

Abstract
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a highly malignant inheritable cardiac channelopathy. The past decade and a half has provided exciting new discoveries elucidating the genetic etiology and pathophysiology of CPVT. This review of the current literature on CPVT aims to summarize the state of the art in our understanding of the genetic etiology and the molecular pathogenesis of CPVT, and how these relate to our current approach to diagnosis and management. We will also shed light on groundbreaking new work that will continue to refine the management of CPVT in the future. As our knowledge of CPVT continues to grow, further studies will yield a better understanding of the efficacy and pitfalls of established diagnostic approaches and therapies as well as help shape newer diagnostic and treatment strategies. Two separate searches were run on the National Center for Biotechnology Information's (NCBI) website. The first used the medical subject headings (MeSH) database using the term “catecholaminergic polymorphic ventricular tachycardia” that was run on the PubMed database using the age filter (birth to 18 years), and it yielded 58 results. The second search using the MeSH database with the search term “catecholaminergic polymorphic ventricular tachycardia,” applying no filters yielded 178 results. The abstracts of all these articles were studied and the articles were categorized and organized. Articles of relevance were read in full. As and where applicable, relevant references and citations from the primary articles were further explored and read in full.
Keywords: Bidirectional ventricular tachycardia (BDVT), catecholaminergic polymorphic ventricular tachycardia (CPVT), polymorphic ventricular tachycardia (PMVT)
INTRODUCTION
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a highly malignant inheritable cardiac channelopathy. As implied by its name, CPVT is associated with the occurrence of potentially life-threatening catecholamine-mediated ventricular arrhythmias (VAs) triggered by stress or exertion.
This review of the current literature on CPVT aims to summarize the state of the art in our understanding of the genetic etiology, the molecular pathogenesis of CPVT, and how these relate to our current approach to diagnosis and management. We will also shed light on groundbreaking new work that will continue to refine the management of CPVT in the future.
DISEASE BURDEN
A case of a 6-year-old girl with no structural abnormality of the heart presenting with bidirectional ventricular tachycardia (BDVT), apparently induced by effort and emotional stress, was first described in 1975.[1] The entity of CPVT, an adrenergic-dependent potentially lethal tachyarrhythmia with no underlying structural heart disease was delineated in a longitudinal case series in 1995.[2] At the time, it was hypothesized to be a possible variant of long QT syndrome with a normal baseline electrocardiogram (ECG).
CPVT is an uncommon condition, accounting for about 12% of autopsy-negative sudden deaths and 1.5% of sudden infant deaths.[3,4] The true prevalence is unknown though a possible prevalence of one in 10,000 has been quoted in the literature.[5,6]
An early study showed that the untreated mortality rate for children with CPVT was approximately 50%.[2] The potential lethality of this condition is further evidenced by studies that have demonstrated that 33-38% of patients presented with cardiac arrest.[7,8] Considering all cardiac events (syncope, aborted cardiac arrest, sudden death), the rate of occurrence is 54-58% in untreated individuals.[7,8,9] Even with treatment, the incidence of cardiac events is close to 30%.[8] More recent data demonstrated a mortality rate of up to 13% for patients undergoing treatment.[8,9]
GENETICS
The genetic basis of CPVT began to be described in 2001.[10,11] The most common genetic mutations responsible for CPVT occur in the gene that encodes the cardiac ryanodine receptor (RyR2).[11] These mutations are usually inherited as autosomal-dominant, and such CPVT cases are designated as CPVT1. Though over 130 RyR2 mutations have been reported, it is expected that approximately 65% of CPVT1 cases could be discovered by the analysis of 16 exons.[5,12]
Less commonly, CPVT is caused by mutations in the gene encoding cardiac calsequestrin (CASQ2), which is inherited as autosomal recessive and is designated CPVT2.[10] A rare third form of CPVT has been mapped to chromosome 7 (7p14-p22) but the underlying gene has not been discovered.[13] Absence of the sarcoplasmic reticulum protein triadin was associated with another rare form of CPVT.[14] Mutations in the gene encoding the potassium inward rectifying channel Kir2.1 (KCNJ2) associated with Anderson-Tawil syndrome have been shown to demonstrate a phenotype similar to CPVT.[15] Mutations in the genes encoding the calcium signaling protein calmodulin (CALM1) have recently been shown to be associated with CPVT.[16]
A number of studies have provided us with an insight into the prevalence of underlying genetic mutations. A comprehensive mutational analysis of RYR2 gene in patients diagnosed with CPVT or long QT syndrome (LQTS) with normal corrected QT (QTc) revealed 63 possible mutations in 47% of the patients.[12] This is consistent with multiple prevalence studies from Europe and Asia, which demonstrate RyR2 mutations in 47-70% of CPVT probands.[9,17,18,19] RyR2 mutations are also detected in 5-38% of the patients with a possible diagnosis of CPVT, and in 15% of asymptomatic relatives.[5,12]
A Japanese study revealed 2% compound heterozygous CASQ2.[19] In a study from France, 7% of CPVT probands were detected to have mutations in CASQ2.[9] Japanese studies have also demonstrated that 2-7% of the patients harbored KCNJ2 mutations.[15,19]
PATHOPHYSIOLOGY
Electrical depolarization leads to myocyte contraction through the process of calcium (Ca2+)-induced Ca2+ release. Ca2+ enters the sarcolemma through L type Ca2+ channels during phase 2 of the action potential, which in turn causes the release of Ca2+ from the sarcoplasmic reticulum (SR) via the cardiac ryanodine channel receptor [See Figure 1]. This exists in a complex with other proteins calsequestrin, triadin, and junctin at the SR junction. Released intracellular Ca2+ binds to troponin C and causes myocardial contraction. During the cardiomyocyte relaxation phase (diastole), Ca2+ release from the SR ceases and Ca2+ is returned to the SR via a Ca2+-ATPase pump, or pumped extracellularly by a sodium/calcium (Na+/Ca2+) exchange channel.[5,20,21]
Figure 1.

Diagram of the cardiomyocyte membrane complex demonstrating the ion channels and currents involved in calcium-induced calcium release and excitation-contraction coupling of the myofibrils
The key feature underlying pathogenesis of CPVT is the aberrant release of Ca2+ into the SR during diastole (termed a transient inward current) leading to diastolic Ca2+ leak, which provides a substrate for delayed afterdepolarizations (DADs), specifically in the setting of β-adrenergic stimulation during stress or exercise. Different hypotheses have been put forth to explain why mutations in the RyR2 gene cause abnormal gating of the ryanodine receptor. The result of these mutations is a gain-of-function in the ryanodine receptor leading to diastolic Ca2+ leak.[21]
Similarly, mutations in the gene encoding calsequestrin may lead to defective protein-protein interactions at the SR membrane complex.[5,6,21]
One of the characteristic electrocardiographic findings in CPVT is BDVT. This is thought to occur due to alternating foci of triggered activity in the right and left ventricles, specifically in the area of the Purkinje system.[20]
CLINICAL PRESENTATION
CPVT has traditionally been considered a childhood disease with most patients presenting with symptoms in the first two decades, with a median age at diagnosis of 15 ± 10 years.[9] However, a recent study suggests the existence of a bimodal distribution of symptom onset in 10-20 years and 32-48 years. The older age distribution had a larger proportion of females and most of them were RyR2-negative.[7,22] Based on these reports, it has been suggested that there may exist “juvenile” and “adult” types of CPVT.
The juvenile type presents in the first two decades of life, is associated with RyR2 mutation, has no gender difference, and has a greater risk of sudden cardiac death. The adult type presents at around 40 years, predominantly in females, is less likely to have RyR2 mutation, and has less risk of sudden cardiac death. It is hypothesized that adult-onset CPVT may be caused by an as yet undiscovered genetic defect, and/or may be associated with female hormonal effect.[7,22]
Unfortunately, CPVT has often been associated with a long delay between initial presentation and eventual diagnosis, up to 9 years in some studies.[23] This is likely due to normal ECG findings at the baseline and a historically low level of awareness about the condition among the general medical community. However, this delay has gradually shortened and most studies estimate a mean delay of about 2-3 years.[17,23] CPVT may also be misdiagnosed as other arrhythmias such as idiopathic ventricular fibrillation or long QT with a normal QT interval.[24]
CLINICAL ASSOCIATIONS
A number of clinical associations with CPVT have been reported in the literature.
Cardiac associations
Sinus node dysfunction has been noted to occur in patients with CPVT. Sinus bradycardia has been reported to occur in 19% and SVT in 16% of the patients with mutations in RyR2.[25] Atrial tachycardia has also been reported as the initial presentation in patients later diagnosed with CPVT.[26] Recent studies in mice models suggest that sinus node dysfunction and atrial tachycardias are concurrent defects caused by the underlying mutation.[27] It appears that the occurrence of VA increases as the sinus rate decreases. Atrial pacing was shown to be effective in preventing ventricular tachycardia. This apparently paradoxical finding is thought to be related to the effect of increased sinus rate on the balance between accelerated SR Ca2+ loading and the diastolic interval. With increasing sinus rates, the diastolic interval decreases, and this may prevent the SR from reaching a critical threshold for spontaneous firing, hence reducing the incidence of spontaneous Ca2+ release and DADs, causing a suppression of ventricular ectopy. Hence, the sinus node dysfunction prevalent in CPVT may contribute to ventricular ectopy by causing the diastolic interval to be relatively long when β-adrenergic stimulation occurs.[27,28]
CPVT has been reported in association with Takotsubo cardiomyopathy.[29] A series of 24 cases from Japan reported the frequent association between left ventricular noncompaction (LVNC) and RyR2 exon 3 deletion, suggesting that detection of this deletion in patients with LVNC may help in prognostication for such patients.[30]
Extracardiac associations
In some cases, CPVT may be misdiagnosed as seizure disorder or refractory epilepsy since patients may present with convulsive syncope. One study found that 15% of the persons carrying CPVT-causing mutations had seizures, with about half of them diagnosed with drug-resistant epilepsy.[9] In such situations, malignant arrhythmias including CPVT must be considered in the differential, especially when the seizures were associated with palpitations or documented tachycardia. Other “red flags” for CPVT presenting as seizures include occurrence with exercise or emotional stress, seizures nonresponsive to antiepileptic medications, a family history of sudden death before the age of 30 years, or family history of recurrent syncope.[31]
DIAGNOSIS
Patients with CPVT do not usually demonstrate ECG abnormalities at rest. The QT interval is not usually prolonged. Sinus bradycardia and supraventricular arrhythmias such as intermittent atrial ectopic tachycardia, sick sinus syndrome, and unspecified supraventricular tachycardia (SVT) are sometimes seen in patients with CPVT.[5,25,26,27]
Holter monitor recording, loop recorder monitoring, and implanted loop recorders may be useful in situations when exercise stress testing is either negative or cannot adequately be performed. These monitors can be diagnostic for patients in whom VT may be related to stress or emotions and not exercise-induced. They can also be helpful to further characterize associated SVT or sinus node dysfunction.[5]
The diagnosis of CPVT most often relies on the demonstration of VA with exercise stress testing though a negative exercise stress test does not exclude CPVT. The VAs often become more pronounced as the sinus rate increases with exercise into the 110-130 beats per minute range, often starting as premature ventricular complexes, progressing to ventricular couplets and triplets (often nonuniform QRS morphologies), and degrading into a ventricular tachycardia [See Figure 2]. The ventricular tachycardia is typically described as bidirectional VT (BDVT) with beat-to-beat alternation of the QRS axis and morphology although they may just appear to be polymorphic ventricular tachycardia (PMVT) [See Figure 3]. The arrhythmias gradually resolve as the exercise ends. Though the classic BDVT occurs in 35% of the probands, the progressive deterioration with increasing exercise is a more common finding.[5,6,32] Studies have revealed that the initial beat of VA comes frequently from the right or left outflow tract region, most frequently suggesting that they are triggered events.[6,7,33]
Figure 2.

Electrocardiogram during exercise stress testing demonstrates increasing frequency of ventricular arrhythmias, degrading from bigeminy to a typical bidirectional ventricular tachycardia
Figure 3.
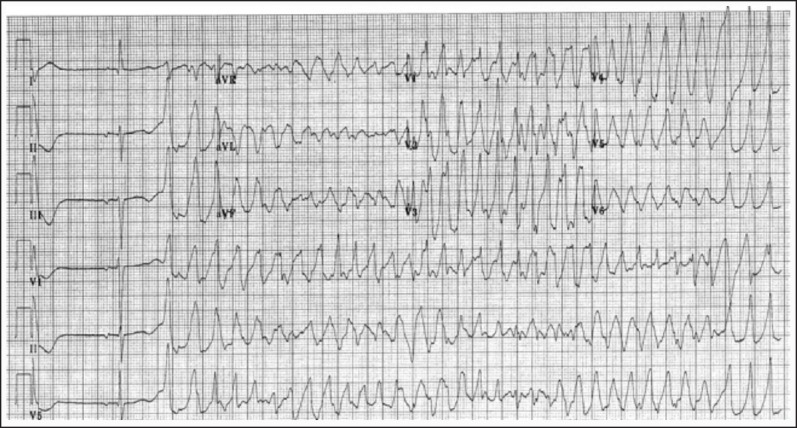
Electrocardiogram demonstrates polymorphic ventricular tachycardia
Factors that have been significantly associated with the failure of premature ventricular contractions (PVCs) to be suppressed with peak exertion during exercise stress testing include a family history of VT or sudden unexplained death (SUD) and the presence of VT on Holter monitor recordings. A history of syncope was not associated with similar exercise test findings.[34]
Due to the catecholamine dependent nature of CPVT, intravenous epinephrine infusion has been used to aid in the diagnosis. In one study of 27 patients with CPVT, exercise testing provoked VT in 63% of the patients and epinephrine in 82%.[7] Though it has been suggested that epinephrine infusion may be an alternative to exercise stress testing, a recent study from Finland demonstrated a sensitivity of 28% and specificity of 98% compared to exercise stress testing.[35]
Electrophysiological studies are not specifically indicated. Associated sinus node dysfunction and SVT can be identified and investigated. A small series of six patients demonstrated that postpacing marked QT prolongation in the first beat after cessation of ventricular pacing was associated with a single mutation in RyR2.[36]
Cardiac imaging including magnetic resonance imaging or computed tomography helps exclude structural diseases that may present similarly including coronary artery anomalies, hypertrophic cardiomyopathy, and arrhythmogenic right ventricular cardiomyopathy.
ASYMPTOMATIC RELATIVES
A growing body of data exist regarding the prevalence of genotypic and phenotypic abnormalities in the asymptomatic relatives of CPVT probands.
Studies have shown that genotype-positive relatives demonstrate a wide variety of phenotypes. In a study of 116 relatives carrying the RYR2 mutation, 50% had a CPVT phenotype on initial cardiac examination including 25% with nonsustained VT. Mutations in the C-terminus of the RYR2 domain had increased odds of nonsustained VT compared with relatives carrying mutations in the N terminus while 19% of the relatives demonstrated sinus bradycardia and 16% demonstrated SVT.[25] In a study from Italy, five out of nine asymptomatic gene carriers for RyR2 mutations in asymptomatic relatives of CPVT probands had exercise-induced arrhythmias on clinical evaluation.
In fact, exercise stress testing can be a useful tool for the evaluation of asymptomatic relatives of CPVT probands. In one study, 25% of the asymptomatic relatives of CPVT probands had positive stress tests, out of whom 95% tested positive for CPVT mutations compared with 32% of asymptomatic relatives who had negative stress tests.
THERAPY
A number of management options are available for CPVT. Traditionally, β-blockers have been the drug of choice and remain so today though new therapeutic options have also emerged.
β-BLOCKERS
Since β-adrenergic stimulation has been intimately connected with the pathophysiology of CPVT, β-blockade has been used to prevent VAs. A meta-analysis encompassing 11 studies looked at the efficacy of β-blocker therapy. Of the 403 patients, 88% received β-blocker therapy. Arrhythmic events in these studies occurred in 0-55% of the patients, with a 4-year event rate of 18.6% and an 8-year event rate of 37.2%. Of these arrhythmic events, 0-40% were near-fatal with a 4-year event rate of 7.7% and an 8-year event rate of 15.3% while 20% were fatal arrhythmic events, with a 4-year event rate of 3.2% and an 8-year event rate of 6.4%.[37] Though β-blocker dosages vary from study to study, there have been no significant differences detected when excluding studies with lower doses.[37]
Regarding the choice of β-blocker, recent studies suggest that nadolol (a nonselective β-blocker) may be more efficacious than metoprolol (a cardioselective β-blocker) with reduced incidence and severity of exercise-induced VAs in patients with CPVT as well as reduced arrhythmic window (the range of heart rates where arrhythmias may occur).[38] Nonselective β-blocker therapy may be preferable for patients without contraindications such as asthma. Regardless of the choice of β-blocker, it is apparent that compliance with drug therapy is extremely important, and that lack of compliance has been implicated in the occurrence of cardiac events in patients with CPVT receiving medical management.[37,39]
CALCIUM CHANNEL BLOCKERS
Calcium channel blockers (CCBs) have also been studied since they can block the influx of Ca2+ into the sarcolemma via the L-type Ca2+ channel. Verapamil use has been used in addition to β-blockade in a small case series of patients, with reduction in exercise-induced VAs.[9,40,41] Recent studies in mice with CASQ2 mutation have suggested that verapamil is most effective for VAs induced by exercise or epinephrine infusion,[42] and this effect has subsequently been demonstrated in a cohort of CASQ2 mutation-positive patients, suggesting a specific role for verapamil in CPVT2.[42]
SODIUM CHANNEL BLOCKERS
Following the seminal discovery by Watanabe et al. that flecainide (a class 1C antiarrhythmic) could block RyR2 in CASQ2 knockout mice, it was successfully used in patients with CPVT1 and CPVT2, reducing VA with exercise.[43] However, the actual mechanisms of Flecainide's action remain uncertain. A possible mechanism of action is that Na+ channel blockade leads to a reduction in the frequency of triggered activity.[44] Bannister et al. reported no direct action of flecainide on RyR2, instead proposing that sodium-dependent modulation of intracellular Ca2+ handling attenuates RyR2 dysfunction in CPVT.[45,46]
Flecainide has been further studied in clinical settings. Patients with CPVT1 received flecainide therapy due to exercise-induced VA despite conventional therapy and 76% demonstrated partial to complete VA suppression, with a dosing range of 100-300 mg.[47] During 12-40 months of follow-up (median 20 months), no arrhythmic event occurred though one patient with low flecainide levels received implantable cardiac defibrillator (ICD) shocks for polymorphic ventricular tachycardia (PVT). A cohort of 10 patients with CPVT2 also demonstrated a similar suppression of exercised-induced VT with the addition of flecainide to conventional therapy.[48] In 12 patients (genotype-negative CPVT) who were recalcitrant to conventional therapy, flecainide therapy reduced VA during exercise testing in eight patients.[49] Flecainide has also been reported to suppress defibrillator-induced storming (which is defined as >3 episodes of VT, VF, or appropriate ICD discharge in a 24-h period) in a patient with CPVT2 who was recalcitrant to other therapies.[50] The long-term efficacy of flecainide, optimal dosing, and its potential role as first-line therapy is still not clear.
IMPLANTABLE CARDIAC DEFIBRILLATOR
Based on the most recent guidelines of the 2006 American College of Cardiology (ACC)/American Heart Association (AHA)/European Society of Cardiology (ESC) guidelines for management of patients with VA and the prevention for sudden cardiac death, ICD placement is a class I recommendation in addition to β-blockade for CPVT patients who have survived a cardiac arrest, and class II a for patients with CPVT who have had syncope and/or documented sustained VT while on β-blockade.[51] Thus, ICDs have been the mainstay of therapy for patients with CPVT who have demonstrated a history concerning the high risk of sudden cardiac death, especially despite β-blocker therapy.
However, ICDs are not free of the risk of significant morbidity in the form of inappropriate shocks and induction of malignant arrhythmias secondary to shock leading to death. Additionally, some patients have died despite appropriate shocks. In a multicenter retrospective analysis of the efficacy of ICDs in pediatric patients with CPVT, 42% of the patients received appropriate shocks.[39] Only 57% of the appropriate shocks led to successful primary termination of VT. All the appropriate shocks were in response to VF and none were in response to PMVT or BDVT. It is speculated that this is due to difference in substrates, BDVT and PMVT arising from focal DADs, and catecholaminergic surges after ICD discharge could cause increased DADs. Inappropriate shocks were experienced by 46% of the patients due to supraventricular tachycardias or due to spontaneous resolution of arrhythmia before the ICD discharge. Overall, 36% of the patients who received shock underwent more malignant arrhythmias, and 29% had electrical storm. Of note, patients were noncompliant with medications at the time of 44% of ICD discharges.[39]
A larger retrospective study from Canada demonstrated appropriate shocks in 46% and inappropriate shocks in 22%. Arrhythmias were terminated with appropriate shock in 55% of the cases. Electrical storming occurred in 18% of the cases.[8]
A study looking at patients with CPVT2 found that none of the four patients with ICDs who received recurrent shocks without tachycardia termination degenerated to VF.[52]
LEFT CARDIAC SYMPATHETIC DENERVATION
Left cardiac sympathetic denervation (LCSD) is an operative procedure that aims to denervate the cardiac sympathetic supply. It has generally been used in cases that are symptomatic despite high dose β-blockade or in patients intolerant of high dose β blockade. Multiple reports of institutional experiences over the years have been published.[53,54,55,56] In general, LCSD has resulted in a marked reduction of arrhythmia burden. Significant complications have been rare. A large case series reported 1-year and 2-year event-free survivals of 87% and 81%, respectively.[53] LCSD has been performed in a minimally invasive fashion with videoscopic assistance, with 77% of the patients having a marked reduction in arrhythmias, 55% completely arrhythmia-free, and 27% nonresponders.[56] Potential complications include Horner's syndrome and pneumothorax.
CATHETER ABLATION
Successful radiofrequency ablation of the PMVT-triggering bidirectional PVCs originating from the Purkinje fibers has been reported in a patient with CPVT, suggesting a potential role for this form of therapy.[57]
EXERCISE THERAPY
A recent study demonstrated that patients with CPVT1 who underwent a 12-week high intensity ergometer bicycle exercise therapy (ET) program had higher threshold heart rates for VA (compared to pre-ET baseline) in comparison to a control group that did not undergo similar ET. This suggests a role for ET as an adjunctive treatment.[58]
SPORTS PARTICIPATION
As the greatest risk for cardiac events for patients with CPVT is during exercise, recommendations for sports participation for these patients have been developed by a number of organizations. The most recently published guidelines from the American Heart Association and the American College of Cardiology recommend that previously symptomatic athletes with CPVT or an asymptomatic CPVT athlete with exercise-induced premature ventricular contractions in bigeminy, couplets, or nonsustained ventricular tachycardia should not participate in competitive sports except for class I a sports (low dynamic and low static sports such as bowling, cricket, curling, golf, and yoga). Exceptions to this limitation should be made only after consultation with a CPVT specialist. Asymptomatic athletes who are genotype-positive for CPVT may participate in all competitive sports with appropriate precautionary measures including electrolyte replenishment, avoidance of dehydration for all, and acquisition of a personal automatic external defifibrillator as part of the athlete's personal sports safety gear and establishment of an emergency action plan with the appropriate school or team officials. An ICD should not be placed just to allow an individual with CPVT to participate in sports.[59]
APPROACH TO MANAGEMENT
Van der Werf et al. propose that all patients with CPVT, whether diagnosed phenotypically, genotypically, or both, should receive therapy. They should be advised against participation in competitive sports, and advised about the importance of medication compliance. β-blockers should be considered first-line therapy at the highest dose tolerated, preferably with nadolol. If β-blocker therapy alone is insufficient additional antiarrhythmic therapy in the form of flecainide (preferred) or verapamil should be used. In cases resistant to combination antiarrhythmic therapy, LCSD or ICD placement must be the next step. Though the ideal medication management after procedures is not clear, β-blocker therapy should be continued.[37]
NEW DEVELOPMENTS FROM THE BENCH
CPVT is an excellent example of a disease where developments from basic science and translational research have shaped our understanding of the disease pathology and affected our management in a profound manner. The world of bench research continues to provide new insights that will affect our management.
Roux Buisson et al. successfully found mutations in genes encoding triadin, a protein partner of RyR2 and CASQ2, in patients with otherwise negative genotype for CPVT.[14]
Autopsy studies have revealed the presence of T cell-mediated inflammation within the stellate (sympathetic) ganglia of patients of CPVT compared to healthy controls, suggesting that this may play a role in increasing adrenergic activity, resulting in VT in genetically susceptible individuals.[60]
Mouse models with CPVT specific mutations have been used to study CPVT. Studies by Katz et al. have demonstrated a specific role for verapamil in CASQ2 mutant mice and demonstrated similar effect in a cohort of human patients.[42] Faggioni et al. have demonstrated a putative protective role for overdrive pacing of the sinus node against VA.[28] α-adrenergic pathway has been identified as contributing to the pathogenesis of catecholamine-induced arrhythmia in mice with CPVT2 mutations, suggesting a possible role for α-adrenergic blockade in humans.[61] Katz et al. also demonstrated that mutant CASQ2 protein levels in certain mutations was inversely related to arrhythmia severity, and prevention of mutant protein degradation may be an avenue of therapy.[62]
The introduction of induced pluripotent stem cells (iPSCs) provided a means of studying patient- and disease-specific cardiac myocytes in vitro. Novak et al. demonstrated that patient-derived mutated cardiomyocytes could be used to study the underlying mechanisms of CPVT.[63] Di Pasquale et al. demonstrated that administration of drug KN-93 to inhibit calmodulin-dependent serine threonine kinase II (CAMKII) reduced the occurrence of DADs in cardiomyocytes carrying RyR2 gene mutation.[64] Jung et al. have demonstrated that dantrolene can reduce frequency and duration of Ca release events and rescue the arrhythmogenic phenotype in IPSCs with RyR2 mutation.[65]
Gene therapy using adeno-associated viral vectors (AAVs) has been reported as a means of cardiac gene transfer in a mouse model of CPVT. Gene transfer using AAV prevented arrhythmogenic phenotype in mutation-positive mice over 12 months. AAV was also shown to rescue the phenotype in adult mice, opening exciting new avenues of treatment in the future.[66,67]
CONCLUSION
The past decade and a half has provided exciting new discoveries elucidating the genetic etiology and pathophysiology of CPVT. Seminal research findings have been successfully translated from the bench to the bedside, shaping our understanding of this disease and guiding our approach to its diagnosis and treatment. As our knowledge of CPVT continues to grow, further studies will yield a better understanding of the efficacy and pitfalls of established diagnostic approaches and therapies as well as help shape newer diagnostic and treatment strategies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Reid DS, Tynan M, Braidwood L, Fitzgerald GR. Bidirectional tachycardia in a child. A study using his bundle electrography. Br Heart J. 1975;37:339–44. doi: 10.1136/hrt.37.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–9. doi: 10.1161/01.cir.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 3.Wilders R. Cardiac ion channelopathies and the sudden infant death syndrome. ISRN Cardiol 2012. 2012:846171. doi: 10.5402/2012/846171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ. Cardiac channel molecular autopsy: Insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin Proc. 2012;87:524–39. doi: 10.1016/j.mayocp.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Werf C, Wilde AA. Catecholaminergic polymorphic ventricular tachycardia: From bench to bedside. Heart. 2013;99:497–504. doi: 10.1136/heartjnl-2012-302033. [DOI] [PubMed] [Google Scholar]
- 6.Liu N, Ruan Y, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Prog Cardiovasc Dis. 2008;51:23–30. doi: 10.1016/j.pcad.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Sy RW, Gollob MH, Klein GJ, Yee R, Skanes AC, Gula LJ, et al. Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2011;8:864–71. doi: 10.1016/j.hrthm.2011.01.048. [DOI] [PubMed] [Google Scholar]
- 8.Roston TM, Vinocur JM, Maginot KR, Mohammed S, Salerno JC, Etheridge SP, et al. Catecholaminergic polymorphic ventricular tachycardia in children: Analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ Arrhythm Electrophysiol. 2015;8:633–42. doi: 10.1161/CIRCEP.114.002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayashi M, Denjoy I, Extramiana F, Maltret A, Buisson NR, Lupoglazoff JM, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009;119:2426–34. doi: 10.1161/CIRCULATIONAHA.108.829267. [DOI] [PubMed] [Google Scholar]
- 10.Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–84. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 12.Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP, et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: A comprehensive open reading frame mutational analysis. J Am Coll Cardiol. 2009;54:2065–74. doi: 10.1016/j.jacc.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhuiyan ZA, Hamdan MA, Shamsi ET, Postma AV, Mannens MM, Wilde AA, et al. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J Cardiovasc Electrophysiol. 2007;18:1060–6. doi: 10.1111/j.1540-8167.2007.00913.x. [DOI] [PubMed] [Google Scholar]
- 14.Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–67. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimura H, Zhou J, Kawamura M, Itoh H, Mizusawa Y, Ding WG, et al. Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet. 2012;5:344–53. doi: 10.1161/CIRCGENETICS.111.962316. [DOI] [PubMed] [Google Scholar]
- 16.Nyegaard M, Overgaard MT, Søndergaard MT, Vranas M, Behr ER, Hildebrandt LL, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–12. doi: 10.1016/j.ajhg.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 18.Andrsova I, Valaskova I, Kubus P, Vit P, Gaillyova R, Kadlecova J, et al. Clinical characteristics and mutational analysis of the RyR2 gene in seven Czech families with catecholaminergic polymorphic ventricular tachycardia. Pacing Clin Electrophysiol. 2012;35:798–803. doi: 10.1111/j.1540-8159.2012.03399.x. [DOI] [PubMed] [Google Scholar]
- 19.Kawamura M, Ohno S, Naiki N, Nagaoka I, Dochi K, Wang Q, et al. Genetic background of catecholaminergic polymorphic ventricular tachycardia in Japan. Circ J. 2013;77:1705–13. doi: 10.1253/circj.cj-12-1460. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe H, Minamino T. Genetics and mechanisms of catecholaminergic polymorphic ventricular tachycardia. Austin J Clin Cardiolog. 2014;1:1011. [Google Scholar]
- 21.Okuda S, Yano M. New data on catecholaminergic polymorphic ventricular tachycardia in Japan: From the bench to the bedside. Circ J. 2013;77:1684–6. doi: 10.1253/circj.cj-13-0623. [DOI] [PubMed] [Google Scholar]
- 22.Sumitomo N. Are there juvenile and adult types in patients with catecholaminergic polymorphic ventricular tachycardia? Heart Rhythm. 2011;8:872–3. doi: 10.1016/j.hrthm.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Kozlovski J, Ingles J, Connell V, Hunt L, McGaughran J, Turner C, et al. Delay to diagnosis amongst patients with catecholaminergic polymorphic ventricular tachycardia. Int J Cardiol. 2014;176:1402–4. doi: 10.1016/j.ijcard.2014.08.020. [DOI] [PubMed] [Google Scholar]
- 24.Siegers CE, Visser M, Loh P, van der Heijden JF, Hassink RJ. Catecholaminergic polymorphic ventricular tachycardia (CPVT) initially diagnosed as idiopathic ventricular fibrillation: The importance of thorough diagnostic work-up and follow-up. Int J Cardiol. 2014;177:e81–3. doi: 10.1016/j.ijcard.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 25.van der Werf C, Nederend I, Hofman N, van Geloven N, Ebink C, Frohn-Mulder IM, et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: Disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ Arrhythm Electrophysiol. 2012;5:748–56. doi: 10.1161/CIRCEP.112.970517. [DOI] [PubMed] [Google Scholar]
- 26.Lawrenz W, Krogmann ON, Wieczorek M. Complex atrial arrhythmias as first manifestation of catecholaminergic polymorphic ventricular tachycardia: An unusual course in a patient with a new mutation in ryanodine receptor type 2 gene. Cardiol Young. 2014;24:741–4. doi: 10.1017/S1047951113001091. [DOI] [PubMed] [Google Scholar]
- 27.Faggioni M, van der Werf C, Knollmann BC. Sinus node dysfunction in catecholaminergic polymorphic ventricular tachycardia: Risk factor and potential therapeutic target? Trends Cardiovasc Med. 2014;24:273–8. doi: 10.1016/j.tcm.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faggioni M, Hwang HS, van der Werf C, Nederend I, Kannankeril PJ, Wilde AA, et al. Accelerated sinus rhythm prevents catecholaminergic polymorphic ventricular tachycardia in mice and in patients. Circ Res. 2013;112:689–97. doi: 10.1161/CIRCRESAHA.111.300076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schimpf R, Meinhardt J, Borggrefe M, Haghi D. Catecholaminergic polymorphic ventricular tachycardia and midventricular Takotsubo cardiomyopathy: A novel association? Herzschrittmacherther Elektrophysiol. 2013;24:63–6. doi: 10.1007/s00399-013-0248-8. [DOI] [PubMed] [Google Scholar]
- 30.Ohno S, Omura M, Kawamura M, Kimura H, Itoh H, Makiyama T, et al. Exon 3 deletion of RYR2 encoding cardiac ryanodine receptor is associated with left ventricular non-compaction. Europace. 2014;16:1646–54. doi: 10.1093/europace/eut382. [DOI] [PubMed] [Google Scholar]
- 31.Hazle MA, Shellhaas RA, Bradley DJ, Dick M, 2nd, Lapage MJ. Arrhythmogenic channelopathy syndromes presenting as refractory epilepsy. Pediatr Neurol. 2013;49:134–7. doi: 10.1016/j.pediatrneurol.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 32.Bat T, Collins KK, Schaffer MS. Syncope during exercise: Just another benign vasovagal event? Curr Opin Pediatr. 2011;23:573–5. doi: 10.1097/MOP.0b013e32834980b6. [DOI] [PubMed] [Google Scholar]
- 33.Sumitomo N, Harada K, Nagashima M, Yasuda T, Nakamura Y, Aragaki Y, et al. Catecholaminergic polymorphic ventricular tachycardia: Electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. 2003;89:66–70. doi: 10.1136/heart.89.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson B, Xie L, Temple J, Octavio J, Srayyih M, Thacker D, et al. Predicting utility of exercise tests based on history/holter in patients with premature ventricular contractions. Pediatr Cardiol. 2015;36:214–8. doi: 10.1007/s00246-014-0988-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marjamaa A, Hiippala A, Arrhenius B, Lahtinen AM, Kontula K, Toivonen L, et al. Intravenous epinephrine infusion test in diagnosis of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol. 2012;23:194–9. doi: 10.1111/j.1540-8167.2011.02188.x. [DOI] [PubMed] [Google Scholar]
- 36.Nof E, Belhassen B, Arad M, Bhuiyan ZA, Antzelevitch C, Rosso R, et al. Postpacing abnormal repolarization in catecholaminergic polymorphic ventricular tachycardia associated with a mutation in the cardiac ryanodine receptor gene. Heart Rhythm. 2011;8:1546–52. doi: 10.1016/j.hrthm.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Werf C, Zwinderman AH, Wilde AA. Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: State of the art and future developments. Europace. 2012;14:175–83. doi: 10.1093/europace/eur277. [DOI] [PubMed] [Google Scholar]
- 38.Leren IS, Saberniak J, Majid E, Haland TF, Edvardsen T, Haugaa KH. Nadolol decreases the incidence and severity of ventricular arrhythmias during exercise stress testing compared with β1-selective β-blockers in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2016;13:433–40. doi: 10.1016/j.hrthm.2015.09.029. [DOI] [PubMed] [Google Scholar]
- 39.Miyake CY, Webster G, Czosek RJ, Kantoch MJ, Dubin AM, Avasarala K, et al. Efficacy of implantable cardioverter defibrillators in young patients with catecholaminergic polymorphic ventricular tachycardia: Success depends on substrate. Circ Arrhythm Electrophysiol. 2013;6:579–87. doi: 10.1161/CIRCEP.113.000170. [DOI] [PubMed] [Google Scholar]
- 40.Swan H, Laitinen P, Kontula K, Toivonen L. Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J Cardiovasc Electrophysiol. 2005;16:162–6. doi: 10.1046/j.1540-8167.2005.40516.x. [DOI] [PubMed] [Google Scholar]
- 41.Rosso R, Kalman JM, Rogowski O, Diamant S, Birger A, Biner S, et al. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:1149–54. doi: 10.1016/j.hrthm.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 42.Katz G, Khoury A, Kurtzwald E, Hochhauser E, Porat E, Shainberg A, et al. Optimizing catecholaminergic polymorphic ventricular tachycardia therapy in calsequestrin-mutant mice. Heart Rhythm. 2010;7:1676–82. doi: 10.1016/j.hrthm.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380–3. doi: 10.1038/nm.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gopinathannair R. Flecainide in CASQ2-mediated catecholaminergic polymorphic ventricular tachycardia: The gift that keeps on giving. Heart Rhythm. 2013;10:1676–7. doi: 10.1016/j.hrthm.2013.08.027. [DOI] [PubMed] [Google Scholar]
- 45.Bannister ML, Thomas NL, Sikkel MB, Mukherjee S, Maxwell C, MacLeod K, et al. The mechanism of flecainide action in CPVT does not involve a direct effect on RyR2. Circ Res. 2015;116:1324–35. doi: 10.1161/CIRCRESAHA.116.305347. [DOI] [PubMed] [Google Scholar]
- 46.Smith GL, MacQuaide N. The direct actions of flecainide on the human cardiac ryanodine receptor: Keeping open the debate on the mechanism of action of local anesthetics in CPVT. Circ Res. 2015;116:1284–6. doi: 10.1161/CIRCRESAHA.115.306298. [DOI] [PubMed] [Google Scholar]
- 47.van der Werf C, Kannankeril PJ, Sacher F, Krahn AD, Viskin S, Leenhardt A, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. 2011;57:2244–54. doi: 10.1016/j.jacc.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khoury A, Marai I, Suleiman M, Blich M, Lorber A, Gepstein L, et al. Flecainide therapy suppresses exercise-induced ventricular arrhythmias in patients with CASQ2-associated catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013;10:1671–5. doi: 10.1016/j.hrthm.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe H, van der Werf C, Roses-Noguer F, Adler A, Sumitomo N, Veltmann C, et al. Effects of flecainide on exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013;10:542–7. doi: 10.1016/j.hrthm.2012.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong RA, Rivera KK, Jittirat A, Choi JJ. Flecainide suppresses defibrillator-induced storming in catecholaminergic polymorphic ventricular tachycardia. Pacing Clin Electrophysiol. 2012;35:794–7. doi: 10.1111/j.1540-8159.2012.03421.x. [DOI] [PubMed] [Google Scholar]
- 51.Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. European Heart Rhythm Association; Heart Rhythm Society, American College of Cardiology; American Heart Association Task Force; European Society of Cardiology Committee for Practice Guidelines. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing committee to develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death) J Am Coll Cardiol. 2006;48:e247–346. doi: 10.1016/j.jacc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 52.Marai I, Khoury A, Suleiman M, Gepstein L, Blich M, Lorber A, et al. Importance of ventricular tachycardia storms not terminated by implantable cardioverter defibrillators shocks in patients with CASQ2 associated catecholaminergic polymorphic ventricular tachycardia. Am J Cardiol. 2012;110:72–6. doi: 10.1016/j.amjcard.2012.02.049. [DOI] [PubMed] [Google Scholar]
- 53.De Ferrari GM, Dusi V, Spazzolini C, Bos JM, Abrams DJ, Berul CI, et al. Clinical management of catecholaminergic polymorphic ventricular tachycardia: The role of left cardiac sympathetic denervation. Circulation. 2015;131:2185–93. doi: 10.1161/CIRCULATIONAHA.115.015731. [DOI] [PubMed] [Google Scholar]
- 54.Coleman MA, Bos JM, Johnson JN, Owen HJ, Deschamps C, Moir C, et al. Videoscopic left cardiac sympathetic denervation for patients with recurrent ventricular fibrillation/malignant ventricular arrhythmia syndromes besides congenital long-QT syndrome. Circ Arrhythm Electrophysiol. 2012;5:782–8. doi: 10.1161/CIRCEP.112.971754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schneider HE, Steinmetz M, Krause U, Kriebel T, Ruschewski W, Paul T. Left cardiac sympathetic denervation for the management of life-threatening ventricular tachyarrhythmias in young patients with catecholaminergic polymorphic ventricular tachycardia and long QT syndrome. Clin Res Cardiol. 2013;102:33–42. doi: 10.1007/s00392-012-0492-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hofferberth SC, Cecchin F, Loberman D, Fynn-Thompson F. Left thoracoscopic sympathectomy for cardiac denervation in patients with life-threatening ventricular arrhythmias. J Thorac Cardiovasc Surg. 2014;147:404–9. doi: 10.1016/j.jtcvs.2013.07.064. [DOI] [PubMed] [Google Scholar]
- 57.Kaneshiro T, Naruse Y, Nogami A, Tada H, Yoshida K, Sekiguchi Y, et al. Successful catheter ablation of bidirectional ventricular premature contractions triggering ventricular fibrillation in catecholaminergic polymorphic ventricular tachycardia with RyR2 mutation. Circ Arrhythm Electrophysiol. 2012;5:e14–7. doi: 10.1161/CIRCEP.111.966549. [DOI] [PubMed] [Google Scholar]
- 58.Manotheepan R, Saberniak J, Danielsen TK, Edvardsen T, Sjaastad I, Haugaa KH, et al. Effects of individualized exercise training in patients with catecholaminergic polymorphic ventricular tachycardia type 1. Am J Cardiol. 2014;113:1829–33. doi: 10.1016/j.amjcard.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 59.Ackerman MJ, Zipes DP, Kovacs RJ, Maron BJ. American Heart Association Electrocardiography and Arrhythmias Committee of the Council on Clinical Cardiology, Council on Cardiovascular Disease in the Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and the American College of Cardiology. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task force 10: The cardiac channelopathies: A scientific statement from the American Heart Association and American College of Cardiology. Circulation. 2015;132:e326–9. doi: 10.1161/CIR.0000000000000246. [DOI] [PubMed] [Google Scholar]
- 60.Rizzo S, Basso C, Troost D, Aronica E, Frigo AC, Driessen AH, et al. T-cell-mediated inflammatory activity in the stellate ganglia of patients with ion-channel disease and severe ventricular arrhythmias. Circ Arrhythm Electrophysiol. 2014;7:224–9. doi: 10.1161/CIRCEP.113.001184. [DOI] [PubMed] [Google Scholar]
- 61.Kurtzwald-Josefson E, Hochhauser E, Bogachenko K, Bogachenko K, Harun-Khun S, Katz G, et al. Alpha blockade potentiates CPVT therapy in calsequestrin-mutant mice. Heart Rhythm. 2014;11:1471–9. doi: 10.1016/j.hrthm.2014.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Katz G, Shainberg A, Hochhauser E, Kurtzwald-Josefson E, Issac A, El-Ani D, et al. The role of mutant protein level in autosomal recessive catecholamine dependent polymorphic ventricular tachycardia (CPVT2) Biochem Pharmacol. 2013;86:1576–83. doi: 10.1016/j.bcp.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Novak A, Barad L, Zeevi-Levin N, Shick R, Shtrichman R, Lorber A, et al. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to β-adrenergic stimulation. J Cell Mol Med. 2012;16:468–82. doi: 10.1111/j.1582-4934.2011.01476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Di Pasquale E, Lodola F, Miragoli M, Denegri M, Avelino-Cruz JE, Buonocore M, et al. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013;4:e843. doi: 10.1038/cddis.2013.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jung CB, Moretti A, Mederos y Schnitzler M, Iop L, Storch U, Bellin M, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012;4:180–91. doi: 10.1002/emmm.201100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hajjar RJ, Lyon AR. Gene therapy for the treatment of catecholaminergic polymorphic ventricular tachycardia. Circulation. 2014;129:2633–5. doi: 10.1161/CIRCULATIONAHA.114.010586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Denegri M, Bongianino R, Lodola F, Boncompagni S, De Giusti VC, Avelino-Cruz JE, et al. Single delivery of an adeno-associated viral construct to transfer the CASQ2 gene to knock-in mice affected by catecholaminergic polymorphic ventricular tachycardia is able to cure the disease from birth to advanced age. Circulation. 2014;129:2673–81. doi: 10.1161/CIRCULATIONAHA.113.006901. [DOI] [PubMed] [Google Scholar]