

Abstract
Background and Purpose
With a prevalence of 1–2%, epilepsies belong to the most frequent neurological diseases worldwide. Although antiepileptic drugs are available since several decades, the incidence of patients that are refractory to medication is still over 30%. Antiepileptic effects of κ opioid receptor (κ receptor) agonists have been proposed since the 1980s. However, their clinical use was hampered by dysphoric side effects. Recently, G‐protein biased κ receptor agonists were developed, suggesting reduced aversive effects.
Experimental Approach
We investigated the effects of the κ receptor agonist U‐50488H and the G‐protein biased partial κ receptor agonist 6′‐GNTI in models of acute seizures and drug‐resistant temporal lobe epilepsy and in the conditioned place avoidance (CPA) test. Moreover, we performed slice electrophysiology to understand the functional mechanisms of 6′‐GNTI.
Key Results
As previously shown for U‐50488H, 6′‐GNTI markedly increased the threshold for pentylenetetrazole‐induced seizures. All treated mice displayed reduced paroxysmal activity in response to U‐50488H (20 mg·kg−1) or 6′‐GNTI (10–30 nmoles) treatment in the mouse model of intra‐hippocampal injection of kainic acid. Single cell recordings on hippocampal pyramidal cells revealed enhanced inhibitory signalling as potential mechanisms causing the reduction of paroxysmal activity. Effects of 6′‐GNTI were blocked in both seizure models by the κ receptor antagonist 5′‐GNTI. Moreover, 6′‐GNTI did not induce CPA, a measure of aversive effects, while U‐50488H did.
Conclusions and Implications
Our data provide the proof of principle that anticonvulsant/antiseizure and aversive effects of κ receptor activation can be pharmacologically separated in vivo.
Abbreviations
- 6′‐GNTI
6′‐guanidinyl‐17‐(cyclopropylmethyl)‐6,7‐dehydro‐4,5α‐epoxy‐3,14‐dihydroxy‐6,7‐2′,3′‐indolomorphinan dihydrochloride
- CPA
conditioned place avoidance
- EKC
ethylketocyclazocine
- mTLE
mesial temporal lobe epilepsy
- PTZ
pentylenetetrazole
- RMP
resting membrane potential
- TLE
temporal lobe epilepsy
Tables of Links
| TARGETS |
|---|
| GPCRs |
| κ opioid receptor |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
With a prevalence of 1–2%, epilepsies belong to the most frequent neurological diseases worldwide (McNamara, 1999). Mesial temporal lobe epilepsy (mTLE) is the most frequent type of epilepsy in humans and is frequently induced by traumatic brain injury. Hippocampal sclerosis and accompanying neurological deficits are key features of mTLE (see Engel, 2001). Despite the introduction of a plethora of anti‐epileptic drugs over the last few decades, the rate of drug‐resistant focal seizures (30% and 70%) did not improve since the study of Coatsworth in 1971 (Coatsworth, 1971; Loscher and Schmidt, 2011). To date, surgical resection of the epileptogenic focus remains as the ultimate option for some of these patients. Even then, in certain subgroups of patients, less than 50% remain seizure free for at least 1 year after removal of the epileptic focus (Spencer and Huh, 2008). Thus, novel targets are needed to develop antiepileptic drugs.
Since the early 1980s, there has been evidence that some endogenous opioids, namely, dynorphin, act as modulators of neuronal excitability in vitro (Henriksen et al., 1982; Siggins et al., 1986). Because of the distribution of dynorphin in the brain, its most probable involvement in epilepsy was postulated in partial complex seizures originating from the limbic system (see Simonato and Romualdi, 1996). In line with this, the deletion of the prodynorphin coding sequence in mice (Loacker et al., 2007) and low dynorphin levels in humans due to mutations in the promoter region (Stögmann et al., 2002; Gambardella et al., 2003) are associated with increased vulnerability to the development of epilepsy. In most animal models of temporal lobe epilepsy (TLE; comprising epilepsies arising cortical = lateral TLE and mTLE), cortical and hippocampal prodynorphin expression is reduced after an initial, short peak of over‐expression (see Simonato and Romualdi, 1996; Schwarzer, 2009). This is in line with significantly increased prodynorphin mRNA in hippocampal granule cells of patients displaying seizures within the last 48 h before surgical removal of the hippocampus, compared with those with a longer seizure‐free period (Pirker et al., 2009) accompanied by an overall reduction of dynorphin immunoreactivity in surgically removed tissue obtained from mTLE patients (de Lanerolle et al., 1997).
Because of the reduction of endogenous dynorphin under epileptic conditions, the κ opioid receptors may be available to bind exogenously applied agonists and the application of κ receptor agonists can suppress experimental seizures (Tortella, 1988; Takahashi et al., 1990; Solbrig et al., 2006; Loacker et al., 2007). Various selective κ receptor agonists applied through different routes yielded time‐dependent and dose‐dependent effects similar to those upon treatment with phenytoin or phenobarbital in models of epilepsy (see Simonato and Romualdi, 1996). We recently demonstrated that the activation of κ receptors promotes the survival of hippocampal and amygdala neurons subsequent to the acute phase after unilateral injection of kainic acid in mice (Schunk et al., 2011).
Along with the promising features of κ receptor activation in epilepsy models, there exists one major drawback. The therapeutic usefulness of full κ receptor agonists is decreased by their dysphoric side effects (Barber and Gottschlich, 1997). Since the failure of clinical trials with centrally active κ receptor agonists, such as spiradoline or enadoline in the 1990s, pharmaceutical companies stepped back from further development of this field. In recent years, it was suggested from animal experiments that the recruitment of β‐arrestin and subsequent phosphorylation of p38 MAPK might be crucial for the aversive effects of κ receptor activation (Bruchas et al., 2007; Bruchas et al., 2011). In contrast, aversive effects of κ receptor stimulation were also observed in β‐arrestin 2 knockout mice (White et al., 2015), suggesting parallel pathways. In parallel, biased κ receptor agonists, which preferentially activate either the G‐protein or the β‐arrestin pathway, were reported from in vitro experiments. A prototypic representative of this type of compound is 6′‐guanidinyl‐17‐(cyclopropylmethyl)‐6,7‐dehydro‐4,5α‐epoxy‐3,14‐dihydroxy‐6,7‐2′,3′‐indolomorphinan dihydrochloride (6′‐GNTI), which was originally reported as a partial κ receptor agonist (Sharma et al., 2001). 6′‐GNTI preferentially activates the G‐protein pathway over the β‐arrestin pathway, compared with the full agonists U‐50488H and ethylketocyclazocine (EKC) (Rives et al., 2012) or U‐69,593 (Schmid et al., 2013). In fact, 6′‐GNTI acts as a partial agonist for the G‐protein activation through κ receptor at low μM concentrations, but does not activate β‐arrestin in the sub‐mM range (Rives et al., 2012). In contrast, RB 64 is a full agonist for G‐protein and β‐arrestin activation, although the potency for β‐arrestin is low (White et al., 2015). Such drugs may open a completely new avenue in the treatment of epilepsy, provided that G‐protein biased κ receptor agonists display anticonvulsant/antiseizure properties with less aversive side effects in the complex in vivo situation.
The aim of this study was to investigate, whether it is possible to separate the anticonvulsant/antiseizure effects from the aversive effects by biased drugs suhc as 6′‐GNTI. For this, we applied in vivo EEG, electrophysiology and behavioural testing. Further, we aimed to compare the aversive effects induced by a full κ receptor agonist and by 6′‐GNTI.
Methods
Animals
All animal care and experimental procedures were approved by the Austrian Animal Experimentation Ethics Board in compliance with the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes ETS no.: 123 and the Canadian Council on Animal Care. Every effort was taken to minimize the number of animals used. The study was designed and is reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
A total of 61 C57BL/6 N wild‐type and 40 prodynorphin knockout (pDyn‐KO) mice were investigated in this study. pDyn‐KO mice were backcrossed onto the C57BL/6 N background over 10 generations (Loacker et al., 2007). For breeding and maintenance, mice were group housed (maximum of five animals per cage) with free access to food and water. Temperature was fixed at 23°C and 60% humidity with a 12 h light–dark cycle (lights on 7 am to 7 pm). Male mice of the same strand and age were arbitrarily sorted into groups, splitting litters into different groups. For the animal experiments, the experimenter was blinded to the treatments of the animals.
Pentylenetetrazole‐induced seizures
Threshold for pentylenetetrazole induced seizures was investigated in four C57BL/6 N wild‐type and 40 pDyn‐KO young adult (about 12–14 weeks; 25 g) male mice. the pentylenetetrazole threshold was measured through infusion of pentylenetetrazole (10 mg·mL in saline) through the tail‐vein in freely moving animals 20 min after intracisternal injection of either saline or different doses of 6′‐GNTI in a fixed volume of 3 μL under light sevofluorane anaesthesia. 5′‐GNTI treatment (also in 3 μL, under light sevofluorane anaesthesia) was performed 10 min prior to 6′‐GNTI in some experiments. Intracisternally saline infused animals, and animals treated with 5′‐GNTI alone, were included as control groups. Infusion was stopped at the first appearance of tonic–clonic seizures and animals were immediately killed by cervical displacement. Threshold was calculated from volume injected and body weight (Loacker et al., 2007). We investigated the effects of 6′‐GNTI in this model of acute seizures because our previous finding showed that the classic κ receptor full agonist U‐50488H was able to restore the reduced pentylenetetrazole‐seizure threshold in pDyn‐KO mice.
Kainic acid injection and electrode implantation
Twenty‐four C57BL/6 N wild‐type young adult (about 12–14 weeks; 25 g) male mice were sedated with ketamine (160 mg·kg−1, i.p.; Graeub Veterinary Products, Switzerland) and then deeply anaesthetized with sevoflurane through a precise vaporizer (Midmark, USA). Mice were injected with 50 nL of a 20 mM kainic acid solution into the left hippocampus as previously described (Loacker et al., 2007). Four electrodes (two cortical and two depth electrodes) were implanted immediately after kainic acid administration. Epoxylite‐coated tungsten depth electrodes (diameter 250 μm; FHC, USA) were placed bilaterally into the hippocampus aimed at the CA1 area (RC −1.80 mm; ML ±1.80 mm; DV −1.60 mm). Surface electrodes were gold‐plated screws placed bilaterally into the skull on top of the motor‐cortex (RC +1.70 mm; ML ±1.6 mm with the bregma as a reference point) to monitor the generalization of abnormal EEG activities. An additional surface electrode was placed on the cerebellum as ground and reference. Electrodes were secured in place with dental acrylate cement (Heraeus Kulzer GmbH, Germany).
For experiments requiring an i.c.v. drug administration, a guide cannula was implanted in the lateral ventricle of the contralateral hemisphere (RC −0.50 mm; ML +1.00 mm; DV +2.00 mm) instead of the contralateral electrode. All animals received meloxicam (2 mg·kg−1) 20 min before and after surgery as an analgesic treatment. The locally injected kainic acid model was chosen because it is a model of trauma‐induced epilepsy that allows the investigation of self‐sustained paroxysmal events, and it was characterized as a model for drug‐resistant epilepsy (Riban et al., 2002).
EEG recording and analysis
The EEG was obtained using a wireless recording device (Neurologger, TSE Systems GmbH, Germany) and automatically analysed using sciworks Software (Datawave Technologies, USA). EEGs were filtered for epileptiform spikes defined as high amplitude discharges (>3 × baseline) lasting less than 70 ms. This definition allows the exclusion of other EEG events induced by hippocampal kainic acid administration, such as high‐voltage sharp waves that last considerably longer (150–200 ms; Riban et al., 2002). Spike trains were defined as the occurrence of at least three spikes with a frequency higher than 1 Hz and lasting for at least 1 s. Prolonged hippocampal paroxysmal discharges (hpds) were defined as spike trains lasting for a minimum of 10 s. Spikes detected in between spike trains and hpds were counted as inter‐ictal spikes. For in vivo EEG, only animals displaying a minimal seizure activity of 100 s during the baseline recording were included. Therefore, group sizes may vary. EEG data are depicted as ratio of events recorded the first hour after treatment compared with the last hour before treatment. For illustration, please see the Supporting Information Fig. S1. Data regarding the U‐50488H treatment in kainic acid model obtained with the datawave software were confirmed with visual analysis.
Conditioned place avoidance
Twenty‐eight C57BL/6 N wild‐type young adult (about 12–14 weeks; 25 g) male mice were used for the conditioned place avoidance (CPA) paradigm. Tests were conducted in a custom‐made, three‐chamber apparatus (Kummer et al., 2011). The conditioning procedure comprised a pre‐test session, four consecutive training days (two training sessions per day, one each for drug or saline) and a CPA test on day six. Pre‐test and CPA test session lengths were 15 min, and the conditioning sessions lasted 30 min. If the pre‐test presented a bias for one of the two chambers, the preferred one was paired with the drug (6′‐GNTI or U‐50488H) to facilitate the detectability of avoidance.
Prior to the conditioning sessions, the animals were treated with one of the drugs tested or saline in a neutral chamber. All experiments were performed with an illumination of 60 Lux in the centre of the conditioning chambers. The time spent in each compartment was analysed with the VideoMot 2 System (TSE Systems GmbH). To analyse the influence of drug treatment on motor activity, the distance travelled in the drug‐paired or saline‐paired chamber was recorded on the first treatment day using the VideoMot 2 System.
Immunohistochemistry
Immunohistochemistry was performed as described elsewhere (Schwarzer et al., 1995). In brief, mice were killed 30 min after drug treatment (n = 4 per group) by an overdose of thiopental (150 mg·kg−1), and brains were fixed by transcardial perfusion with 4% paraformaldehyde in PBS (50 mM PBS, pH 7.2). Immunohistochemistry was performed on coronal 40 μm vibratome sections incubated free‐floating in blocking solution (10% normal goat serum and 0.3% Triton X‐100 in TBS) for 90 min. Primary antibodies against Zif268 (1:2000; Santa Cruz; # sc189) or pERK (1:400; Cell Signalling Technology; #9101) were applied overnight at room temperature followed by horseradish peroxidase conjugated secondary antibodies (1:500, Dako) and 3,3′‐diaminobenzidine for detection. Specificity of antibodies was controlled by avoiding primary antibodies or by pre‐absorbing the primary antibody with the respective blocking peptide (Zif 268, Santa Cruz, # sc189 P; pERK 1, Cell Signalling, #9101; 10 μg mL) overnight at +4°C before the application on the section.
Electrophysiology
Five C57BL/6 N male wild‐type mice (25 g) between 8–16 weeks of age were used for in vitro electrophysiology. Mice were decapitated; their brains rapidly removed and submerged in an icy slurry of artificial CSF (ACSF) optimized for slice preparation (slice solution), containing (in mM) 118 NaCl, 3 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 10 glucose, 26 NaHCO3 and 2.5 CaCl2, which was bubbled continuously with carbogen (95% O2, 5% CO2). Kynurenic acid (1 mM) was also added to the slice solution to reduce glutamate‐mediated excitotoxicity. Mid‐ventral, transverse sections of the hippocampus (300 μm) were cut using a vibrating slicer (Slicer HR2; Sigmann Elektronik). Slices were then transferred to a carbogenated ACSF (bath) solution, which contained the following (in mM): 124 NaCl, 3 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 10 glucose, 26 NaHCO3 and 2.5 CaCl2 (300–305 mOsm·L−1). Slices were stored at room temperature (22°C) in the bath solution for at least 30 min following slicing; the bath solution was also used to perfuse slices for all experiments. Slices were placed into a recording chamber attached to a fixed stage of a moveable upright microscope (Axioskop FS2; Carl Zeiss, Vienna, Austria) and held submerged by a platinum and polyester fibre ‘harp’. Slices were continuously perfused with a warmed (34 ± 0.5°C), carbogenated bath ACSF at a rate of 2–3 mL·min−1 for at least 20 min prior to recording.
Pipettes were pulled from thin‐walled borosilicate glass (TW150F; WPI, Sarasota, FL, USA) with a two‐stage puller (PP‐83; Narishige, Amityville, NY) to a tip resistance of 5–7 MΩ when backfilled with an internal solution containing (in mM) 126 K‐gluconate, 10 HEPES, 4 KCl, 5 MgATP, 0.3 NaGTP, 1 EGTA and 0.3 CaCl2, to which 0.05–0.1% neurobiotin was added, and the pH was adjusted to 7.27–7.30 with KOH, (275–285 mOsm·L−1). Recordings were made using a Multiclamp 700B amplifier data and were acquired using a pCLAMP 10.3 via a Digidata 1322 interface (Molecular Devices, Sunnyvale, CA).
CA3 and CA1 pyramidal neurons were visually identified using infrared differential interference contrast optics and selected for recordings based on characteristic pyramidal morphology and the presence of a large apical dendrite. After obtaining the whole cell patch clamp configuration, neurons were held in a voltage clamp at −60 mV for 5–10 min before beginning experiments and between experimental measurements. Only neurons that showed stable resting membrane potential (RMP), holding a current (in a voltage clamp) in a series of control measurements, and which showed stable access resistance throughout the entire experiment, were selected for analysis.
A stimulation isolation unit (Iso‐Flex, AMPI, Jerusalem) was used to elicit synaptic responses onto recorded neurons. Stimulating electrodes (bath ACSF‐filled patch pipettes) were placed in either the mossy fibre layer for CA3 recordings or the Shaffer collaterals for CA1 recordings, exciting axons from the dentate gyrus and CA3 respectively. Stimulation intensities were adjusted to evoke responses of approximately half‐maximal amplitude. Synaptic responses that were evoked as neurons were held at −40 mV, which allowed inhibitory (likely GABAergic) and excitatory responses to be differentiated as outward and inward currents respectively. At −40 mV, most neurons showed both evoked EPSCs and inhibitory postsynaptic current (IPSCs). Holding neurons at −60 mV eliminated the IPSC component of evoked responses, suggesting the IPSC was largely GABAA receptor‐mediated. Spontaneous IPSCs and EPSCs were also measured during 2 min continuous voltage clamp recordings at −40 mV. The RMP of neurons was measured by averaging the potential over a 30 s period of passive current clamp recording. Drug effects on postsynaptic conductances were measured using a series of hyperpolarizing voltage steps conducted from a holding potential of −40 mV. Eight successive hyperpolarizing steps were used; the initial step hyperpolarized the membrane 10 mV, and each successive step increased by a −10 mV increment such that the final step moved the membrane from −40 to −120 mV. Each successive hyperpolarizing step was shortened by Xms to limit voltage‐mediated damage to the cell membrane. Several (>2) sets of recordings of neuronal and synaptic properties (evoked synaptic responses, etc.) were taken at 5 min intervals before drug application.
6′‐GNTI, dissolved in 10 mL ACSF to a final concentration of 1 μM, was applied via bath perfusion over a period of 2–3 min and then was washed out with ACSF. Recordings were taken immediately prior to drug application, during application, 1 min after application and successively every 5 min until drug effects washed out. The electrophysiology was performed unblinded due to practical reasons.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as mean ± SEM. Following acquisition, electrophysiological recordings were viewed and analysed using pClamp 10.3 (Molecular Devices). Prism 5 for Mac (version 5.0f) was used to perform a statistical analysis of in vivo experiments and to generate figures. For the statistical analysis, a one‐way ANOVA with a Dunnett post hoc test was applied to in vivo experiments. For electrophysiology, the two‐tailed, paired t‐tests were applied for RMP analysis, and a one‐way ANOVA was used to compare drug effects on IPSC. CPA data were analysed using paired t‐test and two‐way ANOVA with a Bonferroni post hoc test. A P value lower than 0.05 was considered significant.
Materials
Diazepam and pentylenetetrazole were purchased from Sigma Aldrich (St. Louis, MO). The κ receptor agonist U‐50488H and antagonist 5′‐GNTI were purchased from Tocris Bioscience, (Bristol, UK) while the G‐protein biased partial κ receptor agonist 6′‐GNTI was provided by the drug supply programme of National Institute on Drug Abuse (NIDA). pentylenetetrazole, U‐50488H, 5′‐GNTI and 6′‐GNTI were dissolved in saline, and the pH was adjusted to 7.2. Diazepam was dissolved in DMSO (10 mg·mL) and further diluted with saline. Diazepam and U‐50488H were applied i.p. For EEG recordings and CPA experiments, 5′‐GNTI (3 or 10 nmoles per 3 μL) and 6′‐GNTI (1–30 nmoles per 3 μL) were given i.c.v., while for the pentylenetetrazole experiments, they were administered intracisternally.
Results
Based on our previous finding that the full κ receptor agonist U‐50488H was able to restore the reduced pentylenetetrazole‐seizure threshold in pDyn‐KO mice, we investigated the effects of 6′‐GNTI in this model of acute seizures. As shown in Figure 1, 6′‐GNTI dose‐dependently increased the seizure threshold in pDyn‐KO mice (one‐way ANOVA, F 8,43 = 11.37). This effect reached a plateau at about 3 nmoles (P < 0.05). Pretreatment of animals with 3 or 10 nmoles of the specific κ receptor antagonist 5′‐GNTI reversed this effect.
Figure 1.
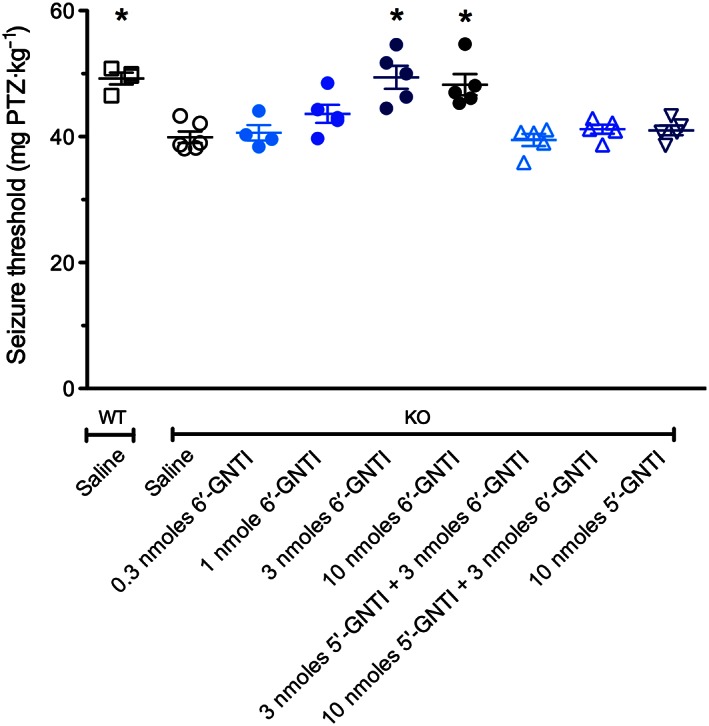
The dose–response curve of 6′‐GNTI on the threshold of pentylenetetrazole‐induced seizures as obtained by tail‐vein infusion. Twenty minutes after i.c.v. injection of 3 (n = 5) or 10 nmoles (n = 5) 6′‐GNTI, the seizure threshold was increased, compared with saline‐treated pDyn‐KO (n = 6) animals. No effect was observed at lower doses (0.3 nmole n = 4; 1 nmole n = 5). The seizure threshold of pDyn‐KO mice treated with 3 or 10 nmoles 6′‐GNTI was undistinguishable from that of saline‐treated wild‐type mice (n = 4). Pretreatment of pDyn‐KO animals with 5′‐GNTI (3 nmoles n = 5; 10 nmoles n = 5) 10 min before 6′‐GNTI entirely reversed this effect, while 5′‐GNTI alone (n = 5) had no effect on the seizure threshold. *P < 0.05, significantly different from saline; one‐way ANOVA with Dunnett's post hoc test. WT, wild type; KO, prodynorphin knockout.
Unilateral injection of kainic acid into the dorsal hippocampus of mice was characterized as a model for drug‐resistant epilepsy based on the non‐responsiveness of paroxysmal discharges to acute carbamazepine, phenytoin or valproate treatment (Riban et al., 2002). This, at least for some individuals, holds true also for modern antiepileptic drugs (Supporting Information Fig. S2). In our study, animals injected with 1 nmole of kainic acid developed hpds within 3–4 weeks after treatment. Generalized seizures were observed rarely (1–3 seizures per day per animal), while hpds occurred 23 ± 3.6 times per hour and lasted for 17.5 ± 1.4 s (n = 21). During hpds, mice displayed behavioural arrest and stereotypies. To investigate the potentials of κ receptor agonists in this model, we first tested whether the full κ receptor agonist U‐50488H affected these EEG abnormalities. As shown in Figure 2A–C, treatment with U‐50488H decreased hpds. A reduction in the number of hpds (one‐way ANOVA, P < 0.05 for 20 mg·kg−1; F 4,29 = 5.081) resulted in a significant reduction of total time spent in hpds (one‐way ANOVA, P < 0.05 for 20 mg·kg−1; F 4,29 = 5.221). In contrast to diazepam‐treated mice, which were not moving (most probably due to the highly sedative effects of diazepam), U‐50488H‐treated animals were actively exploring the cage during this time. The onset of suppression of spike trains is very rapid for both diazepam and U‐50488H, as shown in Figure 2D.
Figure 2.
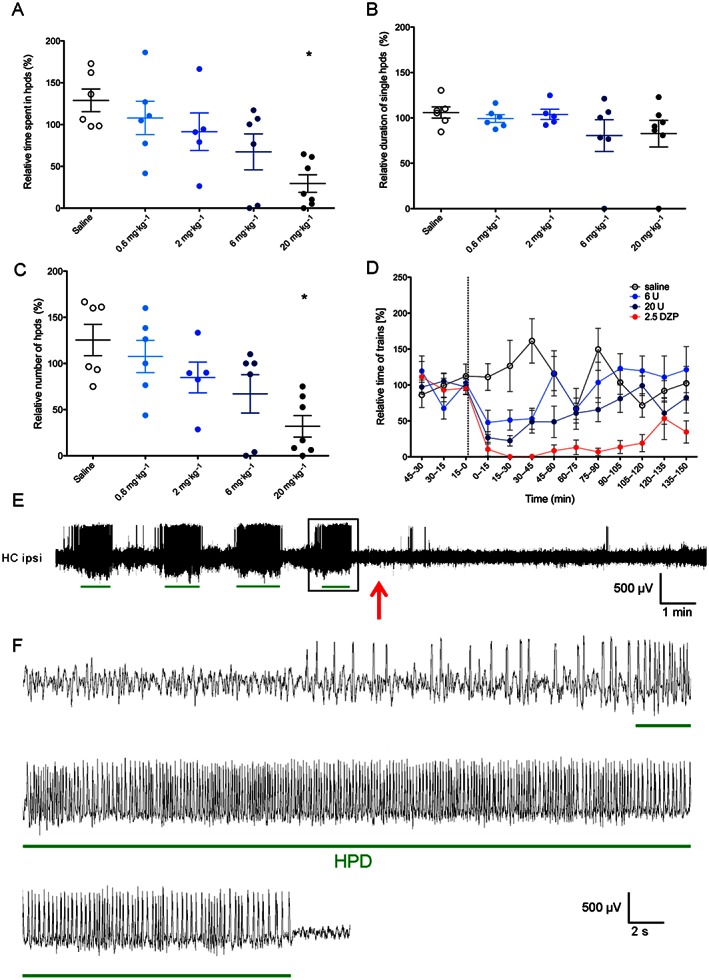
Effects of treatment with U‐50488H on time (A), average duration (B) and number (C) of hpds in kainic acid‐injected mice (n = 10). Ratios of total recorded events 60 min post‐treatment/60 min pretreatment are given. (D) Time course of spike trains after saline, U‐50488H (6 and 20 mg·kg−1) and diazepam treatments. Data are presented as ratio of total time of spike trains at time interval indicated/average time of paroxysmal discharges of the three 15 min intervals before treatment. The time of treatment is indicated by the dotted line. (E) Representative EEG trace showing spike trains recorded from the ipsilateral hippocampus in the 15 min periods preceding and following a 20 mg·kg−1 U‐50488H treatment. Time of U‐50488H administration is indicated with a red arrow. Green lines indicate the EEG parts detected as hpds by the analysis software. (F) Higher magnification of the hpds framed in panel E. *P < 0.05, significantly different from saline; one‐way ANOVA with Dunnett's post hoc test. DZP, diazepam; U, U‐50488H; HC ipsi, ipsilateral hippocampus.
U‐50488H, as a prototypic specific full κ receptor agonist, was appropriate to assess the potential of κ receptors in the kainic acid model. However, U‐50488H is known to produce aversive effects, which need to be overcome to consider the application of κ receptor agonists in humans. These aversive effects were reported to depend on the recruitment of the β‐arrestin and subsequent phosphorylation of p38α‐MAPK (Bruchas et al., 2007; Land et al., 2008) in mice. To examine whether anticonvulsive and antiseizure effects can be elicited through κ receptor activation, independent of the activation of the β‐arrestin pathway, we investigated the G‐protein biased partial κ receptor agonist 6′‐GNTI, which activates G‐protein mediated effects of κ receptor with low recruitment of β‐arrestin (Rives et al., 2012).
As shown in Figure 3A–C, 6′‐GNTI reduced hpds, affecting both duration and number of events. The event duration (one‐way ANOVA, P < 0.05 for 30 nmoles; F 4,31 = 7.582) as well as the number of events (one‐way ANOVA, P < 0.05 for 10 nmoles and P < 0.05 for 30 nmoles; F 4,31 = 9.854) were reduced, resulting in a significant reduction of total time of hpds (one‐way ANOVA, P < 0.05 for 30 nmoles; F 4,31 = 10.59). Although 6′‐GNTI is only a partial κ receptor agonist, animals displayed a strong reduction of spike trains after the treatment with 10 or 30 nmoles (Figure 3D). This effect was fully blocked by the κ receptor antagonist 5′‐GNTI (Supporting Information Fig. S4). Comparable results were obtained when EEGs were analysed for inter‐ictal spikes (Table 1).
Figure 3.
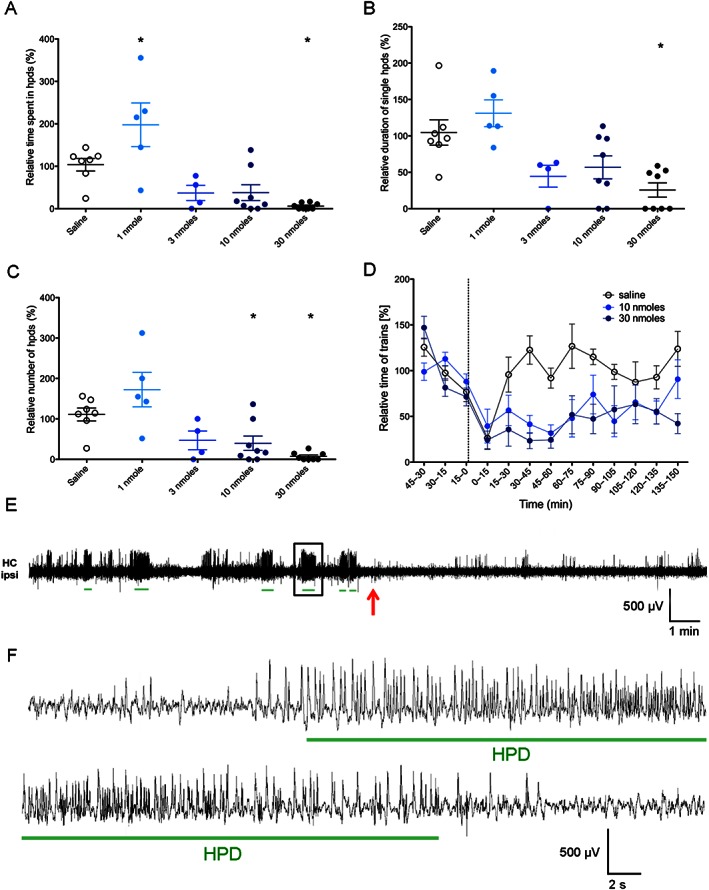
Effects of 6′‐GNTI treatments on time (A), average duration (B) and number (C) of hpds in kainic acid‐injected mice (n = 14). Ratios of total recorded events 60 min post‐treatment/60 min pretreatment are given. (D) Time course of spike trains after saline and 6′‐GNTI (10 and 30 nmoles) treatments. Data are presented as ratio of total time of EEG abnormalities at time interval indicated/average time of paroxysmal discharges of the three 15 min intervals before treatment. The time treatment is indicated by the dotted line. (E) Representative EEG trace showing spike trains recorded from the ipsilateral hippocampus in the 15 min periods preceding and following a 30 nmoles 6′‐GNTI treatment. Time of 6′‐GNTI administration is indicated with a red arrow. Green lines indicate the EEG parts detected as hpds by the analysis software. (F) Higher magnification of the hpds framed in panel E. *P < 0.05, significantly different from saline; one‐way ANOVA with Dunnett's post hoc test. HC ipsi, ipsilateral hippocampus.
Table 1.
Effects of U‐50488H and 6′‐GNTI on inter‐ictal spikes and spike trains. *P <0.05, significantly different from saline.
| Inter‐ictal spikes | ||||
|---|---|---|---|---|
| Saline | 0.6 mg·kg−1 U‐50488H | 2 mg·kg−1 U‐50488H | 6 mg·kg−1 U‐50488H | 20 mg·kg−1 U‐50488H |
| 98.03 ± 8.69% (n = 6) | 106.4 ± 10.1% (n = 7) | 82.57 ± 14.78% (n = 6) | 92.92 ± 11.04% (n = 7) | 103.3 ± 9.95% (n = 7) |
| Saline | 1 nmole 6′‐GNTI | 3 nmole 6′‐GNTI | 10 nmole 6′‐GNTI | 30 nmole 6′‐GNTI |
| 162.3 ± 37.19% (n = 8) | 114.5 ± 28.3% (n = 6) | 76.94 ± 8.79% (n = 8) | 137.7 ± 24.35% (n = 11) | 67.88 ± 8.29% (n = 9)* |
| Spike trains | ||||
| Saline | 0.6 mg·kg−1 U‐50488H | 2 mg·kg−1 U‐50488H | 6 mg·kg−1 U‐50488H | 20 mg·kg−1 U‐50488H |
| 126.2 ± 4.49% (n = 6) | 111.6 ± 11.08% (n = 8) | 95.85 ± 9.796% (n = 7) | 72.35 ± 18.63% (n = 7)* | 38.68 ± 10.34% (n = 8)* |
| Saline | 1 nmole 6′‐GNTI | 3 nmole 6′‐GNTI | 10 nmole 6′‐GNTI | 30 nmole 6′‐GNTI |
| 105.6 ± 4.606% (n = 8) | 123.8 ± 14.31% (n = 7)* | 75.11 ± 12.94% (n = 8) | 62.04 ± 15.38% (n = 10)* | 27.68 ± 10.05% (n = 8)* |
Activation of κ receptors with the full agonist U‐50488H induces strong place avoidance. No such data were available for 6′‐GNTI. Therefore, we applied a CPA paradigm to investigate potential aversive effects of 6′‐GNTI treatment. As expected, U‐50488H (2.5 mg·kg−1) induced strong place avoidance (Figure 4A; saline 68 ± 63.3 s; U‐50488H −123 ± 35.0 s; n = 6; Mann–Whitney P < 0.05). In contrast, 6′‐GNTI induced neither avoidance nor preference for the drug‐conditioned chamber at any tested dose (Figure 4B). Moreover, 6′‐GNTI did not influence motor activity of mice at the doses tested (Figure 4C).
Figure 4.
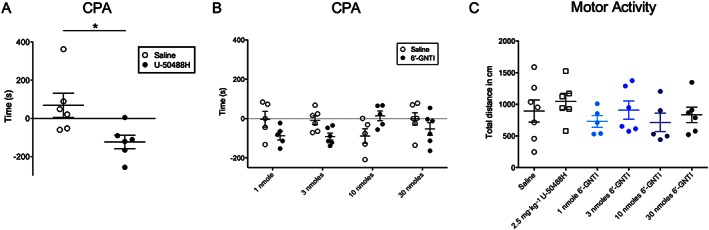
CPA for U‐50488H (A; n = 6) and different doses of 6′‐GNTI (B; each dose n = 5–6). Time is calculated as time spent in the drug‐paired compartment post test minus pre‐test.. *P < 0.05, significantly different from saline; two‐way ANOVA with Bonferroni's post hoc test. (C) Analysis of motor activity in the first 10 min following administration of saline, U‐50488H and different doses of 6′‐GNTI. None of the treatments revealed a statistical significant effect.
To investigate whether lack of aversive effects might be resulting from local application and restricted spread of the drug, we investigated the expression of the protein Zif268 (known to be activated by κ receptors, 30 min after treatment) with saline, 2.5 mg·kg−1 U‐50488H or 30 nmoles 6′‐GNTI. Strong Zif268‐immunoreactivity was observed in various regions of the brain, most prominent in the striatum, nucleus accumbens (Supporting Information Fig. S3) and cortical regions in the 6′‐GNTI group and to a lower level in the U‐50488H group. At this time interval, phosphorylation of ERK1 was hardly detectable. Of note is the fact that several neurons in the dorsal raphe displayed pERK immunoreactivity in the 6′‐GNTI group (Supporting Information Fig. S3).
Although 6′‐GNTI was first described several years ago (Sharma et al., 2001), no data on its effects on hippocampal pyramidal neurons are available. Therefore, we investigated 6′‐GNTI effects on hippocampal slices. Stimulation of hippocampal mossy fibers evoked IPSCs in CA3 pyramidal cells with an amplitude of 134 ± 32 pA. In the presence of 1 μM 6′‐GNTI, the amplitude of evoked IPSC significantly increased to 199 ± 33 pA (Figure 5A; n = 7; one‐way ANOVA, P < 0.05 for 6′‐GNTI vs. control; F 2,20 = 23.28), the effect was reversed upon washout of the drug. In a separate experiment, we tested whether the specific κ receptor antagonist 5′‐GNTI was able to block the increased eIPSC amplitude. After 6′‐GNTI application (demonstrating the responsiveness of cells to 6′‐GNTI), slices were bathed in ACSF containing 5′‐GNTI but not 6′‐GNTI. Under these conditions, eIPSC amplitudes were on control level (Figure 5B). Evoked IPSC amplitude did not change upon reapplication of 6′‐GNTI in the presence of 5′‐GNTI (Figure 5B), suggesting that the κ receptor dependence of the 6′‐GNTI effect on eIPSC amplitude.
Figure 5.
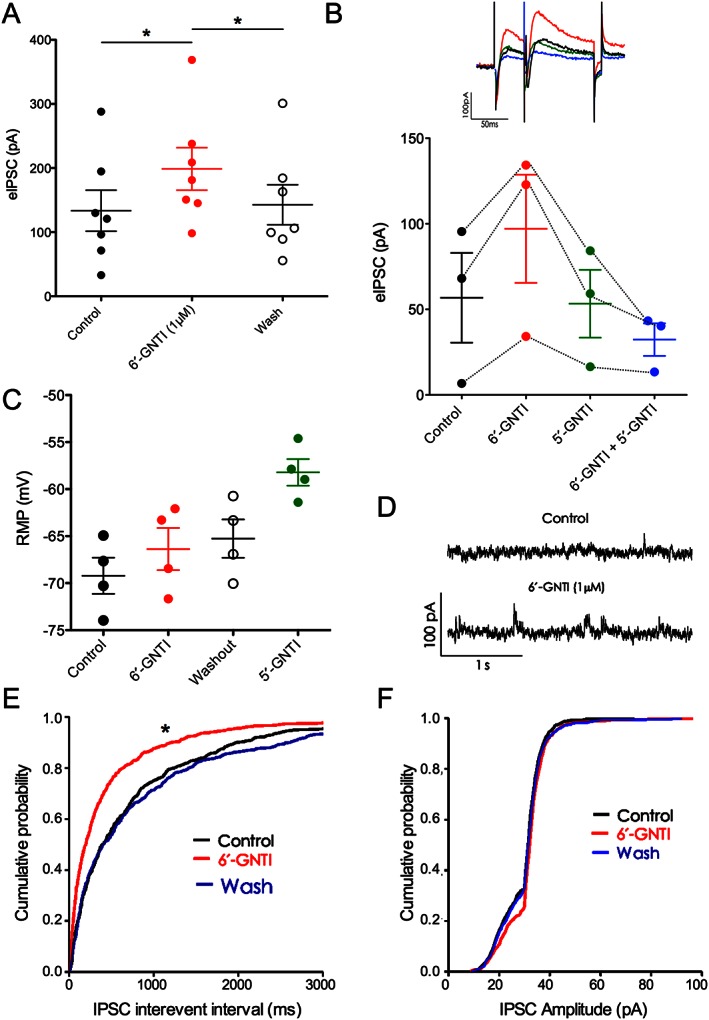
6′‐GNTI effects on hippocampal pyramidal neurons. (A) 6′‐GNTI treatment (1 μM) increased the amplitude of evoked inhibitory post‐synaptic current (eIPSC) in pyramidal cells (n = 7). The effect was reversed upon washout of the drug. *P < 0.05, significantly different from control; one‐way ANOVA with Dunnett's post hoc test. (B) Top: representative traces showing eIPSC under different conditions. Bottom: bath application of 5′‐GNTI alone and in the presence of 6′‐GNTI (n = 3). (C) RMP was not affected by 6′‐GNTI (n = 4). (D) Representative traces showing spontaneous IPSCs in control and 6′‐GNTI treated slices. (E) Increased spontaneous IPSCs frequency in pyramidal cells upon 6′‐GNTI bath perfusion (n = 5). (F) Spontaneous IPSCs amplitudes on pyramidal cells (n = 5). In (E ) and (F), *P < 0.05, significantly different from control; one‐way ANOVA with Kolmogorov–Smirnov test.
In addition, 6′‐GNTI increased the frequency of spontaneous IPSCs in pyramidal cells. Mean frequency was significantly increased by 6′‐GNTI from 1.2 ± 0.1 to 2.1 ± 0.2 Hz (Figure 5E; n = 5; P < 0.05; K‐S test), and reversed on washout. No effect on pyramidal cell spontaneous IPSCs amplitudes was observed (Figure 5F).
In this series of neurons, 6′‐GNTI did not affect the RMP. However, 5′‐GNTI shifted the RMP in 4/5 pyramidal neurons tested from −70.5 ± 1.3 to −58.2 ± 1.4 mV (Figure 5C).
Discussion
Treatment of mice injected with kainic acid with the κ receptor agonist U‐50488H results in reduced neuronal loss (Schunk et al., 2011) and reduces hippocampal paroxysmal discharges (this study). However, general κ receptor agonists induce dysphoria in humans (Barber and Gottschlich, 1997) and aversive effects in mice (Land et al., 2008). These aversive effects were suggested to be dependent on the β‐arrestin binding to κ receptors and subsequent activation of p38 MAPK in mice (Bruchas et al., 2007; Carr and Mague, 2008). In contrast, the full κ receptor agonists U69593 and salvinorin A as well as the G‐protein biased agonist RB‐64 (a salvinorin A derivative) produced pronounced aversive effects also in β‐arrestin 2 knockout mice (White et al., 2015). Activation of p38 MAPK was not addressed in this study, thereby not answering the question whether p38 MAPK could have been activated through other mechanisms (i.e. β‐arrestin 1). RB‐64 is a full agonist for G‐protein and β‐arrestin activation, although the potency for β‐arrestin is low (White et al., 2015). The G‐protein biased partial κ receptor agonist 6′‐GNTI activates the β‐arrestin pathway at an almost negligible level, resulting in a lack of ERK1/2 phosphorylation in striatal neurons (Schmid et al., 2013). The remaining activation of the Akt pathway was suggested to be G‐protein dependent. Interestingly, we observed strong expression of Zif268 in the striatum and other brain regions after application of 6′‐GNTI, but we cannot provide an underlying mechanism for this effect. Regulation of Zif268 expression potentially involves activation of PKA, PKC or CaMK signalling, involving either MAPK/ERK or CREB‐dependent mechanisms (see Bozon et al., 2002).
6′‐GNTI was described as a partial agonist for G‐protein activation and inhibition of adenylate cyclase, with hardly any activation of β‐arrestin compared with full agonists such as U‐50488H and EKC, in HEK293T cells transfected with human κ receptors (Rives et al., 2012). In our experiments, 6′‐GNTI displayed significant elevation of seizure threshold and reduction in hpds activity without induction of CPA, a measure of aversive effects, or motor impairment in vivo. Marked and widespread expression of Zif268 suggests that i.c.v. application of 6′‐GNTI actually activates κ receptors in areas proposed to be responsible for dysphoric effects, thereby suggesting that lack of aversive effects is not due to the local application. In contrast, RB‐64 induced CPA (White et al., 2015). As pointed out earlier, the pharmacological profiles of these two drugs differ. However, the route of administration might also influence this difference because, in our study, 6′‐GNTI was given i.c.v., while White et al. (2015) administered RB‐64 systemically. As CPA measures any type of unpleasant effects, we cannot exclude the possibility that activation of peripheral κ receptors by RB‐64 may cause the CPA. Moreover, it cannot be concluded that biased drugs display the same pharmacodynamic features in vitro and in vivo, as this bias may depend on the microenvironment of the receptor.
The fast onset of response suggests that activation of the G‐protein, but not the β‐arrestin pathway, is essential for anticonvulsant or antiseizure effects of κ receptor agonists. This is in line with the observations that presynaptic activation of κ receptors decreases N‐type, L‐type and P/Q‐type Ca2 + currents (Rusin et al., 1997) most probably through direct interaction with the βγ subunit of the G‐protein, resulting in a reduction of glutamate release. Stimulation of voltage‐gated K+ channels through postsynaptic κ receptors was proposed to occur in pyramidal neurons (Moore et al., 1994; Madamba et al., 1999). Our electrophysiological data do not clearly support these possibilities. By contrast, increased inhibitory signalling may be responsible for seizure suppression. GABAergic interneurons express not only κ receptors (Racz and Halasy, 2002) but also δ opioid receptors (Commons and Milner, 1996). It is unclear whether δ receptor and κ receptors form heterodimers that could bind 6′‐GNTI (Waldhoer et al., 2005) or interfere with excitation of interneurons. Detailed analysis of this aspect, including the question whether the effect results from the direct stimulation of GABAergic neurons or from an indirect stimulation through increased firing of excitatory inputs on GABAergic neurons, is beyond the scope of this study. 6′‐GNTI shows a high affinity for the κ receptor/δ receptor and κ receptor/μ opioid receptor heterodimers, as well as for κ receptor homodimers in vitro (Waldhoer et al., 2005). In line with this, the blockade of κ receptors by the specific antagonist 5′‐GNTI abolished the effect of 6′‐GNTI.
Translatability is always an issue when it comes to animal models. This is especially true in the case of structural differences between the human and the animal drug target. There are differences between human and rodent κ receptors in terms of β‐arrestin recruitment (Schattauer et al., 2012). Importantly, 6′‐GNTI does not induce recruitment of β‐arrestin in HEK cells expressing human κ receptors (Rives et al., 2012). This is in line with the lack of CPA in this study, considering the proposed functional relationship between β‐arrestin recruitment and aversive effects (Bruchas et al., 2007). As observed in rodents, in vitro experiments on human κ receptors expressed in CHO cells demonstrated partial agonist activity of 6′‐GNTI on the binding of [35S]‐GTP‐γS (Schmid et al., 2013), suggesting activation of the G‐protein.
Local injection of kainic acid into the area CA1 of the dorsal mouse hippocampus was described as a drug‐resistant model of TLE (Riban et al., 2002). Trauma‐induced epilepsies often comprise partial complex seizures originating from the limbic system, for which implications of the dynorphinergic system were postulated (see Simonato and Romualdi, 1996). Rodents and humans display a similar pattern of dynorphin expression and κ receptor binding in the hippocampus. Most animal models of TLE reflect the dynamics of post‐ictal overexpression and inter‐ictal reduction in dynorphin mRNA and peptide as described in human mTLE (see Simonato and Romualdi, 1996; Schwarzer, 2009). Alterations in κ receptor binding in epilepsy appear mostly dependent on neuronal loss, but not on a change in expression or trafficking. Thus, patients suffering from mass‐associated TLE or paradoxical TLE did not show marked differences in κ receptor specific [3H]U‐69,593 binding compared with post‐mortem controls. In contrast, hippocampi of mTLE patients displayed reduced binding in area CA1, but not the subiculum, being in line with marked neuronal loss in CA1 but not in the subiculum (de Lanerolle et al., 1997). In addition, in rodents, alterations mainly reflect morphological changes due to the death of κ receptor‐expressing neurons in the hippocampus in epilepsy (see Ben‐Ari, 2001). Reduced amounts of dynorphin but unaltered levels of κ receptors might result in an increased number of receptors available for agonist treatment. Due to the well‐documented and parallel findings in mouse and man, we expect a highly predictive value of experimental data obtained from animal models for functional implications in humans.
There is no general agreement as to which parameter of animal models might have the highest predictive value for human epilepsies. Therefore, we include different settings for the EEG analysis to analyse inter‐ictal spikes and spike trains separately. Moreover, we filtered for seizure‐like events of 10 s or more (hippocampal paroxysmal discharges), as these were suggested to have higher informative values in terms of human seizures. Irrespective of the settings, 6′‐GNTI induced reduction of inter‐ictal spikes and seizure‐like events, at least at the highest dose tested. In contrast, the modern antiepileptic drugs showed significant reduction of spike trains, but were mostly ineffective on hippocampal paroxysmal discharges.
In conclusion, our data provide proof for the principle that anticonvulsant and antiseizure effects can be achieved without inducing place‐aversion by applying a partial κ receptor agonist, which preferentially activates the G‐protein over the β‐arrestin pathway, at least in vitro.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Treatment design for U‐50488H and 6′‐GNTI applied on kainic acidinjected mice (referring to Figures 2 and 3). Data were calculated as ratio of the 60 min following the treatment on the 60 min preceding the treatment. (B) Representation of the automatic detection of spikes, spike trains and hpds. Spikes were defined as high amplitude discharges (>3 x baseline) lasting less than 70 ms. Spike trains were defined as the occurrence of at least three spikes with a frequency higher than 1 Hz and lasting for at least 1 s. Dashed green line =0 μV; green line = baseline; red line = threshold (3 x baseline).
Figure S2 Effects of antiepileptic drug treatments on time of hpds on kainic acid injected mice (n = 12). Ratios of total recorded events 60 min post/60 min pre‐treatment are given. * P < 0.05, significant difference; one‐way ANOVA with Dunnett's post hoc test..
Figure S3 Immunohistochemistry for Zif268 (upper panel, depicting nucleus accumbens) and pERK1 (lower panel, depicting dorsal raphe) was performed 30 minutes after i.c.v. application of saline (a, e; n = 4), 2.5 mg/kg U‐ 50488H (b, f; n = 4) or 30 nmoles 6′‐GNTI (c, d, g, h; n = 4). Note the strong labelling for Zif268 in the nucleus accumbens and the moderate labeling for pERK1 in the dorsal raphe after 6′‐GNTI injection. In d and h the primary antibodies, Zif 268 (n = 4) and pERK 1 (n = 4) respectively, were pre‐absorbed with the blocking peptide, resulting in a lack of labeling as compared to c and g (sections obtained from the same brains). Scale bars in c for a‐d = 50 μm, in g for e‐h = 25 μm.
Figure S4 Effects of 6′‐GNTI (30 nmoles/3 μl; n = 8) and 5′‐GNTI treatments (10 nmoles/3 μl; n = 5) on time of hpds on kainic acid injected mice compared t saline (n = 7). 6′‐GNTI effect was entirely reversed upon coadministration with 5′‐GNTI (n = 4). Instead, 5′‐GNTI alone (n = 5) had no effect on the time spent in hpds. * P <0.05, significant difference; one‐way ANOVA with Bonferroni's post hoc test.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
The authors are grateful for the support of the drug supply programme of NIDA, who supplied us with the 6′‐GNTI. We also want to thank James Wood (PhD) for fruitful discussions and support. This work was financed by the Austrian Science Fund (FWF) grants W1206‐B05 and I‐977‐B24.
Zangrandi, L. , Burtscher, J. , MacKay, J. P. , Colmers, W. F. , and Schwarzer, C. (2016) The G‐protein biased partial κ opioid receptor agonist 6′‐GNTI blocks hippocampal paroxysmal discharges without inducing aversion. British Journal of Pharmacology, 173: 1756–1767. doi: 10.1111/bph.13474.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber A, Gottschlich R (1997). Novel developments with selective, non‐peptidic kappa‐opioid receptor agonists. Expert Opin Investig Drugs 6: 1351–1368. [DOI] [PubMed] [Google Scholar]
- Ben‐Ari Y (2001). Cell death and synaptic reorganizations produced by seizures. Epilepsia 42 (Suppl 3): 5–7. [DOI] [PubMed] [Google Scholar]
- Bozon B, Davis S, Laroche S (2002). Regulated transcription of the immediate‐early gene Zif268: mechanisms and gene dosage-dependent function in synaptic plasticity and memory formation. Hippocampus 12: 570–577. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot AK, Li S et al. (2007). Stress‐induced p38 mitogen‐activated protein kinase activation mediates kappa‐opioid‐dependent dysphoria. J Neurosci 27: 11614–11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB et al. (2011). Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron 71: 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr GV, Mague SD (2008). p38: the link between the kappa‐opioid receptor and dysphoria. J Neurosci 28: 2299–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coatsworth JJ (1971). Studies on the clinical effect of marketed antiepileptic drugs. NINDS Monograph 12. [Google Scholar]
- Commons KG, Milner TA (1996). Cellular and subcellular localization of delta opioid receptor immunoreactivity in the rat dentate gyrus. Brain Res 738: 181–195. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPH, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP . Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle NC, Williamson A, Meredith C, Him JH, Tabuteau H, Spencer DD et al. (1997). Dynorphin and the kappa 1 ligand [3H]U69,593 binding in the human epileptogenic hippocampus. Epilepsy Res 28: 189–205. [DOI] [PubMed] [Google Scholar]
- Engel JJ (2001). Mesial temporal lobe epilepsy: what have we learned? Neuroscientist 7: 340–352. [DOI] [PubMed] [Google Scholar]
- Gambardella A, Manna I, Labate A, Chifari R, Serra P, La Russa A et al. (2003). Prodynorphin gene promoter polymorphism and temporal lobe epilepsy. Epilepsia 44: 1255–1256. [DOI] [PubMed] [Google Scholar]
- Henriksen SJ, Chouvet G, McGinty J, Bloom FE (1982). Opioid peptides in the hippocampus: anatomical and physiological considerations. Ann N Y Acad Sci 398: 207–220. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer K, Klement S, Eggart V, Mayr MJ, Saria A, Zernig G (2011). Conditioned place preference for social interaction in rats: contribution of sensory components. Front Behav Neurosci 5: 80 http://dx.doi.org/10.3389/fnbeh.2011.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C (2008). The dysphoric component of stress is encoded by activation of the dynorphin kappa‐opioid system. J Neurosci 28: 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loacker S, Sayyah M, Wittmann W, Herzog H, Schwarzer C (2007). Endogenous dynorphin in epileptogenesis and epilepsy: anticonvulsant net effect via kappa opioid receptors. Brain 130: 1017–1028. [DOI] [PubMed] [Google Scholar]
- Loscher W, Schmidt D (2011). Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 52: 657–678. [DOI] [PubMed] [Google Scholar]
- Madamba SG, Schweitzer P, Siggins GR (1999). Dynorphin selectively augments the M‐current in hippocampal CA1 neurons by an opiate receptor mechanism. J Neurophysiol 82: 1768–1775. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO (1999). Emerging insights into the genesis of epilepsy. Nature 399: A15–A22. [DOI] [PubMed] [Google Scholar]
- Moore SD, Madamba SG, Schweitzer P, Siggins GR (1994). Voltage‐dependent effects of opioid peptides on hippocampal CA3 pyramidal neurons in vitro. J Neurosci 14: 809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SPH, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Gasser E, Czech T, Baumgartner C, Schuh E, Feucht M et al. (2009). Dynamic up‐regulation of prodynorphin transcription in temporal lobe epilepsy. Hippocampus 19: 1051–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racz B, Halasy K (2002). Kappa opioid receptor is expressed by somatostatin‐ and neuropeptide Y‐containing interneurons in the rat hippocampus. Brain Res 931: 50–55. [DOI] [PubMed] [Google Scholar]
- Riban V, Bouilleret V, Pham‐lê BT, Fritschy JM, Marescaux C, Depaulis A (2002). Evolution of hippocampal epileptic activity during the development of hippocampal sclerosis in a mouse model of temporal lobe epilepsy. Neuroscience 112: 101–111. [DOI] [PubMed] [Google Scholar]
- Rives ML, Rossillo M, Liu‐Chen LY, Javitch JA (2012). 6′‐Guanidinonaltrindole (6′‐GNTI) is a G protein‐biased kappa‐opioid receptor agonist that inhibits arrestin recruitment. J Biol Chem 287: 27050–27054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusin KI, Giovannucci DR, Stuenkel EL, Moises HC (1997). Kappa‐opioid receptor activation modulates Ca2+ currents and secretion in isolated neuroendocrine nerve terminals. J Neurosci 17: 6565–6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattauer SS, Miyatake M, Shankar H, Zietz C, Levin JR, Liu‐Chen LY et al. (2012). Ligand directed signaling differences between rodent and human kappa‐opioid receptors. J Biol Chem 287: 41595–41607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Streicher JM, Groer CE, Munro TA, Zhou L, Bohn LM (2013). Functional selectivity of 6′‐guanidinonaltrindole (6′‐GNTI) at kappa‐opioid receptors in striatal neurons. J Biol Chem 288: 22387–22398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunk E, Aigner C, Stefanova N, Wenning G, Herzog H, Schwarzer C (2011). Kappa opioid receptor activation blocks progressive neurodegeneration after kainic acid injection. Hippocampus 21: 1010–1020. [DOI] [PubMed] [Google Scholar]
- Schwarzer C (2009). 30 years of dynorphins–new insights on their functions in neuropsychiatric diseases. Pharmacol Ther 123: 353–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer C, Williamson JM, Lothman EW, Vezzani A, Sperk G (1995). Somatostatin, neuropeptide Y, neurokinin B and cholecystokinin immunoreactivity in two chronic models of temporal lobe epilepsy. Neuroscience 69: 831–845. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Jones RM, Metzger TG, Ferguson DM, Portoghese PS (2001). Transformation of a kappa‐opioid receptor antagonist to a kappa‐agonist by transfer of a guanidinium group from the 5′‐ to 6′‐position of naltrindole. J Med Chem 44: 2073–2079. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Henriksen SJ, Chavkin C, Gruol D (1986). Opioid peptides and epileptogenesis in the limbic system: cellular mechanisms. Adv Neurol 44: 501–512. [PubMed] [Google Scholar]
- Simonato M, Romualdi P (1996). Dynorphin and epilepsy. Prog Neurobiol 50: 557–583. [DOI] [PubMed] [Google Scholar]
- Solbrig MV, Adrian R, Chang DY, Perng GC (2006). Viral risk factor for seizures: pathobiology of dynorphin in herpes simplex viral (HSV‐1) seizures in an animal model. Neurobiol Dis 23: 612–620. [DOI] [PubMed] [Google Scholar]
- Spencer S, Huh L (2008). Outcomes of epilepsy surgery in adults and children. Lancet Neurol 7: 525–537. [DOI] [PubMed] [Google Scholar]
- Stögmann E, Zimprich A, Baumgartner C, Aull‐Watschinger S, Höllt V, Zimprich F (2002). A functional polymorphism in the prodynorphin gene promotor is associated with temporal lobe epilepsy. Ann Neurol 51: 260–263. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Senda T, Tokuyama S, Kaneto H (1990). Further evidence for the implication of a kappa‐opioid receptor mechanism in the production of psychological stress‐induced analgesia. Jpn J Pharmacol 53: 487–494. [DOI] [PubMed] [Google Scholar]
- Tortella FC (1988). Endogenous opioid peptides and epilepsy: quieting the seizing brain? Trends Pharmacol Sci 9: 366–372. [DOI] [PubMed] [Google Scholar]
- Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E et al. (2005). A heterodimer‐selective agonist shows in vivo relevance of G protein‐coupled receptor dimers. Proc Natl Acad Sci U S A 102: 9050–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Robinson JE, Zhu H, Diberto JF, Polepally PR, Zjawiony JK et al. (2015). The G protein‐biased k‐opioid receptor agonist RB‐64 is analgesic with a unique spectrum of activities in vivo . J Pharmacol Exp Ther 352: 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Treatment design for U‐50488H and 6′‐GNTI applied on kainic acidinjected mice (referring to Figures 2 and 3). Data were calculated as ratio of the 60 min following the treatment on the 60 min preceding the treatment. (B) Representation of the automatic detection of spikes, spike trains and hpds. Spikes were defined as high amplitude discharges (>3 x baseline) lasting less than 70 ms. Spike trains were defined as the occurrence of at least three spikes with a frequency higher than 1 Hz and lasting for at least 1 s. Dashed green line =0 μV; green line = baseline; red line = threshold (3 x baseline).
Figure S2 Effects of antiepileptic drug treatments on time of hpds on kainic acid injected mice (n = 12). Ratios of total recorded events 60 min post/60 min pre‐treatment are given. * P < 0.05, significant difference; one‐way ANOVA with Dunnett's post hoc test..
Figure S3 Immunohistochemistry for Zif268 (upper panel, depicting nucleus accumbens) and pERK1 (lower panel, depicting dorsal raphe) was performed 30 minutes after i.c.v. application of saline (a, e; n = 4), 2.5 mg/kg U‐ 50488H (b, f; n = 4) or 30 nmoles 6′‐GNTI (c, d, g, h; n = 4). Note the strong labelling for Zif268 in the nucleus accumbens and the moderate labeling for pERK1 in the dorsal raphe after 6′‐GNTI injection. In d and h the primary antibodies, Zif 268 (n = 4) and pERK 1 (n = 4) respectively, were pre‐absorbed with the blocking peptide, resulting in a lack of labeling as compared to c and g (sections obtained from the same brains). Scale bars in c for a‐d = 50 μm, in g for e‐h = 25 μm.
Figure S4 Effects of 6′‐GNTI (30 nmoles/3 μl; n = 8) and 5′‐GNTI treatments (10 nmoles/3 μl; n = 5) on time of hpds on kainic acid injected mice compared t saline (n = 7). 6′‐GNTI effect was entirely reversed upon coadministration with 5′‐GNTI (n = 4). Instead, 5′‐GNTI alone (n = 5) had no effect on the time spent in hpds. * P <0.05, significant difference; one‐way ANOVA with Bonferroni's post hoc test.
Supporting info item
Supporting info item
Supporting info item
Supporting info item