

Abstract
Background and Purpose
Apelin‐13, an endogenous ligand for the apelin (APJ) receptor, behaves as a potent modulator of metabolic and cardiovascular disorders. Here, we examined the effects of apelin‐13 on myocardial injury in a mouse model combining ischaemia/reperfusion (I/R) and obesity and explored their underlying mechanisms.
Experimental Approach
Adult male C57BL/6J mice were fed a normal diet (ND) or high‐fat diet (HFD) for 6 months and then subjected to cardiac I/R. The effects of apelin‐13 post‐treatment on myocardial injury were evaluated in HFD‐fed mice after 24 h I/R. Changes in protein abundance, phosphorylation, subcellular localization and mRNA expression were determined in cardiomyoblast cell line H9C2, primary cardiomyocytes and cardiac tissue from ND‐ and HFD‐fed mice. Apoptosis was evaluated by TUNEL staining and caspase‐3 activity. Mitochondrial ultrastructure was analysed by electron microscopy.
Key Results
In HFD‐fed mice subjected to cardiac I/R, i.v. administration of apelin‐13 significantly reduced infarct size, myocardial apoptosis and mitochondrial damage compared with vehicle‐treated animals. In H9C2 cells and primary cardiomyocytes, apelin‐13 induced FoxO1 phosphorylation and nuclear exclusion. FoxO1 silencing by siRNA abolished the protective effects of apelin‐13 against hypoxia‐induced apoptosis and mitochondrial ROS generation. Finally, apelin deficiency in mice fed a HFD resulted in reduced myocardial FoxO1 expression and impaired FoxO1 distribution.
Conclusions and Implications
These data reveal apelin as a novel regulator of FoxO1 in cardiac cells and provide evidence for the potential of apelin‐13 in prevention of apoptosis and mitochondrial damage in conditions combining I/R injury and obesity.
Abbreviations
- FoxO
forkhead box O
- HFD
high‐fat diet
- I/R
ischaemia/reperfusion
- MI
myocardial infarction
- mtDNA
mitochondrial DNA
- HF
heart failure
- DCFHDA
dichlorodihydrofluorescein diacetate
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Enzymes c |
| Bax | Caspase 3 |
| Bcl‐2 | COX1 |
| GPCRs b | Cyclophilin A |
| Apelin receptor |
| LIGANDS | |
|---|---|
| Adiponectin | H2O2 |
| Apelin‐13 | Leptin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a, 2015b, 2015c).
Introduction
Myocardial infarction (MI) due to coronary artery disease is a leading cause of death and disability in obese individuals (Rana et al., 2004). Deprivation of oxygen and energy after coronary artery occlusion causes severe damage in the cardiac tissue and mediates cell death. Cardiomyocyte death was originally considered to occur exclusively via necrosis, an uncontrolled disintegration of the cell through rupture of the plasma membrane and subsequent release of intracellular contents into surrounding tissue. However, it is now clear that apoptosis, or programmed cell death, plays a significant role in the failing heart (Kang and Izumo, 2000; Wencker et al., 2003; Konstantinidis et al., 2012). Because of the limited ability of cardiac myocytes to proliferate, low levels of apoptosis can result in profound structural and functional consequences in the myocardium leading to cardiac dysfunction and heart failure (HF) (Mani, 2008). Wencker et al. demonstrated that induction of apoptosis in just 0.02% of myocytes is sufficient to cause a lethal cardiomyopathy in mice at 9 weeks of age (Wencker et al., 2003), suggesting that cardiac apoptosis may be an important component of HF pathogenesis. However, in situations combining myocardial damage and obesity, the molecular and cellular mechanisms that govern apoptotic machinery are poorly understood.
The FoxO transcription factor family governs fundamental programmes, including apoptosis, stress resistance and mitochondrial activities (Wang et al., 2014). FoxO transcription factors (FoxO1, FoxO3, FoxO4 and FoxO6) belong to the forkhead family of transcriptional regulators, of which FoxO1 is abundantly expressed in the cardiovascular system (Evans‐Anderson et al., 2008; Sengupta et al., 2011). A deficiency of FoxO1 induces embryonic lethality due to impaired vasculogenesis and is important in the aetiology of cardiovascular diseases (Furuyama et al., 2004). Recent studies have demonstrated that FoxO1 is a critical mediator of oxidative stress resistance in cardiac cells (Sengupta et al., 2011). Likewise, defects in FoxO1 specifically in mouse cardiomyocytes lead to increased oxidative damage and decreased myocardial function after acute ischaemia/reperfusion (I/R) or MI (Sengupta et al., 2011). A recent report has provided new in vivo evidence of the role of FoxO1 in cardiomyopathy related to metabolic stress (Battiprolu et al., 2012). However, the exact role of FoxO1 in obesity‐triggered myocardial remodelling processes and related mechanisms remains obscure.
Obesity is associated with altered levels of adipose tissue‐derived factors, commonly referred to as adipokines (Maury et al., 2007). Dysregulation of adipokines such as leptin and adiponectin contributes to the development of metabolic and cardiovascular disease (Bluher, 2013; Nakamura et al., 2014). Circulating levels of leptin are elevated in obesity, hypertension, chronic HF and MI, while diminished level of adiponectin are found in obese, diabetic and coronary artery disease patients (Nakamura et al., 2014). Changes in adipokine production can affect the balance between apoptosis and survival, and ultimately cell fate decisions. We have previously demonstrated that apelin, a recently described adipokine, plays an important role in the regulation of cardiovascular and metabolic homeostasis (Dray et al., 2008; Pchejetski et al., 2012). Apelin, an endogenous ligand for the GPCR, apelin (APJ) receptor, exerts inotropic activity and increases coronary blood flow by vascular dilation (Kleinz et al., 2005). Pyr1‐1apelin‐13 (apelin 13) is the main form circulating in plasma, and it has greater biological activity than apelin‐36 or apelin‐17, measured as the extracellular acidification rate in cultured cells expressing the apelin receptor (Tatemoto et al., 1998; Zhen et al., 2013). In response to stress, apelin‐13 prevents myocardial remodelling and improves cardiac function (Foussal et al., 2010; Pchejetski et al., 2012). The defects in apelin in mice induce age‐dependent progressive cardiac dysfunction, which is prevented by apelin infusion (Kuba et al., 2007), suggesting an important role for the apelinergic system in maintaining cardiac performance. In humans, circulating and cardiac levels of apelin are reduced in subjects with acute MI and established coronary artery disease (Weir et al., 2009; Kadoglou et al., 2010; Tycinska et al., 2010), whereas in obese individuals, plasma apelin concentrations are increased (Kleinz and Davenport, 2005; Dray et al., 2010). The loss of apelin enhances the susceptibility to apoptosis and myocardial injury ex. vivo and in vivo suggesting that apelin plays an important role in cell fate decisions in response to ischaemic stress (Wang et al., 2013). We have previously demonstrated that apelin regulates ROS production during the cardiac remodelling processes. ROS play a dual role in I/R acting as secondary messengers in intracellular signalling cascades, which maintain the cellular ‘redox’ status; however, ROS can also trigger irreversible injury leading to cell death. To date, the precise role of apelin ROS‐dependent cellular responses to I/R in an obese state remains to be determined.
The results of the present study revealed that apelin‐13 prevents myocardial apoptosis, mitochondrial damage and myocardial injury in conditions combining I/R injury and obesity. We demonstrated that apelin‐13 is an important regulator of FoxO1 dynamics in cardiac myocytes in response to stress. Furthermore, we showed that apelin counteracted cell apoptosis and ROS generation via the FoxO1 pathway.
Methods
Reagents and antibodies
Antibodies used in this study are as follows: anti‐FoxO1 (C29H4) and phospho‐FoxO1 (Ser256) antibodies (#9416) from Cell Signalling Technology (Danvers, MA, USA); anti‐tubulin from Santa Cruz Biotechnologies (Dallas, TX, USA); anti‐β‐actin from Sigma (A1978; St Louis, MO, USA). Fluorescent Alexa‐coupled secondary antibodies were from Life Technologies (Carlsbad, CA, USA) and HRP‐coupled secondary antibodies from Cell Signalling Technologies. DAPI was from Life Technologies. Apelin‐13 was purchased from Bachem (Bubendorf, Switzerland) and is referred to as apelin throughout this study. OXPHOS was from Mitosciences (MS604/G2594; Eugene, OR, USA). siRNA against FoxO1 was from Ambion (Foster City, CA, USA). All other chemicals were from Sigma unless otherwise stated.
Animal studies
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 1985) and was performed in accordance with the recommendations of the French Accreditation of the Laboratory Animal Care (approved by the local Centre National de la Recherche Scientifique Ethics Committee). At 8 weeks, C57BL/6J mice were fed a high‐fat diet (HFD, 45% fat) or normal diet (ND, 4% fat) for 6 months. The metabolic profile of HFD‐fed mice is summarized in Table 1. Apelin knock‐out (KO) mice were fed with ND as described previously (Alfarano et al., 2014). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Table 1.
Cardiometabolic profile of ND‐ and HFD‐fed mice
| Metabolic parameters | ND | HFD |
|---|---|---|
| Body weight (g) | 30.2 ± 0.7 | 48.3 ± 0.4* |
| Glucose (mM) | 8.5 ± 0.6 | 11.4 ± 0.8* |
| Insulin (pg·mL−1) | 1364.7 ± 293.2 | 4053.3 ± 518.4* |
| Echocardiographic parameters | ND | HFD |
|---|---|---|
| IVST, cm | 0.08 ± 0.01 | 0.10 ± 0.01* |
| LVID, cm | 0.37 ± 0.01 | 0.39 ± 0.01 |
| LVPWT, cm | 0.08 ± 0.01 | 0.10 ± 0.01* |
| EF, % | 79.15 ± 2.73 | 69.23 ± 0.889* |
| FS, % | 39.43 ± 1.35 | 33.59 ± 0.63* |
Body weight, plasma levels of glucose and insulin, interventricular septum thickness (IVST), left ventricular internal diameter (LVID), left ventricular posterior wall thickness (LVPWT), fractional shortening (FS) and ejection fraction (EF) were measured in mice fed a ND or HFD for 6 months. Data are means ± SEM; n = 5–6 per group.
P < 0.05 versus ND‐fed group.
Experimental protocol
A mouse model of I/R was used as previously described (Pchejetski et al., 2007). In brief, the mice were incubated and placed under mechanical ventilation after undergoing general anaesthesia, induced by i.p. injection of ketamine(35 mg·kg−1) and xylazine (5 mg·kg−1). A left parasternotomy was performed to expose hearts, and a 7–0 silk suture (Softsilk; US Surgical, Norwalk, CT, USA) was placed around the left anterior descending coronary artery. A snare was placed on the suture, and regional myocardial ischaemia was produced by tightening the snare. After 45 min of ischaemia, the occlusive snare was released to initiate reperfusion up to 24 h. Sham‐operated control mice underwent the same surgical procedures except that the snare was not tightened. Animals were randomly divided into four groups: (i) sham vehicle (n = 6); (ii) I/R vehicle (n = 7); (iii) sham apelin (n = 7); and (iv) I/R apelin (n = 7). Apelin‐13 (0.1 μg·kg−1) or vehicle (PBS) was injected into the jugular vein at 5 min of reperfusion in a final volume of 100 μL.
Evaluation of apoptosis
The apoptosis level both in vivo and in vitro was assessed using the DeadEnd Fluorometric TUNEL system according to manufacturer's instructions (Promega, Madison, WI, USA) as described previously (Pchejetski et al., 2012).
Determination of area at risk and infarct size
Determination of area at risk and infarct size was done as described previously (Pchejetski et al., 2012). Briefly, after injection of 1.5% Evans blue into the left ventricular cavity, mice were killed, and the heart was removed. Infarct size was evaluated by triphenyltetrazolium chloride staining and expressed as a percentage of the ischaemic risk area.
Electron microscopy
Ultrastructural studies of cardiac tissues by electron microscopy were performed as described previously (Attane et al., 2012). Briefly, cardiac tissues were fixed in cold 2.5% glutaraldehyde/1% paraformaldehyde, post‐fixed in 2% osmium tetroxide, embedded in resin and sectioned.
Immunolabelling of paraffin sections
Paraformaldehyde‐fixed (4%) and paraffin‐embedded heart sections were deparaffinized and rehydrated; antigen retrieval was performed using a sodium citrate treatment, followed by permeabilization with 0.2% Triton X‐100 for 20 min. After the blocking of non‐specific sites with 1% BSA, the primary antibodies were incubated overnight at 4°C. After labelling with appropriate secondary antibodies, the sections were mounted in Vectashield mounting medium including DAPI (Vector Laboratories, Burlingame, CA, USA) and imaged by confocal microscopy.
Echocardiographic studies
Echocardiography was performed in isoflurane‐anaesthetized mice using a Vivid7 imaging system (General Electric Healthcare, Toulouse, France) equipped with a 14 MHz sectorial probe. Two‐dimensional images were recorded in parasternal long‐axis and short‐axis projections, with guided M‐mode recordings at the midventricular level in both views. Left ventricular (LV) dimensions and wall thickness were measured in at least three beats from each projection and averaged. Interventricular septum, LV posterior wall thickness and LV internal dimensions at diastole (LVID) were measured. Fractional shortening and ejection fraction were calculated from the two‐dimensional images.
Caspase‐3 activity and metabolic plasma measurements
Caspase‐3 activity was assessed with EnzChek Caspase‐3 Assay Kit #1 (Life Technologies) according to the manufacturer's instructions. Insulinemia (Mercodia, Uppsala, Sweden) and glycemia (Accu‐check, Roche Diagnostics, Risch‐Rotkreuz, Switzerland) were measured in fasted state. Body fat mass composition was determined as described previously (Alfarano et al., 2014).
Primary mice cardiomyocytes preparation, cell culture, transfection and treatments
Adult mice ventricular cardiomyocytes were isolated from adult obese mice and maintained as described previously (Alfarano et al., 2014). The rat embryonic cardiomyoblastic cell line H9C2 was cultured in MEM (Gibco 41090‐028, ThermoFischer Scientific, Waltham, MA, USA) supplemented with 10% FBS and 1% penicillin–streptomycin in a 37°C, 5% CO2 incubator. siRNA transfection was performed with Lipofectamine RNAiMAX (Life Technologies) according to manufacturer's instructions. Cells were pretreated for 15 min with apelin (10−7–10−9 mol·L−1) and then subjected to normoxia (5% CO2; 21% O2, balance N2) or hypoxia for 2 h in a hypoxic chamber (5% CO2, 1% O2, balance N2). To measure cell apoptosis induced by hypoxia, the cells were left for 16 h in hypoxic conditions.
Hydrogen peroxyde and superoxide production
Global ROS production in cells was assessed as described previously (Bianchi et al., 2005; Pchejetski et al., 2007) using the oxidation of H2DCFDA (Life Technologies) to DCF (dichlorodihydrofluorescein). Mitochondrial O2 − and H2O2 production in cells was measured by MitoSOX (Life Technologies) and MitoPY1 (Sigma‐Aldrich, St Louis, MO, USA) at 1 μM (on H9C2 cells) or 5 μM (on cardiomyocytes) for 30 min following live‐cell imaging on a confocal microscope equipped with an incubation chamber with temperature control and CO2 enrichment.
Western blotting
Immunoblot analyses were carried out on clarified lysates of cardiac tissues or cells quantified using the BCA (bicinchoninic acid) protein assay (Thermo Scientific; Thermo Fisher Scientific Waltham, MA, USA) and denaturated in Laemmli sample buffer (Sigma‐Aldrich, St Louis, MO, USA). Proteins were separated by SDS‐PAGE, before western blotting on nitrocellulose membranes using the Trans‐Blot Turbo Transfer System (Bio‐Rad, Hercules, CA, USA). Immunoreactive bands were detected by chemiluminescence with the Clarity Western ECL Substrate (Bio‐Rad) on a ChemiDoc MP Acquisition System (Bio‐Rad).
Immunofluorescence
Immunofluorescence was performed essentially as previously described (Boal et al., 2010). Briefly, cells grown on glass coverslips were PFA(paraformaldehyde)‐fixed and permeabilized using TritonX‐100 before incubation with primary and secondary antibodies, mounted in Mowiol and imaged using confocal microscopy on a Zeiss LSM780 microscope. For nuclear FoxO quantification, the fluorescence intensity of FoxO1 protein in the nucleus was quantified and normalized against the fluorescence intensity within the total cell.
Real‐time RT‐PCR analysis
Total RNAs were isolated from cultured mouse cardiac fibroblasts using the RNeasy mini kit (Qiagen, Hilden, Germany). Total RNAs (300 ng) were reverse transcribed using Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA) in the presence of random hexamers. Real‐time quantitative PCR was performed as previously described (Alfarano et al., 2014). The expression levels of Bax and Bcl2 were normalized to GAPDH mRNA expression. The sequences of the primers used are as follow and given in the 5′–3′ orientation: FoxO1, sense GCGGGCTGGAAGAATTCAAT, antisense GTTCCTTCATTCTGCACTCGAATAA; GAPDH, sense TGCACCACCAACTGCTTAGC, antisense GGCATGGACTGTGGTCATGAG; Bax, sense CGGCGAATTGGAGATGAACT, antisense GTCCACGTCAGCAATCATCCT; Bcl‐2, sense TCCCGATTCATTGCAAGTTGTA, antisense GCAACCACACCATCGATCTTC. For mitochondrial DNA (mtDNA) analysis, the content of mtDNA was calculated using real‐time quantitative PCR by measuring the threshold cycle ratio of a mitochondrial‐encoded gene (COX1) and a nuclear‐encoded gene (cyclophilin A) as previously described (Attane et al., 2012).
Statistical analysis
Data are expressed as mean ± SEM. Comparison between two groups was performed by Student's t‐test, while comparison of multiple groups was performed by one‐way ANOVA followed by a Bonferroni's post hoc test using GraphPad prism version 5.00 (GraphPad Software, Inc, La Jolla, CA, USA.). Statistical significance was defined as P < 0.05. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results
Protection of cardiac tissue by apelin‐13 against apoptosis and mitochondrial injury in acute phase of myocardial I/R in obese state
To evaluate the impact of obesity on cardiometabolic parameters, male C57BL/6J mice were fed a ND (4% fat) or HFD (45% fat) for 6 months. As shown in Table 1, in comparison with ND‐fed mice, HFD‐fed animals had increased body weight, plasma glucose and insulin levels. Echocardiographic analysis revealed an increased interventricular septum, LV posterior wall and decreased EF and FS in HFD‐fed mice as compared with ND‐fed animals, suggesting that chronic exposure to a HFD induces cardiac dysfunction.
We next examined the effect of obesity on myocardial apoptosis in cardiac tissue stained using the TUNEL assay. As shown in Figure 1A and B, no differences were observed in the number of apoptotic nuclei between ND‐ and HFD‐fed mice. However, cardiac I/R resulted in increased apoptosis, activation of caspase‐3 and deregulation of pro‐apoptotic proteins in HFD‐fed mice (Figure 1C–F). Importantly, apelin‐13 administration at 5 min of reperfusion significantly reduced the number of TUNEL positive cells, myocardial caspase‐3 activity and expression of Bax as compared with vehicle‐treated mice after 24 h I/R (Figure 1 C–F). In contrast, Bcl‐2 expression level, an anti‐apoptotic protein, was increased in apelin‐treated I/R hearts from HFD‐fed mice (Figure 1G). Apelin‐dependent prevention of apoptosis in HFD‐fed mice after I/R was accompanied by a reduction in infarct size as compared with vehicle‐treated animals (Figure 1H and I). Electron microscopy examination of cardiac tissue from ND‐ and HFD‐fed mice revealed the presence of numerous lipid droplets with a higher proportion of smaller mitochondria (Figure 2A) without changes in mtDNA content (Figure 2B) in obese mice. As shown in Figure 2C, myocardial I/R in HFD‐fed mice induced mitochondrial damage, including swelling and structural disruption. Administration of apelin‐13 prevented I/R‐induced mitochondrial damage (Figure 2C) and stimulated mtDNA content (Figure 2D) as compared with vehicle‐treated HFD‐fed mice. In addition, we found that chronic HFD consumption resulted in a decrease in OXPHOS complex IV, the terminal enzyme in the mitochondrial respiratory chain (Figure 2E), and treatment of HFD‐fed mice with apelin significantly increased cytochrome c oxidase expression in response to cardiac I/R (Figure 2F).
Figure 1.
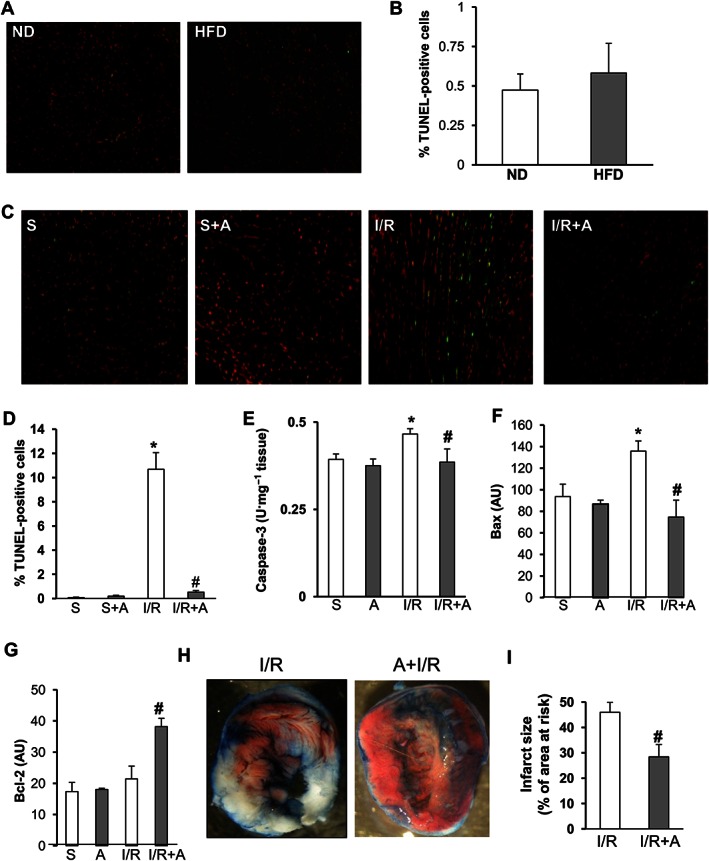
Protection of cardiac tissue by apelin‐13 against apoptosis and necrosis in acute phase of myocardial I/R in obese mice. (A) Representative images and (B) quantification of TUNEL positive cells in the heart sections from ND‐fed and HFD‐fed mice. (C) Representative images of TUNEL analysis of cardiac tissue sections from HFD‐fed mice treated with vehicle or apelin‐13, A, after 24 h of sham‐operation, S, or I/R. (D) Quantification of apoptosis in (C). (E–G), Caspase‐3 activity (E), Bax (F) and Blc‐2 (G) expression levels in hearts from HFD‐fed mice treated as indicated. (H–I) Representative images of triphenyltetrazolium chloride staining of heart sections from HFD‐fed mice treated with vehicle or apelin‐13, A, after 24 h of sham‐operation, S, or I/R. (I) Quantification of infarct size expressed as % of area at risk. *, P < 0.05 versus S; #, P < 0.05 versus I/R.
Figure 2.
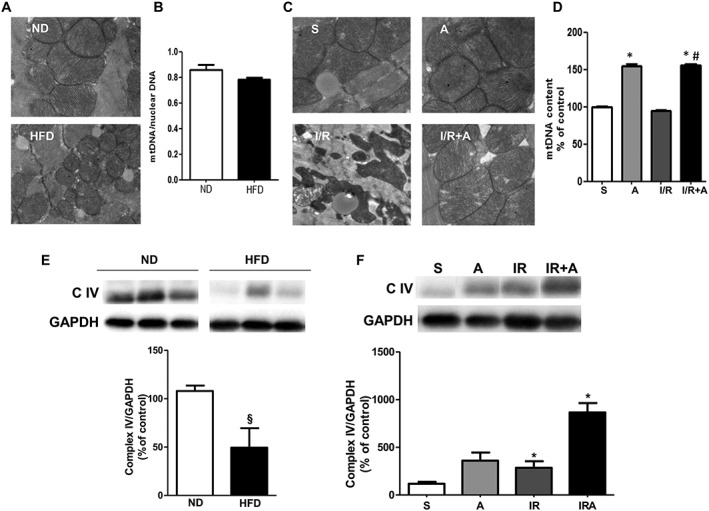
Prevention of mitochondrial damage by apelin‐13 in acute phase of myocardial I/R in obese mice. (A) Typical electron micrographs (original magnifications ×10000) from ND‐ and HFD‐fed mice. (B) Quantitative RT‐PCR analysis of mtDNA content in hearts from ND‐ and HFD‐fed mice. (c) Scanning electron microscope images (original magnification ×10000) in mice treated with vehicle or apelin‐13, A, after 24 h of sham‐operation, S, or I/R. (D) Effect of apelin‐13 treatment on mtDNA content in hearts from HFD‐fed mice subjected to I/R. (E) Western blot analysis of the myocardial OXPHOS complex IV in ND‐ and HFD‐fed mice. (F) Effect of apelin‐13 treatment on expression of OXPHOS complex IV in hearts from HFD‐fed mice subjected to I/R. *, P < 0.05 versus S; #, P < 0.05 versus I/R; §, P < 0.05 versus ND.
Attenuation of FoxO1 nuclear translocation by apelin‐13 in relation to HFD status
Given the importance of FoxO1 transcription factor for governing apoptosis, mitochondrial activity and metabolism, we next examined the effect of apelin on the myocardial activation status of FoxO1 in response to obesity and I/R injury. Activation of FoxO1 was examined by monitoring the phosphorylation of FoxO1 proteins at Ser256 and nucleocytoplasmic distribution in cardiac tissue. Compared with the control ND group, HFD‐fed mice exhibited decreased levels of FoxO1 phosphorylation in whole heart tissue extracts (3a) and increased nuclear localization of FoxO1 (Figure 3B and C). In response to I/R injury, apelin stimulated FoxO1 phosporylation in whole cell extracts from HFD‐fed mouse hearts (Figure 3D) and prevented its nuclear translocation in cardiac tissue (Figure 3E and F). To confirm the role of apelin in FoxO1 dynamics in cardiac tissue, we finally examined its localization in left ventricles from apelin KO obese mice. As shown in Figure 3G, apelin‐deficient mice displayed a FoxO1 nuclear targeting phenotype, as compared with wild‐type (WT) mice. Analysis of the confocal images revealed a significant decrease in the fluorescence intensity of FoxO1 (Figure 3G) in apelin KO mice as compared with WT animals. In addition, myocardial FoxO1 expression level, as assessed by Western blot, was down‐regulated in apelin KO mice as compared with WT (Figure 3H).
Figure 3.
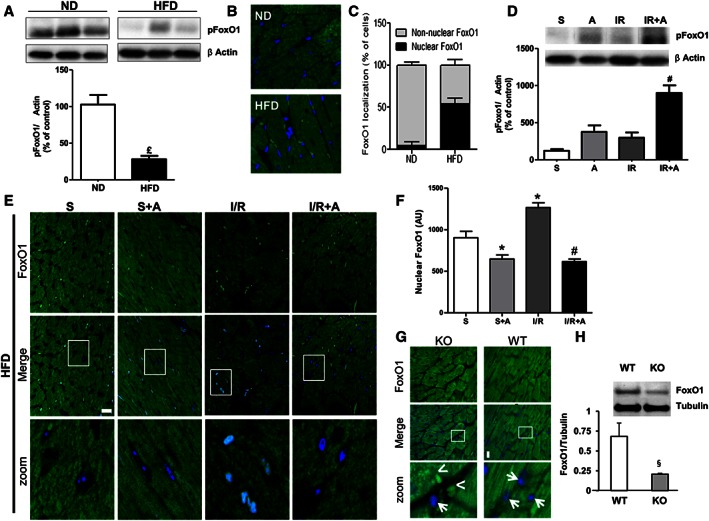
Apelin‐dependent myocardial FoxO1 activation in cardiac tissue from obese mice subjected to cardiac I/R. (A) Myocardial phosphorylation pattern of FoxO1 in ND‐ and HFD‐fed mice. (B) Representative images for nuclear and cytoplasmic localization of FoxO1 in ND‐ and HFD‐fed mice. (C) Whole‐tissue quantification of nuclear and cytoplasmic distribution of FoxO1 in ND‐ and HFD‐fed mice. (D) Effect of apelin‐13 treatment on FoxO1 phosphorylationin cardiac tissue from HFD‐fed mice subjected to I/R. (E) Effect of apelin‐13 treatment on subcellular localization of FoxO1 in cardiac sections from HFD‐fed mice treated with vehicle or apelin‐13, A, after 24 h of sham, S, or I/R operations. (F) Quantification of FoxO1 nuclear translocation was carried out by measuring the mean fluorescence intensity of nuclear FoxO1 in HFD‐fed mice. (G) FoxO1 immunostaining (in green) on heart sections from HFD‐fed WT or apelin KO mice. Nuclei were stained with DAPI (in blue). Bar is 20 μm. Arrows point to nuclei devoid of FoxO1, whereas arrowheads point to nuclear staining. (H) FoxO1 protein expression levels in hearts from WT or apelin KO mice. *, P < 0.05 versus S; £, P < 0.05 versus ND; #, P < 0.05 versus IR; §, P < 0.05 versus WT.
Cardiac cell expression and trafficking of FoxO1 are regulated by apelin‐13
The regulation of expression and trafficking of FoxO factors are critical for the transcriptional control of cell death and mitochondrial function in response to cellular stress (Ullman et al., 1997; Wang et al., 2014). We next examined whether apelin‐13 can regulate FoxO1 in cardiac cells under normoxia and hypoxia. As shown in Figure 4A, treatment of H9C2 cardiomyoblast cells with apelin induced a dose‐dependent increase in FoxO1 mRNA levels under normoxia as detected by quantitative RT‐PCR. Western blot analysis confirmed the increased expression of FoxO1 proteins induced by apelin in H9C2 cells (Figure 4B). As shown in Figure 4C and D, exposure of H9C2 cells to hypoxia (1% O2) for 2 h resulted in noticeable nuclear translocation of FoxO1, as compared with the uniform cytosolic distribution in normoxic cardiomyoblasts. Quantitative analysis of FoxO1 subcellular localization in H9C2 cells revealed that apelin not only maintained FoxO1 in the cytoplasm but also significantly prevented its hypoxia‐induced nuclear retention (Figure 4D).
Figure 4.
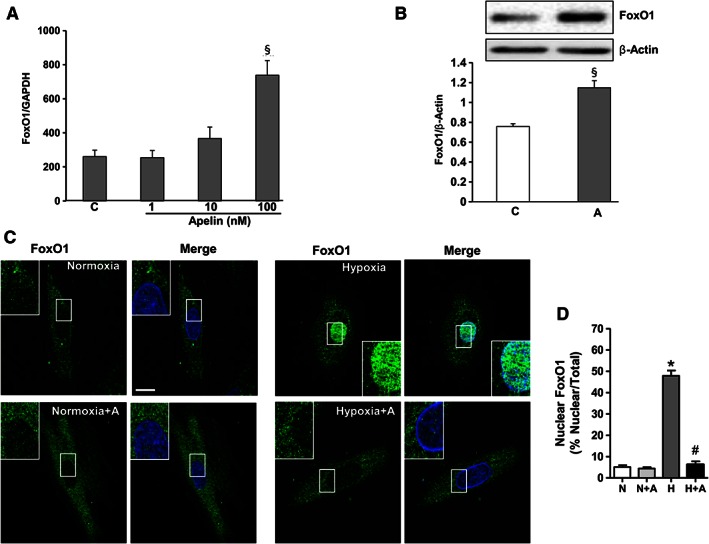
FoxO1 activity is regulated by apelin in H9C2 cardiomyoblasts. (A) Dose‐dependent effect of apelin‐13 on FoxO1 expression levels was assessed by quantitative RT‐PCR in H9C2 cardiomyoblasts treated or not with apelin (1–100 nM) for 24 h. (B) H9C2 cells were treated with apelin (A, 100 nM), and cell lysates were probed with anti‐FoxO1 and anti‐β actin antibodies. FoxO1 protein expression levels were quantified by densitometry and normalized against β actin. (C) Representative confocal images of H9C2 cells pretreated or not with apelin for 15 min (A, 100 nM), submitted to hypoxia, H, or normoxia, N, for 2 h and stained for FoxO1 antibody. FoxO1 proteins are shown in green; nuclei were stained with DAPI (in blue). Bar is 10 μm. (D) Quantification of FoxO1 nuclear translocation is expressed as the fluorescence intensity in the nucleus normalized to the total fluorescence intensity in the cell. *, P < 0.05 versus N; #, P < 0.05 versus H; §, P < 0.05 versus C.
Examination of phosphorylation pattern of FoxO1 expression in cardiomyocytes isolated from ND‐ and HFD‐fed mice indicated that apelin increased FoxO1 phosphorylation in both obese and non‐obese conditions (Figure 5A and B). Analysis of subcellular localization of FoxO1 in isolated cardiomyocytes from HFD‐fed mice demonstrated that in response to hypoxia, FoxO1 translocated massively in the nuclei, as demonstrated in Figure 5C and quantified in Figure 5D. Treatment of cardiomyocytes with apelin‐13 inhibited hypoxia‐induced nuclear translocation of FoxO1 (Figure 5C and D). As shown in Figure 5E, cardiomyocyte protein levels of FoxO1 were decreased under hypoxia, whereas treatment of cells with apelin significantly increased FoxO1 protein expression in response to hypoxia.
Figure 5.
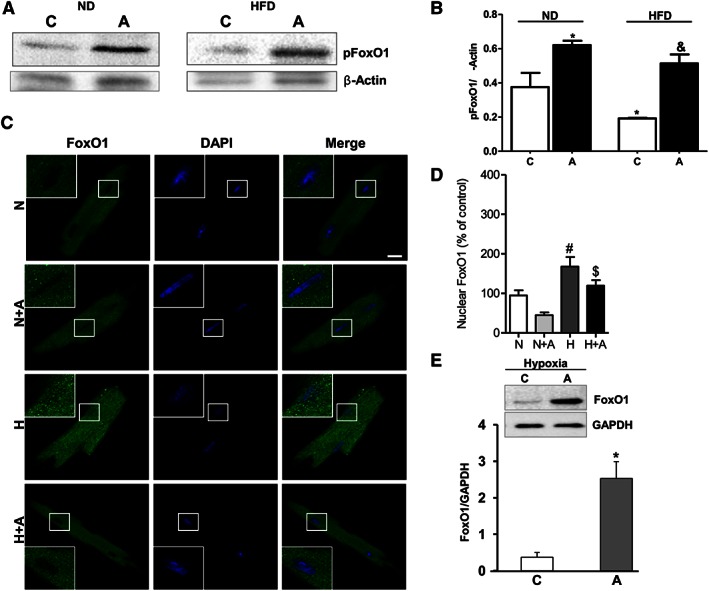
Apelin‐dependent regulation of FoxO1 activity in cardiomyocytes isolated from HFD‐fed mice under hypoxia. (A) Representative western blot images of FoxO1 phosphorylation pattern in cardiomyocytes isolated from ND‐ and HFD‐fed mice. (B) Quantification of FoxO1 phosphorylation in cardiomyocytes isolated from ND‐ and HFD‐fed mice and treated with apelin. *, P < 0.05 versus C (ND); &, P < 0.05 versus C (HFD). (C) Representative confocal images of cardiomyocytes isolated from HFD‐fed mice stimulated with apelin for 15 min (A, 100 nM) under hypoxia, H, or normoxia, N,. Nuclei were stained with DAPI (in blue) and FoxO1 (in green). Bar is 20 μm. (D) Quantification of FoxO1 nuclear translocation is expressed as the fluorescence intensity in the nucleus normalized against the total fluorescence intensity in the cell. (E) Effect of apelin on FoxO1 protein expression levels was assessed by western blot analysis in cardiomyocytes treated with apelin (100 nM) for 24 h. FoxO1 protein expression levels were quantified by densitometry. *, P < 0.05 versus C; #, P < 0.05 versus N; $, P < 0.05 versus H.
Apelin‐13 reduces hypoxia‐induced mitochondrial ROS and apoptosis through a FoxO1 pathway
FoxO1 coordinates the transcriptional programme of cardiomyocyte survival upon induction of oxidative stress (Wang Y et al., 2014). We next examined whether FoxO1 is involved in apelin‐dependent effects on mitochondrial ROS generation and apoptosis. Analysis of mitochondria‐specific ROS generation in response to hypoxia in cardiomyocytes isolated from HFD‐fed mice demonstrated that apelin treatment attenuated hypoxia‐induced intracellular ROS production (Figure 6A and B) measured by DCFDA. At the level of mitochondria, we found that apelin significantly reduced superoxide (O2) (Figure 6C and D) and H2O2 (Figure 6E and F) levels, measured by MitoSOX and MitoPY1 respectively. Moreover, the apoptosis of cardiomyocytes induced by hypoxia was markedly decreased by apelin treatment (Figure 6G and H).
Figure 6.
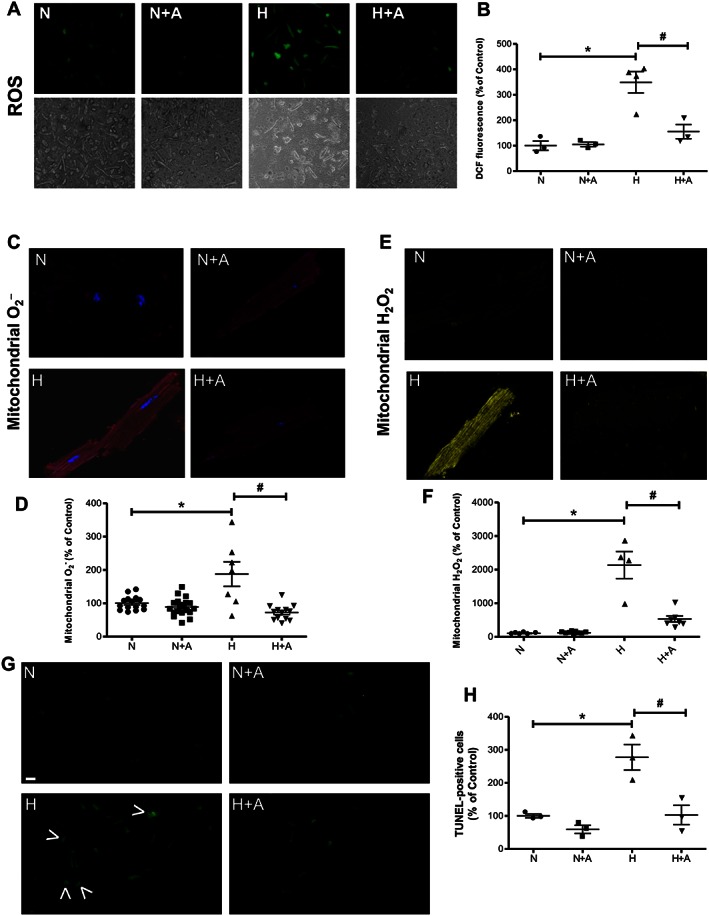
Apelin‐13 treatment attenuates hypoxia‐induced apoptosis and ROS overproduction in cardiomyocytes isolated from HFD‐fed mice. Primary cardiomyocytes isolated from HFD‐fed mice were treated with apelin (A, 100 nM) for 15 min. Representative images and quantification of intracellular ROS (A and B), mitochondrial O2 − (C and D) and H2O2 (E and F) levels measured by the fluorescent probes DCFDA, MitoSOX Red or MitoPY1 respectively. (G) Apoptosis was measured by TUNEL. Arrowheads highlight TUNEL‐positive cells. Bar is 20 μm. (H) Quantification of % of apoptotic TUNEL‐positive cells.*, P < 0.05 versus N; #, P < 0.05 versus H.
In order to elucidate the molecular mechanisms underlying apelin‐mediated protection of cells from apoptosis and ROS overproduction, we transfected H9C2 cells with FoxO1 siRNA. As shown in Figure 7A and B, siRNA knockdown of FoxO1 specifically inhibited its expression by 85% in H9C2 cells. Importantly, we found that silencing of FoxO1 abolished the ability of apelin to attenuate hypoxia‐induced mitochondrial O2 − production (Figure 7C and D). In addition, FoxO1 knockdown drastically inhibited apelin‐mediated anti‐apoptotic activity in response to hypoxia (Figure 7E and F). Taken together, these data suggest that FoxO1 is a crucial target for apelin in the control of oxidative stress and cardiac cell apoptosis.
Figure 7.
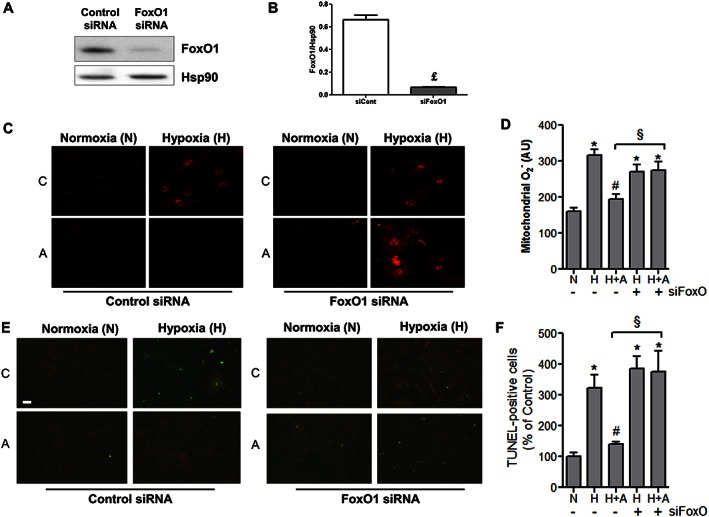
Apelin‐13 treatment regulates cell apoptosis and mitochondrial ROS production through a FoxO1 pathway. (A and B) Quantification of FoxO1 protein expression levels in H9C2 transfected with scramble control siRNA (siRNA Control) or FoxO1siRNA; £, P < 0.05 versus si RNA Control. (C and D) Representative images and quantification of transfected H9C2 cells treated or not, C, with apelin (A, 100 nM for 15 min) under hypoxia, H, or normoxia, N, for 2 h. Mitochondrial ROS production was measured by MitoSOX Red fluorescence (in red). (E and F) Analysis of TUNEL‐positive H9C2 cells transfected with siRNA Control or FoxO1siRNA and treated as in (C–D) Bar is 20 μm. *, P < 0.05 versus N; #, P < 0.001 versus H; §, P < 0.05 between indicated conditions.
Discussion and conclusions
The FoxO1 transcription factor orchestrates a number of cellular processes involved in cell fate decisions in a cell type‐ and environment‐specific manner, including cell resistance to apoptosis and oxidative stress (Medema et al., 2000; Kops et al., 2002). The present study provides evidence for a novel regulatory mechanism of FoxO1 activation in cardiac cells. The main finding of this work is that apelin‐13 regulates myocardial FoxO1 phosphorylation status and nucleocytoplasmic shuttling at the acute phase of cardiac I/R injury in obese mice. Moreover, we demonstrated that apelin promotes cardiomyocyte survival and counteracts excessive mitochondria‐derived ROS formation through the FoxO1 pathway. Using a mouse model combining obesity and I/R injury, we showed that a reduction in apoptosis and mitochondrial damage by apelin‐13 post‐reperfusion treatment is associated with increased levels of phosphorylated FoxO1 protein and the prevention of FoxO1 nuclear translocation in injured cardiac tissue. Finally, we showed that apelin gene‐deficient mice exposed to HFD display impaired FoxO1 expression and trafficking with nuclear translocation in cardiac tissue. These results point to a novel mechanism of FoxO1‐shuttling system in cardiac cells and provide in vitro and in vivo evidence for the therapeutic potential of apelin‐13 to limit I/R‐induced myocardial apoptosis and mitochondrial damage in obese subjects.
There is accumulating evidence that subcellular localization of FoxO transcription factors is critical for various cellular functions (Kowluru and Matti, 2012; Ponugoti et al., 2012). The translocation of FoxO transcriptional factors is regulated by multiple mechanisms, including the inhibition of nuclear import and promotion of nuclear export (Wang et al., 2014). The shuttling of proteins between the nucleus and cytoplasm is a highly regulated process and requires accessory factors (Ullman et al., 1997). At present, the molecular mechanisms of FoxO1 regulation in cardiac cells remain obscure. In the present study, we demonstrated that subcellular distribution of FoxO1 may be modulated by apelin in cardiomyocytes. Analysis of cardiac cells by confocal laser scanning microscopy showed that obesity promoted the translocation of FoxO1 into the nucleus in cardiomyocytes. Phosphorylation modification is a critical mechanism to regulate the nuclear/cytoplasmic shuttling of FoxOs and their transcriptional activity (Zhao et al., 2011). Here, we demonstrated a significant decrease in FoxO1 phosphorylation at Ser256 in cardiac tissue from HFD‐fed mice. These data are in line with a recent report showing FoxO1 dephosphorylation in whole cell extracts from HFD‐fed mouse hearts (Battiprolu et al., 2012). In response to hypoxia, we observed FoxO1 translocation into the nucleus in cardiomyocytes isolated from HFD‐fed mice. Our findings are consistent with results reported by recent studies showing that FoxO1 is mainly localized in the nucleus under hypoxic conditions in a variety of cell lines (Battiprolu et al., 2012; Awad et al., 2014). Analysis of subcellular localization of FoxO1 revealed that apelin‐13 promotes the retention of FoxO1 in the cytoplasm by preventing its nuclear translocation in response to oxygen deprivation. Furthermore, we found that apelin prevents hypoxia‐induced FoxO1 dephosphorylation in cardiac myocytes. In a mouse model of cardiac injury induced by I/R, we showed that an injection of apelin at the early phase of reperfusion stimulates the phosphorylation of FoxO1 proteins and inhibits its nuclear import confirming apelin‐dependent regulation of myocardial FoxO1 status. Consistent with these data, we revealed cardiac nuclear translocation of FoxO1 in apelin KO mice, providing strong evidence for a key role of apelin in the control of nucleocytoplasmic distribution of FoxO1 in the heart.
FoxO transcription factors sense cellular stress and govern fundamental processes including cell death and ROS production. Because the heart is constantly adapting to different stresses, FoxOs appear to play an extremely important role in cardiac physiology and pathophysiology. In mice, FoxO1 deficiency specifically in cardiomyocytes results in increased myocardial cell death, reduced cardiac performance and increased scar formation following myocardial ischaemia (Sengupta et al., 2011). Increased cardiac I/R injury in mice with cardiac FoxO deficiency is accompanied by reduced expression of antioxidants, DNA repair enzymes and anti‐apoptotic genes (Sengupta et al., 2011). In our study, we demonstrated for the first time that activation of FoxO1 by apelin prevents hypoxia‐induced mitochondrial O2 − and H2O2 generation in cultured cardiomyoblasts exposed to hypoxia. Given the importance of the FoxO family in the regulation of oxidative stress, FoxO1 may be an important indicator of mitochondrial oxidative stress in cardiac cells. This notion is corroborated by the recent study demonstrating that FoxO3 is an early biomarker of oxidative stress in conditions of metabolic stress (Raju et al., 2013). However, ROS might play a dual role acting as secondary messengers in intracellular signalling cascades and as highly reactive molecules causing irreversible oxidative damage to mtDNA, proteins and lipids (Braunersreuther et al. 2012). Excessive mitochondrial ROS production triggers activation of cellular apoptotic programmes leading to cell death (Wang, 2001). Limiting mitochondrial ROS production by apelin may be particularly important in cardiac protection against I/R damage. Indeed, we showed that apelin‐dependent reduction in hypoxia‐induced ROS production is associated with inhibition of apoptotic cell death in response to oxygen deprivation. Importantly, silencing of FoxO1 by siRNA in cardiomyoblasts attenuated apelin‐mediated anti‐apoptotic and mitochondrial ROS reduction activities, suggesting that apelin may control cell fate decisions through FoxO1 pathways. One of the major limitations of our in vitro models is their inability to fully mimic the in vivo conditions. In order to adopt the in vivo experiments and protocol treatment that more closely approximate the clinical situation, we have examined the effects of apelin‐13 post‐treatment on myocardial damage and oxidative stress in HFD‐fed mice subjected to I/R. Our results suggest that apelin administration after 5 min of reperfusion reduced myocardial apoptosis and oxidative stress in conditions combining obesity and cardiac I/R injury. Moreover, apelin‐13 post‐treatment decreased myocardial expression of pro‐apototic protein Bax and increased the level of anti‐apoptotic Bcl‐2 leading to reduced myocardial cell death. In addition, we found apelin‐dependent attenuation of I/R‐induced mitochondrial damage and increased mtDNA content in the myocardium, suggesting an accelerated mitochondrial biogenesis in the context of a well‐preserved mitochondrial ultrastructure after apelin administration. Regulators of mitochondrial activity play an important role in coordinating cellular adaptation to stress (Ferber et al., 2011). Further studies are required to determine whether the protection provided by apelin on acute I/R injury could improve cardiac function in the late phase of the reperfusion period. Together, these studies reveal a major role for apelin in the regulation of myocardial FoxO1 activation and offer a new treatment option for cardiac I/R injury in obese subjects.
Author contributions
P.V., A.P. and O.K. conceived and designed the study. F.B. and J.R. performed in vivo experiments. F.B., H.T., A.T. and O.K. performed in vitro experiments. D.C. performed microsurgery procedures on mice. J.R. and R.A. performed and analysed quantitative RT‐PCR. C.A. and J.R. performed histomorphological analysis. F.B., H.T. and O.K. wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Acknowledgements
This work was supported by grants from the National Institute of Health and Medical Research (INSERM), Fondation Lefoulon‐Delalande, Fondation de France, Région Midi‐Pyrénées and Fondation pour la Recherche Médicale (FRM) and ERASMUS MUNDUS MEDEA project.
Boal, F. , Timotin, A. , Roumegoux, J. , Alfarano, C. , Calise, D. , Anesia, R. , Parini, A. , Valet, P. , Tronchere, H. , and Kunduzova, O. (2016) Apelin‐13 administration protects against ischaemia/reperfusion‐mediated apoptosis through the FoxO1 pathway in high‐fat diet‐induced obesity. British Journal of Pharmacology, 173: 1850–1863. doi: 10.1111/bph.13485.
References
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfarano C, Foussal C, Lairez O, Calise D, Attané C, Anesia R et al. (2014). Transition from metabolic adaptation to maladaptation of the heart in obesity: role of apelin. Int J Obes (Lond) 39: 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attane C, Foussal C, Le Gonidec S, Benani A, Daviaud D, Wanecq E et al. (2012). Apelin treatment increases complete Fatty Acid oxidation, mitochondrial oxidative capacity, and biogenesis in muscle of insulin‐resistant mice. Diabetes 61: 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad H, Nolette N, Hinton M, Dakshinamurti S (2014). AMPK and FoxO1 regulate catalase expression in hypoxic pulmonary arterial smooth muscle. Pediatr Pulmonol 49: 885–897. [DOI] [PubMed] [Google Scholar]
- Battiprolu P, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M et al. (2012). Metabolic stress‐induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest 122: 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L et al. (2005). Oxidative stress by monoamine oxidase mediates receptor‐independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 112: 3297–3305. [DOI] [PubMed] [Google Scholar]
- Bluher M (2013). Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract Res Clin Endocrinol Metab 27: 163–177. [DOI] [PubMed] [Google Scholar]
- Boal F, Guetzoyan L, Sessions RB, Zeghouf M, Spooner RA, Lord JM et al. (2010). LG186: An inhibitor of GBF1 function that causes Golgi disassembly in human and canine cells. Traffic 11: 1537–1551. [DOI] [PubMed] [Google Scholar]
- Braunersreuther V, Mach F, Montecucco F (2012). Reactive oxygen‐induced cardiac intracellular pathways during ischemia and reperfusion. Curr Signal Transduct Ther 7: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray C, Debard C, Jager J, Disse E, Daviaud D, Martin P et al. (2010). Apelin and APJ regulation in adipose tissue and skeletal muscle of type 2 diabetic mice and humans. Am J Physiol Endocrinol Metab 298: E1161–E1169. [DOI] [PubMed] [Google Scholar]
- Dray C, Knauf C, Daviaud D, Waget A, Boucher J, Buléon M et al. (2008). Apelin stimulates glucose utilization in normal and obese insulin‐resistant mice. Cell Metab 8: 437–445. [DOI] [PubMed] [Google Scholar]
- Evans‐Anderson H, Alfieri C, Yutzey E (2008). Regulation of cardiomyocyte proliferation and myocardial growth during development by FOXO transcription factors. Circ Res 102: 686–694. [DOI] [PubMed] [Google Scholar]
- Ferber E, Peck B, Delpuech O, Bell GP, East P, Schulze A et al. (2011). FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ 19: 968–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foussal C, Lairez O, Calise D, Pathak A, Guilbeau‐Frugier C, Valet P et al. (2010). Activation of catalase by apelin prevents oxidative stress‐linked cardiac hypertrophy. FEBS Lett 584: 2363–2370. [DOI] [PubMed] [Google Scholar]
- Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida‐Araki K et al. (2004). Abnormal angiogenesis in Foxo1 (Fkhr)‐deficient mice. J Biol Chem 279: 34741–34749. [DOI] [PubMed] [Google Scholar]
- Kadoglou N, Lampropoulos S, Kapelouzou A, Gkontopoulos A, Theofilogiannakos EK, Fotiadis G et al. (2010). Serum levels of apelin and ghrelin in patients with acute coronary syndromes and established coronary artery disease–KOZANI STUDY. Transl Res 155: 238–246. [DOI] [PubMed] [Google Scholar]
- Kang P, Izumo S (2000). Apoptosis and heart failure a critical review of the literature. Circ Res 86: 1107–1113. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinz MJ, Davenport AP (2005). Emerging roles of apelin in biology and medicine. Pharmacol Ther 107: 198–211. [DOI] [PubMed] [Google Scholar]
- Kleinz MJ, Skepper JN, Davenport AP (2005). Immunocytochemical localisation of the apelin receptor, APJ, to human cardiomyocytes, vascular smooth muscle and endothelial cells. Regul Pept 126: 233–240. [DOI] [PubMed] [Google Scholar]
- Konstantinidis K, Whelan R, Kitsis R (2012). Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol 32: 1552–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops G, Dansen T, Polderman P, Saarloos I, Wirtz K, Coffer P et al. (2002). Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419: 316–321. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Matti A (2012). Hyperactivation of protein phosphatase 2 A in models of glucolipotoxicity and diabetes: potential mechanisms and functional consequences. Biochem Pharmacol 84: 591–597. [DOI] [PubMed] [Google Scholar]
- Kuba K, Zhang L, Imai Y, Arab S, Chen M, Maekawa Y et al. (2007). Impaired heart contractility in Apelin gene‐deficient mice associated with aging and pressure overload. Circ Res 101: e32–e42. [DOI] [PubMed] [Google Scholar]
- Mani K (2008). Programmed cell death in cardiac myocytes: strategies to maximize post‐ischemic salvage. Heart Fail Rev 13: 193–209. [DOI] [PubMed] [Google Scholar]
- Maury E, Ehala‐Aleksejev K, Guiot Y, Detry R, Vandenhooft A, Brichard S (2007). Adipokines oversecreted by omental adipose tissue in human obesity. Am J Physiol Endocrinol Metab 293: E656–E665. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema R, Kops G, Bos J, Burgering B (2000). AFX‐like Forkhead transcription factors mediate cell‐cycle regulation by Ras and PKB through p27kip1. Nature 404: 782–787. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Fuster J, Walsh K (2014). Adipokines: a link between obesity and cardiovascular disease. J Cardiol 63: 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pchejetski D, Foussal C, Alfarano C, Lairez O, Calise D, Guilbeau‐Frugier C et al. (2012). Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur Heart J 33: 2360–2369. [DOI] [PubMed] [Google Scholar]
- Pchejetski D, Kunduzova O, Dayon A, Calise D, Seguelas MH, Leducq N et al. (2007). Oxidative stress‐dependent sphingosine kinase‐1 inhibition mediates monoamine oxidase A‐associated cardiac cell apoptosis. Circ Res 100: 41–49. [DOI] [PubMed] [Google Scholar]
- Ponugoti B, Dong G, Graves D (2012). Role of forkhead transcription factors in diabetes‐induced oxidative stress. Exp Diabetes Res 2012: 939751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana J, Mukamal K, Morgan J, Muller J, Mittleman M (2004). Obesity and the risk of death after acute myocardial infarction. Am Heart J 147: 841–846. [DOI] [PubMed] [Google Scholar]
- Raju I, Kannan K, Abraham E (2013). FoxO3a Serves as a Biomarker of Oxidative Stress in Human Lens Epithelial Cells under Conditions of Hyperglycemia. PLoS One 8: e67126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta A, Molkentin J, Paik J, DePinho R, Yutzey K (2011). FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J Biol Chem 286: 7468–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou M et al. (1998). Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun 251: 471–476. [DOI] [PubMed] [Google Scholar]
- Tycinska A, Sobkowicz B, Mroczko B, Sawicki R, Musial W, Dobrzycki S et al. (2010). The value of apelin‐36 and brain natriuretic peptide measurements in patients with first ST‐elevation myocardial infarction. Clin Chim Acta 411: 2014–2018. [DOI] [PubMed] [Google Scholar]
- Ullman K, Powers M, Forbes D (1997). Nuclear export receptors: from importin to exportin. Cell 90: 967–970. [DOI] [PubMed] [Google Scholar]
- Wang W, McKinnie S, Patel V, Haddad G, Wang Z, Zhabyeyev P et al. (2013). Loss of apelin exacerbates myocardial infarction adverse remodeling and ischemia–reperfusion injury: therapeutic potential of synthetic apelin analogues. J Am Heart Assoc 2: e000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X (2001). The expanding role of mitochondria in apoptosis. Genes Dev 15: 2922–2933. [PubMed] [Google Scholar]
- Wang Y, Zhou Y, Graves D (2014). FOXO transcription factors: their clinical significance and regulation. Biomed Res Int 2014; Article ID 925350, 13 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir R, Chong K, Dalzell J, Petrie C, Murphy C, Steedman T et al. (2009). Plasma apelin concentration is depressed following acute myocardial infarction in man. Eur J Heart Fail 11: 551–558. [DOI] [PubMed] [Google Scholar]
- Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM et al. (2003). A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 111: 1497–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang Y, Zhu WG (2011). Applications of post‐translational modifications of FoxO family proteins in biological functions. J Mol Cell Biol 3: 276–282. [DOI] [PubMed] [Google Scholar]
- Zhen EY, Higgs RE, Gutierrez AJ (2013). Pyroglutamyl apelin‐13 identified as the major apelin isoform in human plasam. Anal Biochem 442: 1–9. [DOI] [PubMed] [Google Scholar]