for experimental support, Drs. Sina Barysch and Ulrike Winter for critical reading of the manuscript, Dr. Monika Langlotz (ZMBH Flow Cytometry & FACS Core Facility) for help with FACS sorting, the Metabolic Core Technology Platform at the University of Heidelberg for HPLC‐based GSH measurement, Dr. Stefan Jentsch, Martinsried, and Dr. Ludger Hengst, Innsbruck for antibodies, Dr. Oliver Gruss for hTERT‐RPE1 cell line, Dr. Christopher Lima, NYC, for sharing unpublished data and all laboratory members for sharing reagents and advice. N.S.V. was supported by a fellowship from the Fondation pour la Recherche Médicale (FRM) and the Marie Curie Training Network “Ubiregulator” (MRTN CT‐2006‐034555). This project received funding from the German Research Society (SFB 1036, TP15).
The EMBO Journal (2016) 35: 1312–1329
References
- Baldus IB, Grater F (2012) Mechanical force can fine‐tune redox potentials of disulfide bonds. Biophys J 102: 622–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barysch SV, Dittner C, Flotho A, Becker J, Melchior F (2014) Identification and analysis of endogenous SUMO1 and SUMO2/3 targets in mammalian cells and tissues using monoclonal antibodies. Nat Protoc 9: 896–909 [DOI] [PubMed] [Google Scholar]
- Barzilai A, Yamamoto K (2004) DNA damage responses to oxidative stress. DNA Repair (Amst) 3: 1109–1115 [DOI] [PubMed] [Google Scholar]
- Bekker‐Jensen S, Mailand N (2010) Assembly and function of DNA double‐strand break repair foci in mammalian cells. DNA Repair (Amst) 9: 1219–1228 [DOI] [PubMed] [Google Scholar]
- Bossis G, Chmielarska K, Gartner U, Pichler A, Stieger E, Melchior F (2005) A fluorescence resonance energy transfer‐based assay to study SUMO modification in solution. Methods Enzymol 398: 20–32 [DOI] [PubMed] [Google Scholar]
- Bossis G, Melchior F (2006) Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol Cell 21: 349–357 [DOI] [PubMed] [Google Scholar]
- Bossis G, Sarry JE, Kifagi C, Ristic M, Saland E, Vergez F, Salem T, Boutzen H, Baik H, Brockly F, Pelegrin M, Kaoma T, Vallar L, Recher C, Manenti S, Piechaczyk M (2014) The ROS/SUMO axis contributes to the response of acute myeloid leukemia cells to chemotherapeutic drugs. Cell Rep 7: 1815–1823 [DOI] [PubMed] [Google Scholar]
- Brandes N, Schmitt S, Jakob U (2009) Thiol‐based redox switches in eukaryotic proteins. Antioxid Redox Signal 11: 997–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo F, Vegliante R, Ghibelli L (2012) Redox modulation of the DNA damage response. Biochem Pharmacol 84: 1292–1306 [DOI] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D (2008) ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9: 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Autreaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813–824 [DOI] [PubMed] [Google Scholar]
- Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB (2002) A thiol peroxidase is an H2O2 receptor and redox‐transducer in gene activation. Cell 111: 471–481 [DOI] [PubMed] [Google Scholar]
- Doris KS, Rumsby EL, Morgan BA (2012) Oxidative stress responses involve oxidation of a conserved ubiquitin pathway enzyme. Mol Cell Biol 32: 4472–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T (2011) Signal transduction by reactive oxygen species. J Cell Biol 194: 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotho A, Melchior F (2013) Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem 82: 357–385 [DOI] [PubMed] [Google Scholar]
- Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP (2009) Mammalian SUMO E3‐ligases PIAS1 and PIAS4 promote responses to DNA double‐strand breaks. Nature 462: 935–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareau JR, Lima CD (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11: 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies J, Hochstrasser M (2012) A new class of SUMO proteases. EMBO Rep 13: 284–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees‐Miller SP, Khanna KK (2004) Autophosphorylation of ataxia‐telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J 23: 4451–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12: 931–947 [DOI] [PubMed] [Google Scholar]
- Groitl B, Jakob U (2014) Thiol‐based redox switches. Biochim Biophys Acta 1844: 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT (2010) ATM activation by oxidative stress. Science 330: 517–521 [DOI] [PubMed] [Google Scholar]
- Halliwell B (2007) Oxidative stress and cancer: have we moved forward? Biochem J 401: 1–11 [DOI] [PubMed] [Google Scholar]
- Huang C, Han Y, Wang Y, Sun X, Yan S, Yeh ET, Chen Y, Cang H, Li H, Shi G, Cheng J, Tang X, Yi J (2009) SENP3 is responsible for HIF‐1 transactivation under mild oxidative stress via p300 de‐SUMOylation. EMBO J 28: 2748–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA (2008) Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem 77: 755–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, Sadofsky MJ, Zhou MM, Rauscher FJ 3rd (2007) PHD domain‐mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell 28: 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Durocher D (2013) Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell 49: 795–807 [DOI] [PubMed] [Google Scholar]
- Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G (1997) The ubiquitin‐like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J 16: 5509–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Wu H, Collier‐Hyams LS, Hansen JM, Li T, Yamoah K, Pan ZQ, Jones DP, Neish AS (2007) Commensal bacteria modulate cullin‐dependent signaling via generation of reactive oxygen species. EMBO J 26: 4457–4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin MF (2007) ATM and the Mre11 complex combine to recognize and signal DNA double‐strand breaks. Oncogene 26: 7749–7758 [DOI] [PubMed] [Google Scholar]
- Lavin MF (2008) Ataxia‐telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 9: 759–769 [DOI] [PubMed] [Google Scholar]
- Leitao BB, Jones MC, Brosens JJ (2011) The SUMO E3‐ligase PIAS1 couples reactive oxygen species‐dependent JNK activation to oxidative cell death. FASEB J 25: 3416–3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Wuerzberger‐Davis SM, Miyamoto S (2006) PIASy mediates NEMO sumoylation and NF‐kappaB activation in response to genotoxic stress. Nat Cell Biol 8: 986–993 [DOI] [PubMed] [Google Scholar]
- Manza LL, Codreanu SG, Stamer SL, Smith DL, Wells KS, Roberts RL, Liebler DC (2004) Global shifts in protein sumoylation in response to electrophile and oxidative stress. Chem Res Toxicol 17: 1706–1715 [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ (2000) Ataxia telangiectasia‐mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci USA 97: 10389–10394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak A, Muller S (2014) SUMO‐specific proteases/isopeptidases: SENPs and beyond. Genome Biol 15: 422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive PL, Banath JP (2006) The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1: 23–29 [DOI] [PubMed] [Google Scholar]
- Olsen SK, Capili AD, Lu X, Tan DS, Lima CD (2010) Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature 463: 906–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SK, Lima CD (2013) Structure of a ubiquitin E1‐E2 complex: insights to E1‐E2 thioester transfer. Mol Cell 49: 884–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT (2015) Mechanisms of ATM Activation. Annu Rev Biochem 84: 711–738 [DOI] [PubMed] [Google Scholar]
- Pereira G, Tanaka TU, Nasmyth K, Schiebel E (2001) Modes of spindle pole body inheritance and segregation of the Bfa1p‐Bub2p checkpoint protein complex. EMBO J 20: 6359–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peuget S, Bonacci T, Soubeyran P, Iovanna J, Dusetti NJ (2014) Oxidative stress‐induced p53 activity is enhanced by a redox‐sensitive TP53INP1 SUMOylation. Cell Death Differ 21: 1107–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichler A, Gast A, Seeler JS, Dejean A, Melchior F (2002) The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108: 109–120 [DOI] [PubMed] [Google Scholar]
- Pichler A, Knipscheer P, Saitoh H, Sixma TK, Melchior F (2004) The RanBP2 SUMO E3 ligase is neither HECT‐ nor RING‐type. Nat Struct Mol Biol 11: 984–991 [DOI] [PubMed] [Google Scholar]
- Polo SE, Jackson SP (2011) Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25: 409–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psakhye I, Jentsch S (2012) Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151: 807–820 [DOI] [PubMed] [Google Scholar]
- Rhee SG (2006) Cell signaling. H2O2, a necessary evil for cell signaling. Science 312: 1882–1883 [DOI] [PubMed] [Google Scholar]
- Saitoh H, Hinchey J (2000) Functional heterogeneity of small ubiquitin‐related protein modifiers SUMO‐1 versus SUMO‐2/3. J Biol Chem 275: 6252–6258 [DOI] [PubMed] [Google Scholar]
- Sarangi P, Zhao X (2015) SUMO‐mediated regulation of DNA damage repair and responses. Trends Biochem Sci 40: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seufert W, Futcher B, Jentsch S (1995) Role of a ubiquitin‐conjugating enzyme in degradation of S‐ and M‐phase cyclins. Nature 373: 78–81 [DOI] [PubMed] [Google Scholar]
- Sobotta MC, Liou W, Stocker S, Talwar D, Oehler M, Ruppert T, Scharf AN, Dick TP (2015) Peroxiredoxin‐2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol 11: 64–70 [DOI] [PubMed] [Google Scholar]
- Stankovic‐Valentin N, Kozaczkiewicz L, Curth K, Melchior F (2009) An in vitro FRET‐based assay for the analysis of SUMO conjugation and isopeptidase cleavage. Methods Mol Biol 497: 241–251 [DOI] [PubMed] [Google Scholar]
- Sulli G, Di Micco R, d'Adda di Fagagna F (2012) Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer 12: 709–720 [DOI] [PubMed] [Google Scholar]
- Tatham MH, Chen Y, Hay RT (2003) Role of two residues proximal to the active site of Ubc9 in substrate recognition by the Ubc9.SUMO‐1 thiolester complex. Biochemistry 42: 3168–3179 [DOI] [PubMed] [Google Scholar]
- Tonks NK (2005) Redox redux: revisiting PTPs and the control of cell signaling. Cell 121: 667–670 [DOI] [PubMed] [Google Scholar]
- Veal EA, Day AM, Morgan BA (2007) Hydrogen peroxide sensing and signaling. Mol Cell 26: 1–14 [DOI] [PubMed] [Google Scholar]
- de la Vega L, Grishina I, Moreno R, Kruger M, Braun T, Schmitz ML (2012) A redox‐regulated SUMO/Acetylation switch of HIPK2 controls the survival threshold to oxidative stress. Mol Cell 46: 472–483 [DOI] [PubMed] [Google Scholar]
- Werner A, Moutty MC, Moller U, Melchior F (2009) Performing in vitro sumoylation reactions using recombinant enzymes. Methods Mol Biol 497: 187–199 [DOI] [PubMed] [Google Scholar]
- Winyard PG, Moody CJ, Jacob C (2005) Oxidative activation of antioxidant defence. Trends Biochem Sci 30: 453–461 [DOI] [PubMed] [Google Scholar]
- Wood MJ, Storz G, Tjandra N (2004) Structural basis for redox regulation of Yap1 transcription factor localization. Nature 430: 917–921 [DOI] [PubMed] [Google Scholar]
- Wu CS, Ouyang J, Mori E, Nguyen HD, Marechal A, Hallet A, Chen DJ, Zou L (2014) SUMOylation of ATRIP potentiates DNA damage signaling by boosting multiple protein interactions in the ATR pathway. Genes Dev 28: 1472–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Lam LS, Lam LH, Chau SF, Ng TB, Au SW (2008) Molecular basis of the redox regulation of SUMO proteases: a protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J 22: 127–137 [DOI] [PubMed] [Google Scholar]
- Yan S, Sun X, Xiang B, Cang H, Kang X, Chen Y, Li H, Shi G, Yeh ET, Wang B, Wang X, Yi J (2010) Redox regulation of the stability of the SUMO protease SENP3 via interactions with CHIP and Hsp90. EMBO J 29: 3773–3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Aslund F, Storz G (1998) Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 279: 1718–1721 [DOI] [PubMed] [Google Scholar]
- Zhou W, Ryan JJ, Zhou H (2004) Global analyses of sumoylated proteins in Saccharomyces cerevisiae. Induction of protein sumoylation by cellular stresses. J Biol Chem 279: 32262–32268 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File