

Abstract
Background and Purpose
Sphingosine1‐phosphate (S1P) receptors mediate multiple events including lymphocyte trafficking, cardiac function, and endothelial barrier integrity. Stimulation of S1P1 receptors sequesters lymphocyte subsets in peripheral lymphoid organs, preventing their trafficking to inflamed tissue sites, modulating immunity. Targeting S1P receptors for treating autoimmune disease has been established in clinical studies with the non‐selective S1P modulator, FTY720 (fingolimod, Gilenya™). The purpose of this study was to assess RPC1063 for its therapeutic utility in autoimmune diseases.
Experimental Approach
The specificity and potency of RPC1063 (ozanimod) was evaluated for all five S1P receptors, and its effect on cell surface S1P1 receptor expression, was characterized in vitro. The oral pharmacokinetic (PK) parameters and pharmacodynamic effects were established in rodents, and its activity in three models of autoimmune disease (experimental autoimmune encephalitis, 2,4,6‐trinitrobenzenesulfonic acid colitis and CD4+CD45RBhi T cell adoptive transfer colitis) was assessed.
Key Results
RPC1063 was specific for S1P1 and S1P5 receptors, induced S1P1 receptor internalization and induced a reversible reduction in circulating B and CCR7+ T lymphocytes in vivo. RPC1063 showed high oral bioavailability and volume of distribution, and a circulatory half‐life that supports once daily dosing. Oral RPC1063 reduced inflammation and disease parameters in all three autoimmune disease models.
Conclusions and Implications
S1P receptor selectivity, favourable PK properties and efficacy in three distinct disease models supports the clinical development of RPC1063 for the treatment of relapsing multiple sclerosis and inflammatory bowel disease, differentiates RPC1063 from other S1P receptor agonists, and could result in improved safety outcomes in the clinic.
Abbreviations
- CD
Crohn's disease
- EAE
experimental autoimmune encephalitis
- IBD
inflammatory bowel disease
- PK
pharmacokinetic
- RMS
relapsing multiple sclerosis
- S1P
sphingosine‐1‐phosphate
- TNBS
2,4,6‐trinitrobenzenesulfonic acid
- UC
ulcerative colitis
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Catalytic receptors b |
| CCR7 | Integrins |
| S1P1 receptor | Enzymes c |
| S1P2 receptor | Adenylyl cyclase |
| S1P3 receptor | |
| S1P4 receptor | |
| S1P5 receptor |
| LIGANDS | |
|---|---|
| cAMP | IFNγ |
| Cycloheximide | IL‐1β |
| Cyclosporine | IL‐6 |
| CYM5442 | IL‐8 (CXCL8) |
| Forskolin | IL‐10 |
| FTY720‐P | RPC1063 (ozanimod) |
| GDP | Sphingosine 1‐phosphate (S1P) |
| GTPγS | TNFα |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a, 2015b, 2015c).
Introduction
Relapsing multiple sclerosis (RMS) is a chronic autoimmune disease of the CNS, characterized by neurological dysfunction including disruption of the blood–brain barrier, nerve cell demyelination, axonal severing and loss, and astrogliosis. The two major clinically defined forms of inflammatory bowel disease (IBD) include ulcerative colitis (UC) and Crohn's disease (CD) both characterized as autoimmune diseases. UC manifests as superficial inflammation of the mucosa of the colon. CD is characterized by transmural inflammation that primarily occurs in the terminal ileum and proximal colon, but can develop anywhere along the gastrointestinal tract. Though RMS, UC and CD are distinct autoimmune diseases with unique pathologies, they are characterized by the trafficking and accumulation of autoimmune‐driven T and B lymphocytes in inflamed tissues (reviewed in Davenport and Munday, 2007). Agents that block immune cell trafficking demonstrate efficacy in these autoimmune diseases. Natalizumab (Tysabri®), an antibody that targets the α4 integrin and restricts leukocyte trafficking, has been shown to be effective in the treatment of RMS and CD. Similarly, the α4β7 integrin‐directed anti‐leukocyte trafficking antibody vedolizumab (Entyvio®) and the β7 integrin‐selective antibody etrolizumab have demonstrated significant activity in patients with UC and CD (Danese and Panes, 2014).
Sphingosine 1‐phosphate (S1P) is a sphingolipid whose physiological functions include controlling lymphocyte trafficking, heart rate and rhythm, and vascular tone (reviewed in Brinkmann, 2007). These activities are mediated through interaction with five G‐protein coupled receptors (S1P1‐5 receptors) that have varying expression on diverse cell types including lymphocytes, atrial myocytes, endothelial cells and all cell types within the CNS (Martin and Sospedra, 2014). Multiple studies have suggested that direct effects on S1P receptors in CNS cells can promote neuronal protection (reviewed in Groves et al., 2013).
For CCR7+ lymphocytes that circulate through peripheral lymphoid tissues, the S1P1 receptor provides the appropriate exit signal by sensing the S1P chemotactic gradient between peripheral lymphoid organs and the circulation. Supraphysiological and pharmacological activation of the S1P1 receptor induces rapid and persistent receptor internalization and desensitization, rendering these cells unable to respond to a S1P gradient (Mullershausen et al., 2009). Lymphocytes that traffic through lymph nodes, such as naïve and central memory T cells, and B cells remain sequestered in peripheral lymphoid organs. Autoreactive lymphocytes are also thought to be sequestered thus preventing their trafficking to sites of inflammation where they contribute to immune‐mediated pathology. Circulating effector memory lymphocyte levels are unaffected as they do not traffic through peripheral lymphoid tissues, and thus remain in the periphery to maintain immune‐surveillance.
The utility of targeting S1P receptors for the treatment of autoimmune diseases has been well established in both preclinical disease models and in clinical studies. FTY720 (fingolimod, Gilenya™), an analogue of S1P, is a prodrug that is phosphorylated in vivo by sphingosine kinase‐2 generating the pharmacologically active FTY720‐P, a non‐selective potent agonist of S1P1,3,4,5R (Mandala et al., 2002). FTY720 has demonstrated activity in animal models of experimental autoimmune encephalitis (EAE), IBD, rheumatoid arthritis, systemic lupus erythematosus and asthma (Okazaki et al., 2002; Mizushima et al., 2004; Deguchi et al., 2006; Idzko et al., 2006; Balatoni et al., 2007; Wang et al., 2007), and is approved for treatment of RMS. A related compound, KRP203 ameliorated disease in an IL‐10 knockout mouse model of IBD (Song et al., 2008). FTY720 has limiting properties including a long circulatory half‐life resulting in a prolonged reduction in peripheral lymphocytes following cessation of dosing, and broad agonist activity across S1P receptor subtypes that probably contribute to safety‐related side effects. Other, more selective S1P receptor‐targeting agents have demonstrated activity in RMS (Selmaj et al., 2013; Olsson et al., 2014; Krosser et al., 2015) and psoriasis (Vaclavkova et al., 2014).
We developed a S1P receptor agonist with improved receptor selectivity and optimal PK properties, including a short circulatory half‐life resulting in rapid lymphocyte repopulation kinetics. RPC1063 is a potent, orally available small molecule S1P1 and S1P5 receptor‐selective agonist that induces significant S1P1 receptor internalization and a robust but rapidly reversible reduction in circulating lymphocytes in animals. We demonstrate that this receptor selectivity profile is sufficient for RPC1063 to be as effective as FTY720 in experimental autoimmune encephalitis (EAE), a preclinical model of multiple sclerosis. RPC1063 also demonstrated robust efficacy, comparable to anti‐TNF antibody therapy, in the naïve CD4+CD45RBhi T cell adoptive transfer model of Crohn's disease, and TNBS‐induced colitis, with efficacy correlating with the magnitude of lymphocyte reduction and suppression of inflammatory cytokines.
Methods
Animals and compounds
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). A total of 295 animals were used in the experiments described here. All rodents were purchased from Harlan Laboratories (Indianapolis, IN, USA) or Simonsen Laboratories (Gilroy, CA, USA). Animals were housed in an ALAAC accredited facility, acclimatized for 3–7 days under standard light‐ and climate‐controlled conditions with free access to food and water. The research was approved by the facility's Institutional Animal Care and Use Committee. FTY720, FTY720‐P, KRP203 KRP203‐P were from Cayman Chemicals (Ann Arbor, MI, USA). BAF312 was as described (Gergely et al., 2012). Cyclosporine was from Paddock Labs (Minneapolis, MN, USA). Hamster anti‐mouse TNF (TN3–19.12) was from ThermoFisher Scientific (Waltham, MA, USA). All other chemicals and reagents were from Sigma‐Aldrich (St Louis, MO, USA).
In vitro pharmacology assays
Cell signalling assays used the LiveBLAzer™‐FRET B/G assay (Life Technologies, Carlsbad, CA) to detect cAMP (S1P1 receptor) or β‐arrestin signalling (S1P4 receptor). Assays were performed in 384‐well plates in triplicate according to the manufacturer's instructions. Compound stocks were stored at 10 mM in 100% DMSO at −80°C, and initially diluted 1:10 with 20% (2‐hydoxypropyl)‐β‐cyclodextrin. A 10‐point dose‐response curve was generated at 40‐times the final assay concentration in 10 mM HEPES pH 7.4, containing 0.1% pluronic F‐127. For the S1P1 receptor assay, 80 μM forskolin was included in the diluent. Briefly, 104 cells/well were incubated with a dose response of ligand at 37°C for 4 hrs. CC4‐AM substrate and probenecid were added and incubated at 37°C for a further 2 h and analysed with a SpectramaxM5 (Molecular Devices, Sunnyvale, CA, USA). For S1P1 receptor cAMP assays, data were normalized to the maximum fluorescence generated by 2 μM forskolin. For GTPγS binding assays, 1‐5 μg per well of membrane protein was incubated with 10 μM GDP, 100–500 μg per well Wheat Germ Agglutinin PVT SPA beads (Perkin Elmer) in 50 mM HEPES, 100 mM NaCl, 10 mM MgCl2, 20 μg·mL‐1 saponin and 0.1% fatty acid free bovine serum albumin (BSA) for 15 min in 96‐well plates. After the addition of compound and 200 pM [35S]‐ GTPγS (Perkin Elmer, 1250 Ci ·mmol‐1), the plates were incubated for 120 min and centrifuged at 300 g for 5 min. Radioactivity was detected with a TopCount Instrument (Packard Instruments, Waltham, MA, USA). All data were fit with a four parameter variable slope non‐linear regression (GraphPad Prism) to generate half‐maximal effective concentration (EC50) and maximum efficacy relative to S1P.
MOG‐induced EAE model in C57Bl6 mice
Female C57BL/6 mice (60 total, 10 weeks of age) were immunized with Myelin Oligodendrocyte Glycoproteins (MOG)35–55 peptide with complete Freund's adjuvant on day 0, and pertussis toxin was administered 2 h and 24 h later. At the first instance of clinical symptoms of EAE (limp tail), mice were randomized into treatment groups (n = 10 per group) and administered the test compound, p.o., once daily for 14 days. Mice that developed disease earlier than 9 days post‐immunization were not enrolled, as these often develop fulminant disease that does not respond to therapy. Test compounds were 0.2 and 0.6 mg·kg−1 RPC1063, 3 mg·kg−1 FTY720 or vehicle (5% DMSO, 5% Tween‐20, 90% 0.1 N HCl). Animals were monitored daily for body weight and clinical symptoms and scored according to standard methods as described in the Supplemental Materials. At the end of the study, mice were anaesthetized via inhalation of isoflurane bubbled with oxygen at 10‐20 kPa, blood drawn via cardiac puncture to exsanguination and analysed by a veterinary haemoanalyser. One mouse died in the 0.6 mg·kg−1 RPC1063 group, possibly due to aspiration of test compound, and was eliminated from the analysis.
TNBS model of inflammatory bowel disease
Male Sprague Dawley rats (48 total;180–200 g) were fasted overnight, lightly anaesthetized by inhalation of isoflurane, as described above, intra‐rectally administered 64 mg TNBS in 50% ethanol and held inverted for 1 min to ensure distribution throughout the colon. Oral gavage of the test compound (5 mL·kg−1) began 2 h after TNBS treatment and continued daily for 7 days. Treatment groups included 0.1, 0.3 and 1 mg·kg−1 RPC1063, 10 mg·kg−1 prednisolone and vehicle (5% DMSO, 5% Tween‐20, 90% 0.1 N HCl). Body weight and clinical observations were monitored daily. At the end of the study, rats were anaesthetized via inhalation of isoflurane as described above, and blood drawn via cardiac puncture to exsanguination 24 h after the final dose for lymphopenia analysis by flow cytometry. Colon disease score was a sum of the number of adhesions, strictures and ulcers combined with ulcer size and colon wall thickness as described in the Supplemental Materials. Colons were resected, flushed of faecal material and the colon length and weights recorded.
Naïve CD4+CD45Rbhi T cell adoptive transfer model in SCID mice
Female SCID mice (54 total, 12 weeks of age and 18–24 g) were injected i.p. with 4x105 CD4+CD45Rbhi T cells from age‐matched female Balb/c mice (70 total) on day 0, and body weight was measured every 3 days. Three weeks after adoptive transfer, mice began to show clinical signs of colitis, including reduced body weight and perianal staining. Mice were administered the test substance p.o. by gavage (5 mL·kg−1) once daily for 21 days. Treatment groups included 1.2 mg·kg−1 RPC1063, 50 mg·kg−1 cyclosporine or vehicle. A separate group for comparison received 300 μg hamster anti‐mouse TNF antibody (TN3–19.12) i.p., once weekly. At the end of the study, mice were anaesthetized via inhalation of isoflurane as described above, and blood drawn via cardiac puncture to exsanguination. Colons were resected, flushed of faecal material and the colon length and weight recorded. Colons were fixed in formalin, paraffin embedded and sections stained with haematoxylin and eosin for histopathology assessment. The scoring system is described in the Supplemental Materials. In a separate study, mice were treated for 21 days with vehicle, 50 mg·kg−1 cyclosporine, or 0.3 mg·kg−1, 0.6 mg·kg−1, 1.2 mg kg−1 RPC1063, and a 2 cm piece of distal colon snap‐frozen for cytokine analysis.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Scoring for in vivo studies was done in a blinded fashion. Some of the data were normalized to control for unwanted sources of variation. Statistical analysis was performed using GraphPad Prism V6, using either Student's t‐test or one‐way ANOVA with Dunnett's comparison for experiments with more than two groups. Statistical significance is defined and reported as P > 0.05.
Other methods
Details for animals and reagents, receptors and cell lines, S1P1 receptor internalization assays, flow cytometry, cytokine analysis and PK of RPC1063 are in Supplemental Methods.
Results
RPC1063 is an agonist of S1P1 and S1P5 receptors
RPC1063 is a bi‐aryl oxadiazole whose full chemical name is (S)‐5‐(3‐(1‐((2‐hydroxyethyl) amino)‐2,3‐dihydro‐1H‐inden‐4‐yl)‐1,2,4‐oxadiazol‐5‐yl)‐2‐isopropoxybenzonitrile hydrochloride (Figure 1). The S1P1 receptor is a Gi/o coupled receptor whose activation inhibits adenylyl cyclase, thus reducing cAMP production. The potency and selectivity of RPC1063 for the S1P receptor family was assessed through inhibition of forskolin‐induced cAMP production (S1P1 receptor), [35S]‐ GTPγS binding assays (S1P1,2,3,5 receptors), or β‐arrestin signalling (S1P4 receptor). RPC1063 demonstrated potent agonist activity of S1P1 receptors. The EC50 values were subnanomolar for S1P1 receptors whether measuring inhibition of cAMP generation (160 ± 60 pM) or [35S]‐ GTPγS binding (410 ± 160 pM; Figure 2A and Table 1). RPC1063 also demonstrated agonist activity at the S1P5 receptor [11 ± 4.3 nM and 83% Emax (percentage of maximum stimulation)]. This represents a 27‐fold selectivity for S1P1 over S1P5 receptors. Selectivity for S1P1 over S1P2,3,4 receptors was significantly greater than 10,000‐fold. RPC1063 receptor selectivity and potency were compared to FTY720, KRP203, and BAF312 (Siponimod), other compounds known to interact with S1P receptors. FTY720 and KRP203 are pro‐drugs that are phosphorylated in vivo, so comparison was made to the biologically active phosphorylated species, FTY720‐P and KRP203‐P, respectively. RPC1063 was very similar in potency at S1P1 receptors when compared to FTY720‐P, BAF312 and KRP203‐P (Table 1). In addition to S1P1 receptors, FTY720‐P potently stimulated S1P3,4,5 receptors, BAF312 stimulated the S1P5R receptor, while KRP203‐P stimulated S1P4, 5 receptors and demonstrated inverse agonist activity on S1P3 receptors (Table 1). None of the compounds demonstrated activity on S1P2 receptors. These studies established that RPC1063 was a potent agonist of the S1P1 receptor with additional agonist activity on the S1P5 receptor, and that activity on the S1P1 receptor was similar to other known, less selective S1P receptor agonists.
Figure 1.
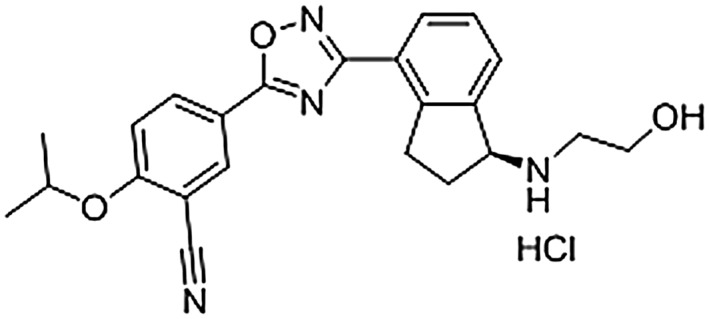
Chemical structure of RPC1063.
Figure 2.
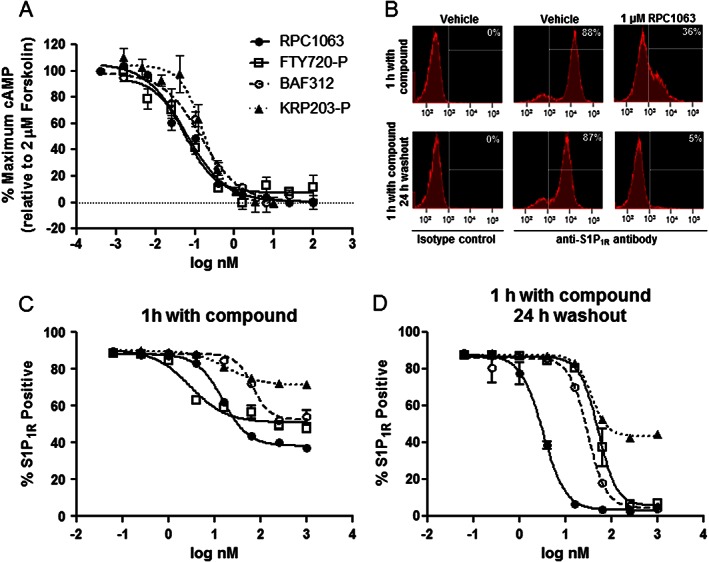
RPC1063 is a potent S1P1 receptor (S1P1R) agonist and induces sustained receptor internalization. (A) S1P1 receptor CREbla CHO‐K1 cells were incubated with 2 μM forskolin in the presence of increasing doses of compound for 4 h. Inhibition of forskolin‐induced cAMP generation is indicative of agonist activity on Gi/o coupled S1P1 receptors. (B) Representative histograms of transfected HEK293T cells expressing S1P1 receptors incubated with vehicle control or 1 μM RPC1063. HEK293T cells were incubated with increasing doses of compound for 1 h (C), or for 1 h followed by extensive washing to remove compound, then a 24 h recovery period (D) and cell surface receptor expression was monitored. Error bars represent SEM of triplicate assays.
Table 1.
In vitro activity of RPC1063 on the S1P receptor family compared to other S1P1 receptor agonists in clinical development
| S1P1R cAMP | S1P1R GTPγS | S1P2R GTPγS | S1P3R GTPγS | S1P4R b Β‐arrestin | S1P5R GTPγS | ||
|---|---|---|---|---|---|---|---|
| RPC1063 | EC50 (nM) | 0.16 ± 0.06 | 0.41 ± 0.16 | >10,000 | >10,000 | >7,865 ± 3697 | 11 ± 4.3 |
| Emaxa (%) | 100 | 97 | 70 | 109 | 21 | 83 | |
| FTY720‐P (Gilenya) | EC50 (nM) | 0.27 ± 0.02 | 0.27 ± 0.04 | >10,000 | 0.90 ± 0.50 | 345 ± 39 | 0.50 ± 0.08 |
| Emaxa (%) | 100 | 85 | 22 | 57 | 130 | 61 | |
| BAF312 (Siponimod) | EC50 (nM) | 0.21 ± 0.02 | 0.39 ± 0.29 | >10,000 | >10,000 | 920 ± 215 | 0.38 ± 0.09 |
| Emaxa (%) | 100 | 75 | 15 | 97 | 38 | 89 | |
| KRP203‐P | EC50 (nM) | 0.27 ± 0.10 | 0.28 ± 0.11 | >10,000 | 22 (IC50) | 11 ± 3.6 | 3 ± 1.2 |
| Emaxa (%) | 100 | N.D. | 9 | −55b | 107 | 36 |
Data represent n = 3–5 independent experiments for all receptors in all signalling pathways.
Emax is relative to the maximal signalling produced by S1P.
S1P1,2,3,5 receptors were assessed by GTPγS binding. S1P4 receptor activity was assessed using β‐arrestin.
KRP203‐P is an inverse agonist on S1P3 receptors.
N.D. = Not Determined
RPC1063 induces sustained S1P1 receptor internalization and degradation
Upon stimulation with potent agonist compounds, the S1P1 receptor has been shown to be internalized and degraded via ubiquitin‐proteosome degradation pathways (Jo et al., 2005). To determine whether RPC1063 induced sustained S1P1 receptor internalization, S1P1 receptor‐HEK293T cells were incubated with different doses of RPC1063 in the presence of 10 μM cycloheximide to prevent translation of new S1P1 receptor protein. Cells were analysed after 1 h treatment, or, after the 1 h treatment washed thoroughly to remove RPC1063 and incubated with 1 μM cycloheximide for a further 24 h. After a 1 h treatment RPC1063 induced significant loss of S1P1 receptor cell surface expression (Figure 2B), similar in magnitude and potency to that seen with FTY720‐P‐treated cells (Figure 2C). Higher concentrations of BAF312 were required to induce a similar magnitude of loss of cell surface receptor expression, while KRP203‐P induced a more modest effect (Figure 2C). Following 1 h of treatment and a 24 h washout period, RPC1063 demonstrated a dose‐dependent effect on S1P1 receptor re‐expression on the cell surface, with near complete and sustained loss of cell surface receptor expression at concentrations above 10 nM (Figure 2D). Loss of cell surface S1P1 receptors was similar to previous reports for BAF312 and FTY720‐P (Mullershausen et al., 2009; Gergely et al., 2012), and RPC1063 was 10‐fold more potent than all three comparators. In a separate immunoblot experiment, continuous exposure to RPC1063 confirmed that the total cellular pool of S1P1 receptors decreased by up to 70% over 18 h (Supporting Information Fig. S1) consistent with receptor internalization followed by ubiquitin‐proteosome dependent degradation as reported for FTY720‐P (Jo et al., 2005; Oo et al., 2007). Finally, pretreatment of S1P1 receptor‐transfected HUVEC cells with RPC1063 rendered them biochemically unresponsive to further stimulation with FTY720‐P (Supporting Information Fig. S2), as measured by phosphorylated Extracellular Signal‐Regulated Kinase (pERK) activation. In contrast, pretreatment of cells with the natural ligand S1P, a relatively weak S1P1 receptor agonist did not affect FTY720‐P activity, consistent with S1P having limited ability to induce sustained receptor internalization and degradation (Jo et al., 2005).
RPC1063 is orally bioavailable and induces a dose‐dependent reduction in peripheral lymphocytes
Administration of RPC1063 p.o. demonstrated dose linear PK (data not shown) with a t 1/2 of 4.7 h and 5.1 h in mice and rats, respectively (Table 2). RPC1063 also demonstrated a high volume of distribution in both species, 10 L·kg−1 and 13 L·kg−1, and effectively partitioned to the brain, with a brain to blood ratio of 10:1 and 16:1 in mice and rats, respectively (Table 2).
Table 2.
Pharmacokinetic parameters in blood after a single oral dose of RPC1063
| Dose | Cmax (μM)a | AUC0‐infinity (μM*h)a | t 1/2 (h) | Clearance (ml min−1 kg−1) | Vd (L kg−1) | Tmax (h) | Brain: blood | |
|---|---|---|---|---|---|---|---|---|
| C57Bl/6 Mice | 1 mg·kg−1 | 0.16 ± 0.03 | 1.50 ± 0.15 | 4.7 | 28 | 10 | 3 | 10 |
| Sprague–Dawley Rats | 0.5 mg·kg−1 | 0.06 ± 0.003 | 0.70 ± 0.07 | 5.1 | 30 | 13 | 3 | 16b |
Mean ± SEM; n = 3 animals per time point
Five days of QD dosing.
A pharmacodynamic effect of S1P1 receptor agonism is the rapid reduction in the number of circulating lymphocytes, thought to be a consequence of receptor internalization and subsequent inability of lymphocytes to respond to an S1P blood/lymph node concentration gradient, thus resulting in their sequestration in peripheral lymphoid tissues (Brinkmann, 2007). In both mice and rats RPC1063 reduced circulating T lymphocytes in an exposure‐dependent manner, establishing a direct relationship between compound PK and the pharmacodynamic response (Figure 3A, B). A maximal effect, 81–82% decrease in circulating T lymphocytes, was observed 6 h after dosing, with a maximal blood concentration of 159 nM and 45 nM in mice and rats respectively. B lymphocytes (B220+) were similarly reduced (Supporting Information Fig. S3). In agreement with the findings obtained with other S1P1 receptor agonists, RPC1063 selectively reduced CD4+CCR7+ and CD8+ CCR7+ T cells (Figure 3C), with significantly less effect on CCR7− lymphocytes (Mehling et al., 2008).
Figure 3.
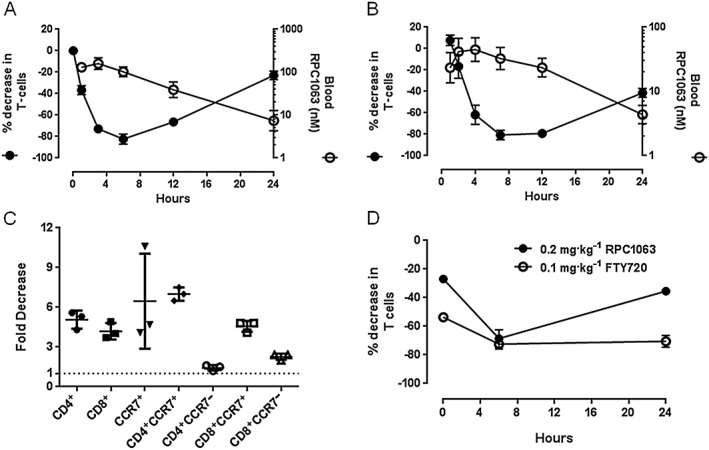
RPC1063 induces dose‐dependent, selective lymphopenia and rapid lymphocyte repopulation kinetics. (A) C57BL/6 mice (n = 3 per time‐point) were dosed 1 mg·kg−1 RPC1063 p.o. and blood samples collected at 1, 3, 6, 12 and 24 h post dose. (B) Sprague Dawley rats (n = 6) were dosed 0.5 mg kg−1 RPC1063 p.o. and blood samples collected at 1, 2, 4, 7, 12 and 24 h post dose. (C) C57BL/6 mice (n = 3) were dosed 1 mg·kg−1 RPC1063 p.o. and blood samples were collected at 6 h post dose. Lymphocyte subsets were then evaluated by flow cytometry. (D) Sprague Dawley rats (n = 3) were administered 0.2 mg·kg−1 RPC1063 p.o. or 0.1 mg·kg−1 FTY720 p.o. daily for 5 days. Blood samples were taken 0, 6 and 24 h after the final dose. RPC1063 was measured in plasma and circulating T lymphocytes quantified by flow cytometry. Error bars represent SEM.
Given the shorter half‐life of RPC1063 relative to FTY720 (25–34 h in mice (Brown, 2009) and 15–30 h in rats (Meno‐Tetang et al., 2006)), we predicted that, as RPC1063 was cleared from circulation, lymphocytes would rapidly repopulate the periphery. In single dose studies, lymphocytes began to repopulate peripheral blood once RPC1063 fell below ~100 nM and ~30 nM in mice and rats respectively (Figure 3A,B). Lymphocyte repopulation kinetics were also evaluated following 5 daily doses of FTY720 or RPC1063 in rats at doses that achieved an equivalent reduction in lymphocytes at 6 h following the final dose. Maximum sequestration of lymphocytes was sustained for at least 24 h after the final dose of FTY720. With RPC1063, significant repopulation of circulating lymphocytes occurred within 24 h of the final dose (Figure 3D).
Therapeutic administration of RPC1063 suppresses clinical symptoms in MOG(35–55) peptide‐induced EAE in a dose‐dependent manner
The MOG35–55 peptide‐induced EAE model was used to evaluate the therapeutic potential of RPC1063. MOG35–55 peptide‐immunized mice develop disease symptoms 9–12 days post‐immunization, and at first signs of clinically relevant symptoms (limp tail), were administered therapy by oral gavage. Control vehicle‐treated mice developed a progressive disease that manifests as weight loss and progressive paraplegia. Disease scores peaked in these animals 4 days after vehicle treatment began, with a clinical score of 2.9 ± 0.46. After 14 days of vehicle treatment, animals had a clinical score of 2.5 ± 0.85 (Figure 4A and B). RPC1063‐treated mice showed a reduction in disease score in a dose‐dependent manner with both doses (0.2 or 0.6 mg·kg−1·day−1) achieving statistical significance over vehicle‐treated animals at the end of the study (Figure 4B). Efficacy achieved by RPC1063 was very similar to that seen with high dose, maximally efficacious FTY720 treatment. Vehicle‐treated animals lost >10% of their body weight over the course of the experiment. All interventions reduced weight loss associated with EAE, while the high dose RPC1063‐ and FTY720‐treated animals actually gained weight during the course of the experiment, a finding consistent with a reduction in disease severity (Figure 4C).
Figure 4.
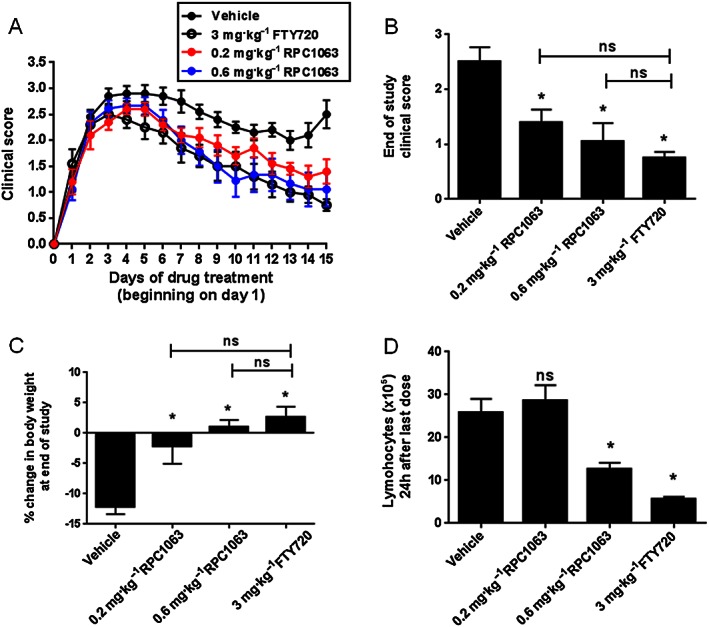
RPC1063 treatment reduces ongoing disease symptoms in EAE. EAE was induced in mice by immunization with MOG35–55 peptide and at the first signs of clinical disease (limp tail) individual mice were randomized and dosed with test compound for 14 days. Clinical disease scores were assessed daily (A) and at the end of the study (B). Body weights (C) were assessed at the end of the study. The number of circulating lymphocytes was assessed by differential count 24 h after the final dose (D). n = 9–10 per group. Error bars represent SEM; *P < 0.05 by One‐way ANOVA.
Analysis of circulating lymphocytes showed that 24 h after the final dose, 0.6 mg·kg−1 RPC1063 reduced the number of lymphocytes by 51% while FTY720 treatment reduced the number by 78% (Figure 4D). Interestingly, although 0.2 mg·kg−1 doses of RPC1063 produced a significant improvement in disease scores and body weight, the lymphocyte reduction induced by this dose was transient, as the number of circulating lymphocytes had returned to baseline 24 h after the final dose.
Therapeutic administration of RPC1063 inhibits clinical and histological disease scores in IBD models
To determine the therapeutic benefit of RPC1063 in other inflammatory conditions we assessed the efficacy of RPC1063 in two preclinical models of inflammatory bowel disease: TNBS‐induced colitis in Sprague Dawley rats and naïve T cell adoptive transfer into Severe Combined Immunodeficiency (SCID) mice.
Colitis was induced by rectal administration of TNBS into Sprague Dawley rats. A dose range of RPC1063 was administered once daily for 7 days in therapeutic mode with the first dose 2 h after TNBS administration. Two rats in the TNBS vehicle‐treated group died from intestinal rupture on day 6 and 7 of the study in line with the acute nature of this disease model. However, none of the RPC1063‐ or prednisolone‐treated animals died, consistent with improved intestinal health. TNBS vehicle‐treated mice showed an 18 ± 5% decrease in body weight at the end of the study (Figure 5A) and did not produce faecal pellets indicating loss of colon function. RPC1063 reduced weight loss in a dose‐dependent manner, achieving statistical significance for the 1 mg·kg−1 group (Figure 5A), and restored colon function as demonstrated by faecal pellets in the cage. RPC1063 also reduced the colon weight:length ratio, a surrogate measure of colon inflammation, with the mid and high dose cohorts (0.3 and 1 mg·kg−1) demonstrating similar efficacy to prednisolone (Figure 5B). The overall macroscopic colon disease score improved in a RPC1063 dose‐dependent manner (Figure 5C). Finally, the reduction in circulating lymphocytes correlated with the degree of efficacy (Figure 5D).
Figure 5.
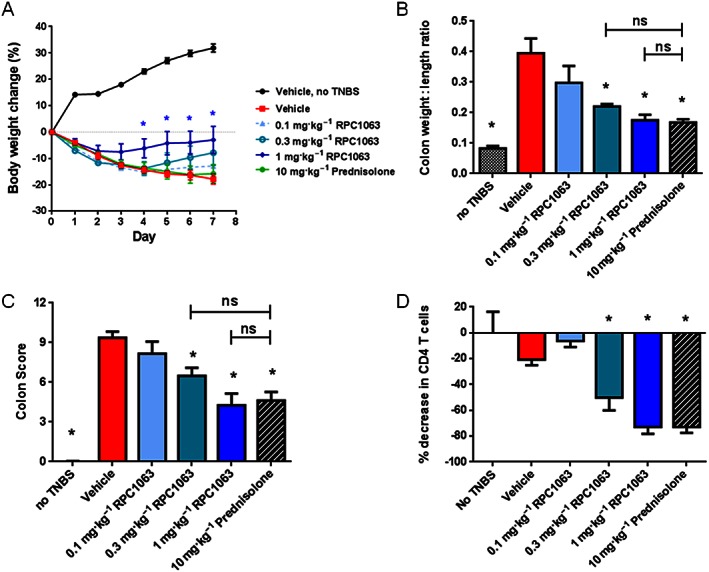
RPC1063 treatment reduces disease activity in TNBS‐induced colitis. Colitis was induced by rectal instillation of TNBS followed 2 h later by daily oral drug treatment. Body weight was assessed daily (A) and *P < 0.05 (determined using Student's t‐test). Colon weight to length ratio (B) and macroscopic colon disease score (C) were determined at the end of the study. The percentage of CD4+ T cells in circulation relative to vehicle treated healthy animals was assessed 24 h after the final dose (D). n = 6–8 per group. Error bars represent SEM. (B‐D) *P < 0.05 by one‐way ANOVA.
We further assessed the efficacy of RPC1063 in a T cell adoptive transfer colitis model with pathological features resembling human Crohn's disease (Ostanin et al., 2006; Maxwell et al., 2009), where agents including anti‐TNF, anti‐IL‐12p40 and anti‐α4β7 antibodies have demonstrated efficacy (Lindebo Holm et al., 2012). As a mechanistic comparator we chose KRP203 which is in clinical development for the treatment of patients with inflammatory bowel disease and has shown efficacy in the IL‐10−/− mouse model of colitis (Song et al., 2008). The selected doses of RPC1063 (1.2 mg·kg−1) and KRP203 (0.3 mg·kg−1) equivalently reduced circulating lymphocytes in C57BL/6 mice after five daily doses of drug (Figure 6A). Immune deficient SCID mice received CD4+CD45Rbhi T cells purified from the spleen of Balb/c mice. Three weeks post‐transfer, mice began to lose weight indicating disease activity, at which point treatment was initiated. During the 20‐day treatment period, vehicle‐treated mice lost 10.5 ± 3.3% body weight at the peak of the disease (day 35 post adoptive transfer), while animals that did not receive adoptive transfer maintained a stable body weight (Figure 6B, D). Both cyclosporine and anti‐TNF antibody therapy were effective at reducing disease severity as assessed by both body weight and colon weight:length ratio (Figure 6B,C). Daily administration of RPC1063 p.o. significantly reduced weight loss for the majority of the treatment period (Figure 6D). In addition, RPC1063 significantly reduced the colon weight:length ratio similar to anti‐TNF‐ and cyclosporine‐treated animals (Figure 6C). By comparison, KRP203‐treated animals demonstrated a colon weight:length ratio and weight loss similar to vehicle‐treated control animals (Figure 6C, D).
Figure 6.
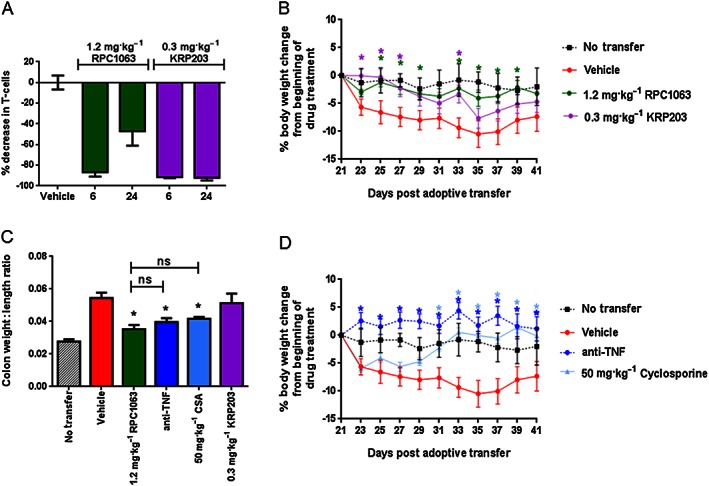
Therapeutic administration of RPC1063 preserves body weight and reduces inflammation in a naïve T‐cell adoptive transfer model of colitis. (A) Lymphopenia induced by RPC1063 and KRP203 in C57Bl/6 mice after 5 days of oral dosing, as assessed by measuring circulating CD4+ and CD8+ T cells, 6 and 24 h after the final dose and compared to vehicle treated animals. (B‐D) Colitis was induced by adoptive transfer of CD4+CD45Rbhi T cells into SCID mice. Daily drug treatment began 21 days post‐transfer, when the first signs of weight loss were observed. (B and D) Body weight was assessed every other day and *P < 0.05 (determined using Student's t‐test). Data are from the same study but displayed on different graphs for clarity. (C) Colon weight to length ratio was determined at the end of the study. n = 10 per group. Error bars represent SEM; *P < 0.05 by one‐way ANOVA.
Histological assessment of colon tissue revealed that RPC1063 treatment significantly reduced disease severity as assessed by measuring the degree of inflammation, gland loss, hyperplasia, neutrophil infiltrate and mucosal thickness (Figure 7A,C). Reduction in disease severity was similar to that seen with cyclosporine and anti‐TNF therapy, and superior to KRP203 treatment. A significant reduction in inflammatory cell infiltrate to the lamina propria of the colon and preservation of colon tissue architecture after treatment with RPC1063 can be seen in Figure 7B. In a separate T cell adoptive transfer study, RPC1063 was administered at 0.1, 0.6 and 1.2 mg·kg−1 and colon tissue extracts were assessed for expression of a selection of inflammatory cytokines (Figure 8). RPC1063 treatment reduced the expression of IFNγ, IL‐10, IL‐1β, IL‐6, IL‐8 and TNFα in a dose responsive manner, with the highest RPC1063 dose (1.2 mg·kg−1) demonstrating similar cytokine reducing effects to cyclosporine.
Figure 7.
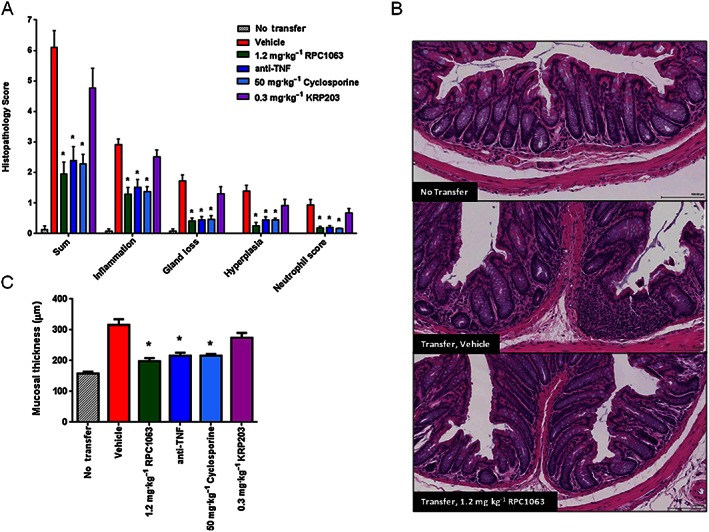
Therapeutic administration of RPC1063 reduces histological disease scores in a naïve T‐cell adoptive transfer model of colitis. (A) End of study histopathological assessment of disease measuring multiple parameters. Scoring system is described in Supplemental Information. (B) Representative images of H&E‐stained distal colon tissue sections demonstrating disruption of gut architecture and mononuclear cell infiltration into the lamina propria of diseased animals, but absent in RPC1063‐treated animals. (C) Mucosal thickness was measured at the end of the study. n = 10 per group. Error bars represent SEM; *P < 0.05 by one‐way ANOVA.
Figure 8.
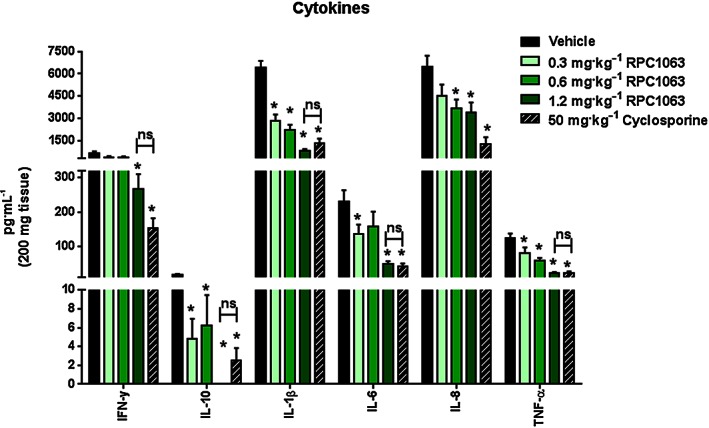
RPC1063 dose‐dependently reduces inflammatory cytokine expression in a naïve T‐cell adoptive transfer model of colitis. At the end of the study, protein was extracted from a 200 mg section of the distal colon and analysed in duplicate for cytokines using Meso Scale Discoveries Electrochemiluminescence system. n = 10 per group. Error bars represent SEM; *P < 0.05 by one‐way ANOVA.
Discussion and conclusions
This study characterizes RPC1063, a novel S1P1 and S1P5 receptor selective agonist. RPC1063 induced durable S1P1 receptor internalization and degradation, resulting in pharmacodynamic effects characterized by a significant but rapidly reversible reduction in circulating lymphocyte subsets. RPC1063 also demonstrated excellent PK and pharmaceutical properties; having high oral bioavailability, a circulating half‐life that supports daily dosing in humans, and also effectively crossing the blood–brain barrier resulting in exposure in the brain. RPC1063 was effective in reducing established disease in mouse EAE thus supporting the development of RPC1063 for the treatment of RMS. RPC1063 also demonstrated significant efficacy in models of IBD, suggesting that inhibition of lymphocyte trafficking via S1P1 receptor engagement may be an effective strategy for treating patients with IBD. RPC1063 is currently in clinical development for the treatment of RMS and IBD.
Multiple assays were performed to explore the potency, specificity and signalling properties of RPC1063. RPC1063 potently induced S1P1 receptor‐mediated induction of GTPγ35S binding and inhibition of cAMP generation, similar to that observed for other known S1P1 receptor agonists FTY720‐P, BAF312, and KRP203‐P. RPC1063 also demonstrated S1P5 agonist activity and no activity on S1P2,3,4 receptors, making it more selective than FTY720‐P.
Unlike FTY720‐P, RPC1063 does not engage S1P3 receptors. This is relevant to the clinical safety profile of this class of compounds. In man, FTY720 has demonstrated cardiac conduction abnormalities including QT interval prolongation (Gilenya, unpubl. data). This was not observed with RPC1063 at supratherapeutic doses (J Hartung unpublished observations). In mice, the S1P3 receptor is expressed on cardiac Purkinje fibers, and FTY720 induces cardiac conduction abnormalities, effects that can be reversed with a selective S1P3 receptor antagonist, and importantly are not observed in S1P3 receptor knock‐out animals, or with S1P1 receptor–selective compounds (H Rosen, personal communication). FTY720 also induces hypertension in both humans and mice, probably through engagement of S1P3 receptors on vascular smooth muscle cells (Fryer et al., 2012). RPC1063 does not induce hypertension in humans (data not shown). Finally, FTY720‐P demonstrates pro‐fibrotic activity, including induction of myofibroblast differentiation, Smad3 signalling, and up‐regulation of connective tissue growth factor and type IV collagen, apparently via S1P3 receptor engagement (Xin et al., 2006; Keller et al., 2007). Fibrosis in heart and lungs was also reported in preclinical toxicology studies with FTY720 (Siarey, 2009). In contrast, there was no evidence of fibrosis following 6–9 month, RPC1063 toxicology studies in either rats or monkeys (data not shown), suggesting that FTY720‐related profibrotic activity is due to S1P3 receptor activation.
Internalization of the S1P1 receptor drives reversible and selective reductions in lymphocytes that modulate the immune system (Thangada et al., 2010). Lymphocyte egress from thymus, lymph nodes and Peyer's patch is dependent upon an S1P gradient between the lymphoid tissue (nanomolar S1P) and plasma (micromolar S1P). Down‐regulation of the lymphocyte‐expressed S1P1 receptor prevents their ability to respond to the S1P gradient and egress from peripheral lymphoid tissues. This “functional antagonism” reduces circulating lymphocytes, including auto‐reactive lymphocytes, preventing trafficking to sites of inflammation and contributing to disease. This effect is primarily observed on naïve and central memory B and T lymphocytes expressing the chemokine receptor CCR7 (Pham et al., 2008). A single oral dose of RPC1063 reduced circulating lymphocytes in a rapidly reversible manner as the compound was cleared from the circulation (Figure 2). At doses of RPC1063 and FTY720 that induced similar reductions in lymphocytes, RPC1063‐treated animals experienced faster recovery towards baseline. This was probably because RPC1063 has a shorter half‐life (t 1/2 is ~5 h in mice) than FTY720 (t 1/2 is 25–34 h in mice; (Meno‐Tetang et al., 2006)). In humans RPC1063 has a 19 h half‐life and 3‐day lymphocyte count recovery into the normal range (Phase I clinical study, unpublished observations). This is in contrast to FTY720 which has a very long half‐life of 168 h in humans (Kovarik et al., 2004) and 4–8 week lymphocyte recovery period (Gilenya Prescribing Information). Having a shorter half‐life and rapid peripheral lymphocyte recovery may provide RPC1063 with significant advantages, including flexibility in treatment with other immune‐modulating agents as needed or allowing for a rapid switch to alternative therapies if the patients disease flares while on therapy. A shorter time to lymphocyte recovery would also offer advantages should opportunistic infections or other treatment‐related complications occur, or if female patients wanted to become pregnant, as targeting the S1P1 receptor pathway has been shown to be teratogenic (Liu et al., 2000; Allende et al., 2003).
Oral administration of RP 1063 following establishment of disease (therapeutic dosing) demonstrated a dose‐dependent reduction in disease scores in EAE, a mouse model of multiple sclerosis. Efficacy achieved was comparable to FTY720 and was consistent with previous findings (Kataoka et al., 2005). Amelioration of EAE disease activity by RPC1063 appears to be due to sequestration of auto‐reactive lymphocytes in peripheral lymphoid tissues, preventing trafficking to the brain and limiting inflammation and tissue damage. However, we found that RPC1063 was efficacious in the EAE model at doses (0.2 mg·kg−1) that produced only transient lymphocyte reductions, consistent with previous studies with CYM5442, a brain penetrating S1P1 receptor selective agonist (Gonzalez‐Cabrera et al., 2012, 2008). Collectively, this suggests there may be other direct CNS effects in addition to restriction of auto‐reactive lymphocyte trafficking that contribute to disease amelioration.
The S1P1 receptor is expressed on multiple cell types in the CNS, including neurons and astrocytes, and engagement of the S1P1 receptor promotes neuronal protection (reviewed in (Dev et al., 2008). In vitro studies demonstrated that FTY720 and SEW2871 (S1P1 receptor‐specific), promoted oligodendrocyte precursor cell survival and differentiation (Coelho et al., 2007; Miron et al., 2008), as well as astrocyte signalling and migration in a S1P1 receptor‐dependent manner (Mullershausen et al., 2007; Osinde et al., 2007). In vivo, FTY720 and SEW2871 reduced infarction size in an ischaemia model (Hasegawa et al., 2010), and astrocyte S1P1 receptor expression was required for FTY720 efficacy in a mouse EAE model (Choi et al., 2011), arguing for direct engagement of S1P1 receptor in the CNS for disease amelioration. Galicia‐Rosas and colleagues made similar observations with AUY954 (S1P1 receptor ‐specific) in EAE (Galicia‐Rosas et al., 2012). The S1P5 receptor is also expressed on various CNS cells. Through engagement of S1P5 receptors on oligodendrocytes and brain endothelial cells, FTY720‐P supports oligodendrocyte progenitor process extension and survival (Miron et al., 2008), and contributes to blood brain barrier integrity (van Doorn et al., 2012). Thus engagement of S1P1 and S1P5 receptors in the CNS may contribute to important neuroprotective mechanisms of clinical significance, supported by the finding that non‐selective FTY720 reduces brain volume loss in RMS patients (Barkhof et al., 2014). RPC1063 effectively partitions to the brain (Table 2) and in the EAE model achieves concentrations 146‐fold and 6‐fold above the EC50 for S1P1 and S1P5 receptors respectively (J. Brooks, unpublished observations). This suggests RPC1063 engages both S1P1 and S1P5 receptors in the CNS.
FTY720 reduces disease in multiple animal models of IBD (Mizushima et al., 2004; Deguchi et al., 2006; Fujii et al., 2006; Daniel et al., 2007). Natalizumab, vedolizumab and etrolizumab are antibodies that target integrins and block the trafficking of various leukocyte populations to sites of inflammation, and are effective drugs in the treatment of patients with RMS and IBD (reviewed in Davenport and Munday, 2007; Danese and Panes, 2014). Cell populations targeted by vedolizumab, including B cells and naïve and central memory CD4+ and CD8+ T cells, overlap significantly with those targeted by RPC1063 (Soler et al., 2009). Indeed, therapeutic dosing of RPC1063 in both IBD models demonstrated significant dose‐dependent disease amelioration of all disease parameters measured. Importantly, RPC1063 was as efficacious as an anti‐TNF antibody in the adoptive transfer model of IBD. This has clinical relevance because several biologics targeting the function of TNF are indicated to treat IBD (reviewed in Croft et al., 2013). Interestingly, in the adoptive transfer model RPC1063 was significantly more effective than KRP203, despite both agents achieving similar levels of lymphocyte reduction. The reason for this is unclear, though it should be noted that KRP203 and RPC1063 have different structural features, receptor specificities and PK properties.
In conclusion, RPC1063 has demonstrated favourable specificity for S1P1 and S1P5 receptors, PK and pharmacodynamic properties. RPC1063 has also demonstrated significant disease ameliorating activity in animal models of multiple sclerosis and IBD. Given the dose‐dependent effects of RPC1063 in these disease models, lymphocyte subset sequestration in peripheral lymphoid tissues probably plays an important immunomodulatory role in controlling disease pathology. However, other effects may also be relevant to treating disease, including direct S1P1‐ and S1P5 receptor‐mediated neuroprotective effects on various CNS cell types in RMS patients. Consistent with preclinical observations, early RPC1063 clinical trial data in healthy volunteers established a favourable PK/PD and safety profile, including limited effects on heart rate and no effect on QT interval prolongation. Also consistent with preclinical EAE studies, RPC1063 recently completed a clinical trial in patients with RMS, demonstrating a compelling safety and efficacy profile (Cohen et al., 2014). Finally, and also consistent with preclinical disease model observations, RPC1063 induced clinical remission and clinical responses during the induction period of a clinical study in patients with ulcerative colitis. Thus the mechanism of action of RPC1063 allows for the treatment of patients with very different disease pathologies but with similar underlying autoimmune‐driven mechanisms.
Author contributions
F.L.S., B.C., J.B., R.P., H.D. and H.G.D. performed the research; F.L.S., G.A.T. and R.J.P. designed the research study; E.B., E.M., M.F.B., H.R. and E.R. contributed essential reagents or tools. All authors contributed to analysis of the data and the drafting and revising of the manuscript. All authors have approved the final version of the work and are accountable for all aspects of the work.
Conflict of interest
F.L.S., B.C., J.B., E.B., R.P., H.D., H.G.D., G.A.T., E.M., M.F.B. and R.J.P. are employees and share holders in Receptos Inc. H.R. and E.R. are scientific founders and share holders in Receptos Inc. The authors have no conflict of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. HEK293 stable cell line expressing S1P1R‐GFP was incubated with 1 μM cycloheximide and a dose response of compound for 18 hrs. Cells were lysed in modified RIPA buffer containing Complete Protease Inhibitor cocktail (Roche). (A) Lysates were separated on 4‐12% SDS PAGE using 6M urea in the loading buffer, transferred to nitrocellulose membrane and blotted with rabbit anti‐GFP(Santa Cruz Biotechnology) and mouse anti‐HSP90 (BD Biosciences) antibodies, followed by Licor Odyssey imaging. (B) Normalized total S1P1‐GFP expression relative to HSP90.
Figure S2. HUVEC cells expressing endogenous S1P1R were used to show that the functional consequence of receptor internalization is loss of response to S1P1R agonist. Cells were preincubated for 1hr with 1 μM RPC1063, FTY720‐P or S1P to induce receptor internalization. Compound was washed out for 5hrs, and cells were re‐stimulated with an S1P1R agonist (FTY720‐P). pERK1/2 activation was assessed using rabbit anti‐Phospho‐ERK1/2 (Cell Signaling #4377) and normalized to mouse anti‐ERK1/2 (Cell Signaling #9107) by in cell western analysis and Licor Odyssey imaging. Data represents mean of triplicate data points. Error bars represent SEM.
Figure S3. C57Bl/6 mice were treated with vehicle or 1 mg kg−1 RPC1063 and blood was taken by cardiac puncture 6 hours later. Blood was incubated with antibodies to the indicated cell surface markers according to Materials and Methods. Data represents mean of three individual mice. Error bars represent SEM.
Supporting info item
Supporting info item
Acknowledgements
We acknowledge Suzana Marusic for assistance with EAE studies; Phillip Bendele for assistance with adoptive transfer colitis studies; Sean O′Neill for TNBS colitis studies; Stephan Miller and Brett Skolnick for assistance with manuscript preparation. FLS, BC, JB, EB, RP, HD, HGD, GAT, EM, MFB and RJP are employees of, and shareholders in, Receptos Inc. HR and ER are founders and shareholders of Receptos Inc. This work was supported by Receptos Inc.
Scott, F. L. , Clemons, B. , Brooks, J. , Brahmachary, E. , Powell, R. , Dedman, H. , Desale, H. G. , Timony, G. A. , Martinborough, E. , Rosen, H. , Roberts, E. , Boehm, M. F. , and Peach, R. J. (2016) Ozanimod (RPC1063) is a potent sphingosine‐1‐phosphate receptor‐1 (S1P1) and receptor‐5 (S1P5) agonist with autoimmune disease‐modifying activity. British Journal of Pharmacology, 173: 1778–1792. doi: 10.1111/bph.13476.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allende ML, Yamashita T, Proia RL (2003). G‐protein‐coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood 102: 3665–3667. [DOI] [PubMed] [Google Scholar]
- Balatoni B, Storch MK, Swoboda EM, Schonborn V, Koziel A, Lambrou GN et al. (2007). FTY720 sustains and restores neuronal function in the DA rat model of MOG‐induced experimental autoimmune encephalomyelitis. Brain Res Bull 74: 307–316. [DOI] [PubMed] [Google Scholar]
- Barkhof F, de Jong R, Sfikas N, de Vera A, Francis G, Cohen J (2014). The influence of patient demographics, disease characteristics and treatment on brain volume loss in Trial Assessing Injectable Interferon vs FTY720 Oral in Relapsing–Remitting Multiple Sclerosis (TRANSFORMS), a phase 3 study of fingolimod in multiple sclerosis. Mult Scler 20: 1704–1713. [DOI] [PubMed] [Google Scholar]
- Brinkmann V (2007). Sphingosine 1‐phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther 115: 84–105. [DOI] [PubMed] [Google Scholar]
- Brown P (2009). Gilenya™ (Fingolimod HCl) Pharmacology Review(s).
- Choi JW, Gardell SE, Herr DR, Rivera R, Lee CW, Noguchi K et al. (2011). FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1‐phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A 108: 751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho RP, Payne SG, Bittman R, Spiegel S, Sato‐Bigbee C (2007). The immunomodulator FTY720 has a direct cytoprotective effect in oligodendrocyte progenitors. J Pharmacol Exp Ther 323: 626–635. [DOI] [PubMed] [Google Scholar]
- Cohen J, Arnold DL, Comi G, Bar‐Or A, Gujrathi S, Hartung JP et al. (2014). Phase 2 results of the RADIANCE trial: a randomized, double‐blind, placebo‐controlled trial of oral RPC1063 in relapsing multiple sclerosis. Mult scler J 20: 497–500. [Google Scholar]
- Croft M, Benedict CA, Ware CF (2013). Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov 12: 147–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese S, Panes J (2014). Development of Drugs to Target Interactions Between Leukocytes and Endothelial Cells and Treatment Algorithms for Inflammatory Bowel Diseases. Gastroenterology 147: 981–989. [DOI] [PubMed] [Google Scholar]
- Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM (2007). FTY720 ameliorates Th1‐mediated colitis in mice by directly affecting the functional activity of CD4 + CD25+ regulatory T cells. J Immunol 178: 2458–2468. [DOI] [PubMed] [Google Scholar]
- Davenport RJ, Munday JR (2007). Alpha4‐integrin antagonism–an effective approach for the treatment of inflammatory diseases? Drug Discov Today 12: 569–576. [DOI] [PubMed] [Google Scholar]
- Deguchi Y, Andoh A, Yagi Y, Bamba S, Inatomi O, Tsujikawa T et al. (2006). The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncol Rep 16: 699–703. [PubMed] [Google Scholar]
- Dev KK, Mullershausen F, Mattes H, Kuhn RR, Bilbe G, Hoyer D et al. (2008). Brain sphingosine‐1‐phosphate receptors: implication for FTY720 in the treatment of multiple sclerosis. Pharmacol Ther 117: 77–93. [DOI] [PubMed] [Google Scholar]
- Fryer RM, Muthukumarana A, Harrison PC, Nodop Mazurek S, Chen RR, Harrington KE et al. (2012). The clinically‐tested S1P receptor agonists, FTY720 and BAF312, demonstrate subtype‐specific bradycardia (S1P(1)) and hypertension (S1P(3)) in rat. PLoS One 7.e52985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii R, Kanai T, Nemoto Y, Makita S, Oshima S, Okamoto R et al. (2006). FTY720 suppresses CD4 + CD44highCD62L‐ effector memory T cell‐mediated colitis. Am J Physiol Gastrointest Liver Physiol 291: G267–G274. [DOI] [PubMed] [Google Scholar]
- Galicia‐Rosas G, Pikor N, Schwartz JA, Rojas O, Jian A, Summers‐Deluca L et al. (2012). A sphingosine‐1‐phosphate receptor 1‐directed agonist reduces central nervous system inflammation in a plasmacytoid dendritic cell‐dependent manner. J Immunol 189: 3700–3706. [DOI] [PubMed] [Google Scholar]
- Gergely P, Nuesslein‐Hildesheim B, Guerini D, Brinkmann V, Traebert M, Bruns C et al. (2012). The selective S1P receptor modulator BAF312 redirects lymphocyte distribution and has species‐specific effects on heart rate: translation from preclinical to clinical studies. Br J Pharmacol 167: 1035–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Cabrera PJ, Cahalan SM, Nguyen N, Sarkisyan G, Leaf NB, Cameron MD et al. (2012). S1P(1) receptor modulation with cyclical recovery from lymphopenia ameliorates mouse model of multiple sclerosis. Mol Pharmacol 81: 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Cabrera PJ, Jo E, Sanna MG, Brown S, Leaf N, Marsolais D et al. (2008). Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P‐like head‐group interactions. Mol Pharmacol 74: 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves A, Kihara Y, Chun J (2013). Fingolimod: direct CNS effects of sphingosine 1‐phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J Neurol Sci 328: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Y, Suzuki H, Sozen T, Rolland W, Zhang JH (2010). Activation of sphingosine 1‐phosphate receptor‐1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke 41: 368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idzko M, Hammad H, van Nimwegen M, Kool M, Muller T, Soullie T et al. (2006). Local application of FTY720 to the lung abrogates experimental asthma by altering dendritic cell function. J Clin Invest 116: 2935–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo E, Sanna MG, Gonzalez‐Cabrera PJ, Thangada S, Tigyi G, Osborne DA et al. (2005). S1P1‐selective in vivo‐active agonists from high‐throughput screening: off‐the‐shelf chemical probes of receptor interactions, signaling, and fate. Chem Biol 12: 703–715. [DOI] [PubMed] [Google Scholar]
- Kataoka H, Sugahara K, Shimano K, Teshima K, Koyama M, Fukunari A et al. (2005). FTY720, sphingosine 1‐phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol 2: 439–448. [PubMed] [Google Scholar]
- Keller CD, Rivera Gil P, Tolle M, van der Giet M, Chun J, Radeke HH et al. (2007). Immunomodulator FTY720 induces myofibroblast differentiation via the lysophospholipid receptor S1P3 and Smad3 signaling. Am J Pathol 170: 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010) NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160:1577‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik JM, Schmouder R, Barilla D, Wang Y, Kraus G (2004). Single‐dose FTY720 pharmacokinetics, food effect, and pharmacological responses in healthy subjects. Br J Clin Pharmacol 57: 586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krosser S, Wolna P, Fischer TZ, Boschert U, Stoltz R, Zhou M et al. (2015). Effect of ceralifimod (ONO‐4641) on lymphocytes and cardiac function: Randomized, double‐blind placebo‐controlled trial with an open‐label fingolimod arm. J Clin Pharmacol 55: 1051–1060. [DOI] [PubMed] [Google Scholar]
- Lindebo Holm T, Poulsen SS, Markholst H, Reedtz‐Runge S (2012). Pharmacological Evaluation of the SCID T Cell Transfer Model of Colitis: As a Model of Crohn's Disease. Int J Inflam 2012: 412178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP et al. (2000). Edg‐1, the G protein‐coupled receptor for sphingosine‐1‐phosphate, is essential for vascular maturation. J Clin Invest 106: 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J et al. (2002). Alteration of lymphocyte trafficking by sphingosine‐1‐phosphate receptor agonists. Science 296: 346–349. [DOI] [PubMed] [Google Scholar]
- Martin R, Sospedra M (2014). Sphingosine‐1 phosphate and central nervous system. Curr Top Microbiol Immunol 378: 149–170. [DOI] [PubMed] [Google Scholar]
- Maxwell JR, Brown WA, Smith CL, Byrne FR, Viney JL (2009). Methods of inducing inflammatory bowel disease in mice. Curr Protoc Pharmacol Chapter 5: Unit5 58. doi: 10.1002/0471141755.ph0558s47 [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehling M, Brinkmann V, Antel J, Bar‐Or A, Goebels N, Vedrine C et al. (2008). FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology 71: 1261–1267. [DOI] [PubMed] [Google Scholar]
- Meno‐Tetang GM, Li H, Mis S, Pyszczynski N, Heining P, Lowe P et al. (2006). Physiologically based pharmacokinetic modeling of FTY720 (2‐amino‐2[2‐(−4‐octylphenyl)ethyl]propane‐1,3‐diol hydrochloride) in rats after oral and intravenous doses. Drug Metab Dispos 34: 1480–1487. [DOI] [PubMed] [Google Scholar]
- Miron VE, Jung CG, Kim HJ, Kennedy TE, Soliven B, Antel JP (2008). FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann Neurol 63: 61–71. [DOI] [PubMed] [Google Scholar]
- Mizushima T, Ito T, Kishi D, Kai Y, Tamagawa H, Nezu R et al. (2004). Therapeutic effects of a new lymphocyte homing reagent FTY720 in interleukin‐10 gene‐deficient mice with colitis. Inflamm Bowel Dis 10: 182–192. [DOI] [PubMed] [Google Scholar]
- Mullershausen F, Craveiro LM, Shin Y, Cortes‐Cros M, Bassilana F, Osinde M et al. (2007). Phosphorylated FTY720 promotes astrocyte migration through sphingosine‐1‐phosphate receptors. J Neurochem 102: 1151–1161. [DOI] [PubMed] [Google Scholar]
- Mullershausen F, Zecri F, Cetin C, Billich A, Guerini D, Seuwen K (2009). Persistent signaling induced by FTY720‐phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol 5: 428–434. [DOI] [PubMed] [Google Scholar]
- Okazaki H, Hirata D, Kamimura T, Sato H, Iwamoto M, Yoshio T et al. (2002). Effects of FTY720 in MRL‐lpr/lpr mice: therapeutic potential in systemic lupus erythematosus. J Rheumatol 29: 707–716. [PubMed] [Google Scholar]
- Olsson T, Boster A, Fernandez O, Freedman MS, Pozzilli C, Bach D et al. (2014). Oral ponesimod in relapsing–remitting multiple sclerosis: a randomised phase II trial. J Neurol Neurosurg Psychiatry 85: 1198–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo ML, Thangada S, Wu MT, Liu CH, Macdonald TL, Lynch KR et al. (2007). Immunosuppressive and anti‐angiogenic sphingosine 1‐phosphate receptor‐1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem 282: 9082–9089. [DOI] [PubMed] [Google Scholar]
- Osinde M, Mullershausen F, Dev KK (2007). Phosphorylated FTY720 stimulates ERK phosphorylation in astrocytes via S1P receptors. Neuropharmacology 52: 1210–1218. [DOI] [PubMed] [Google Scholar]
- Ostanin DV, Pavlick KP, Bharwani S, D'Souza D, Furr KL, Brown CM et al. (2006). T cell‐induced inflammation of the small and large intestine in immunodeficient mice. Am J Physiol Gastrointest Liver Physiol 290: G109–G119. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. NC‐IUPHAR(2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham TH, Okada T, Matloubian M, Lo CG, Cyster JG (2008). S1P1 receptor signaling overrides retention mediated by G alpha i‐coupled receptors to promote T cell egress. Immunity 28: 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmaj K, Li DK, Hartung HP, Hemmer B, Kappos L, Freedman MS et al. (2013). Siponimod for patients with relapsing–remitting multiple sclerosis (BOLD): an adaptive, dose‐ranging, randomised, phase 2 study. Lancet Neurol 12: 756–767. [DOI] [PubMed] [Google Scholar]
- Siarey R (2009). Gilenya (Fingolimod HCL) Pharmacology/Toxicology NDA Review and Evaluation.
- Soler D, Chapman T, Yang LL, Wyant T, Egan R, Fedyk ER (2009). The binding specificity and selective antagonism of vedolizumab, an anti‐alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases. J Pharmacol Exp Ther 330: 864–875. [DOI] [PubMed] [Google Scholar]
- Song J, Matsuda C, Kai Y, Nishida T, Nakajima K, Mizushima T et al. (2008). A novel sphingosine 1‐phosphate receptor agonist, 2‐amino‐2‐propanediol hydrochloride (KRP‐203), regulates chronic colitis in interleukin‐10 gene‐deficient mice. J Pharmacol Exp Ther 324: 276–283. [DOI] [PubMed] [Google Scholar]
- Thangada S, Khanna KM, Blaho VA, Oo ML, Im DS, Guo C et al. (2010). Cell‐surface residence of sphingosine 1‐phosphate receptor 1 on lymphocytes determines lymphocyte egress kinetics. J Exp Med 207: 1475–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaclavkova A, Chimenti S, Arenberger P, Hollo P, Sator PG, Burcklen M et al. (2014). Oral ponesimod in patients with chronic plaque psoriasis: a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet 384: 2036–2045. [DOI] [PubMed] [Google Scholar]
- van Doorn R, Nijland PG, Dekker N, Witte ME, Lopes‐Pinheiro MA, van het Hof B et al. (2012). Fingolimod attenuates ceramide‐induced blood–brain barrier dysfunction in multiple sclerosis by targeting reactive astrocytes. Acta Neuropathol 124: 397–410. [DOI] [PubMed] [Google Scholar]
- Wang F, Tan W, Guo D, He S (2007). Reduction of CD4 positive T cells and improvement of pathological changes of collagen‐induced arthritis by FTY720. Eur J Pharmacol 573: 230–240. [DOI] [PubMed] [Google Scholar]
- Xin C, Ren S, Eberhardt W, Pfeilschifter J, Huwiler A (2006). The immunomodulator FTY720 and its phosphorylated derivative activate the Smad signalling cascade and upregulate connective tissue growth factor and collagen type IV expression in renal mesangial cells. Br J Pharmacol 147: 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. HEK293 stable cell line expressing S1P1R‐GFP was incubated with 1 μM cycloheximide and a dose response of compound for 18 hrs. Cells were lysed in modified RIPA buffer containing Complete Protease Inhibitor cocktail (Roche). (A) Lysates were separated on 4‐12% SDS PAGE using 6M urea in the loading buffer, transferred to nitrocellulose membrane and blotted with rabbit anti‐GFP(Santa Cruz Biotechnology) and mouse anti‐HSP90 (BD Biosciences) antibodies, followed by Licor Odyssey imaging. (B) Normalized total S1P1‐GFP expression relative to HSP90.
Figure S2. HUVEC cells expressing endogenous S1P1R were used to show that the functional consequence of receptor internalization is loss of response to S1P1R agonist. Cells were preincubated for 1hr with 1 μM RPC1063, FTY720‐P or S1P to induce receptor internalization. Compound was washed out for 5hrs, and cells were re‐stimulated with an S1P1R agonist (FTY720‐P). pERK1/2 activation was assessed using rabbit anti‐Phospho‐ERK1/2 (Cell Signaling #4377) and normalized to mouse anti‐ERK1/2 (Cell Signaling #9107) by in cell western analysis and Licor Odyssey imaging. Data represents mean of triplicate data points. Error bars represent SEM.
Figure S3. C57Bl/6 mice were treated with vehicle or 1 mg kg−1 RPC1063 and blood was taken by cardiac puncture 6 hours later. Blood was incubated with antibodies to the indicated cell surface markers according to Materials and Methods. Data represents mean of three individual mice. Error bars represent SEM.
Supporting info item
Supporting info item