

Abstract
ALG9-CDG is one of the less frequently reported types of CDG. Here, we summarize the features of six patients with ALG9-CDG reported in the literature and report the features of four additional patients. The patients presented with drug-resistant infantile epilepsy, hypotonia, dysmorphic features, failure to thrive, global developmental disability, and skeletal dysplasia. One patient presented with nonimmune hydrops fetalis. A brain MRI revealed global atrophy with delayed myelination. Exome sequencing identified a novel homozygous mutation c.1075G>A, p.E359K of the ALG9 gene. The results of our analysis of these patients expand the knowledge of ALG9-CDG phenotype.
Keywords: ALG9-CDG, Congenital disorders of glycosylation, Epilepsy, Hydrops fetalis, Skeletal dysplasia, Whole exome sequencing
Introduction
Congenital disorders of glycosylation (CDG) are genetic disorders caused by defects in the synthesis of the glycans of glycoproteins as well as glycolipids. More than 80 inborn errors of metabolism have been described due to congenital defects in protein N-linked glycosylation. CDG affect multiple organ systems. The severity of symptoms is highly variable and shows a broad clinical spectrum with considerable overlap. An increasing number of disorders have been discovered using new techniques that combine glycobiology with next-generation sequencing (Jaeken 2013; Timal et al. 2012). The majority of CDG known to date represent defects of the dolichol-linked oligosaccharide assembly classified as CDG-I. In 2004, a novel form of CDG, ALG9-CDG, was described caused by a deficiency of alpha-1,2-mannosyltransferase due to a defect in the ALG9 gene (Frank et al. 2004). Here, we describe the clinical, biochemical, and molecular features of four new cases of ALG9-CDG.
Methods
We retrospectively reviewed the clinical and genetic data of the patients. HPLC analysis of serum transferrin isoforms was performed as described (Arndt et al. 2007).The ALG9 mutation was identified using the inborn errors of metabolism gene panel, part of the Mendeliome assay (Saudi Mendeliome Group 2015). Gene panel analysis consisted of removing all non-exonic and non-splice site variants, then utilizing population frequency from our in-house Saudi database to exclude benign polymorphisms. Once the patients were confirmed to have the mutation, the parents were tested. All four patients had a skeletal survey, a brain MRI, US of the abdomen and kidneys, an echocardiogram, and an EEG.
Results
We have evaluated four patients from a large consanguineous family (pedigree). The main clinical features are summarized in Table 1.
Table 1.
Clinical and molecular characteristics of reported and present patients with ALG9-CDG
| Patient (Frank et al. 2004) | Patient (Weinstein et al. 2005) | Patient (Vleugels et al. 2009) | Patients (3 patients) (Tham et al. 2015) | Patient 1 | Patient 2 | Patient 3 | Patient 4 (25 days old) | |
|---|---|---|---|---|---|---|---|---|
| Epilepsy | + | + | + | + | + | + | − | |
| Developmental disability | + | + | + | + | + | + | − | |
| Microcephaly | + | + | ? | + | + | + | − | |
| Dysmorphism | NA | + | + | + | + | + | + | + |
| Skeletal dysplasia | NA | NA | NA | Severe | Mild | Mild | Mild | + |
| Hepatomegaly | + | + | + | Mild | Mild | Mild | Mild | |
| Failure to thrive | NA | + | + | + | + | + | − | |
| Hydrops fetalis | − | − | − | − | − | − | − | + |
| Echocardiography | NA | Pericardial effusion | Pericardial effusion | CHD (1 patient): large ventricular septum defect and a double outlet right ventricle with anomalous outflow tracts No CHD in 2 patients |
Mild tricuspid regurgitation | Normal | Mild pericardial effusion | Atrial septal defect, mild dilatation of right ventricle |
| US kidneys | NA | Polycystic kidneys | NA | Polycystic kidneys | Normal | Normal | Normal | Normal |
| Brain MRI | NA | Cerebral and cerebellar atrophy, delayed myelination | Normal (CT) | Cerebral and cerebellar atrophy, delayed myelination | Cerebral and cerebellar atrophy, delayed myelination | Cerebral and cerebellar atrophy, delayed myelination | US head normal | |
| ALG9 mutation | c.1567G>A (p.E523K) | c.860A>G (p.Y286C) | c.860A>G (p.Y286C) | c.1173+2T>A (p.V340Afs*57) | c.1588G>A (p.E530K) | c.1588G>A (p.E530K) | c.1588G>A (p.E530K) | c.1588G>A (p.E530K) |
NA information not available, CHD congenital heart disease
Pease note that in Saudi Mendeliome Group (2015), there are three patients
The Mendeliome assay revealed a previously reported homozygous missense mutation (NM_024740.2:c.1588G>A:p.E530K) in the ALG9 gene. Sanger sequencing confirmed this mutation. The parents were tested and found to be heterozygous for the mutation.
The index case (IV:5) is a 6-year-old female. The mother mentioned decreased fetal movements during pregnancy. The patient was the product of a full-term normal vertex delivery with a birth weight of 3,170 g, length of 49 cm, and head circumference of 34 cm. Apgar score was 8 at 1 min and 9 at 5 min. She was diagnosed since birth to have hip dislocation, dysmorphic features, and congenital heart disease in the form of minor tricuspid regurgitation.
At the age of 4 months, the patient started to develop frequent seizure attacks in the form of frequent clusters of clonic and tonic movements with up-rolling of the eyes. She was frequently admitted for uncontrolled seizures and unexplained febrile episodes. The EEG showed bursts of spikes/sharp waves in the left parieto-central area with slow background activity in favor of epileptic encephalopathy. After the first 5 years of life, the patient became seizure-free with a normal EEG.
The patient showed global developmental disability. At the age of 6 years, she can sit without support, reach for objects, make incomprehensible sounds, turn her head toward a voice, and recognize her family.
On examination at the age of 6 years, all growth parameters were at the 5th percentile. She was fixing but not following. She was hypotonic and showed exaggerated deep tendon reflexes but without clonus.
Facial dysmorphism includes frontal bossing, depressed nasal bridge, low seated ears, large mouth, and hypertelorism. Other associated abnormalities were inverted widely spaced nipples, abnormal distribution of fat on the buttocks, cutis marmorata, cutis aplasia congenita, and broad thumbs (Fig. 1). No organomegaly was noted.
Fig. 1.
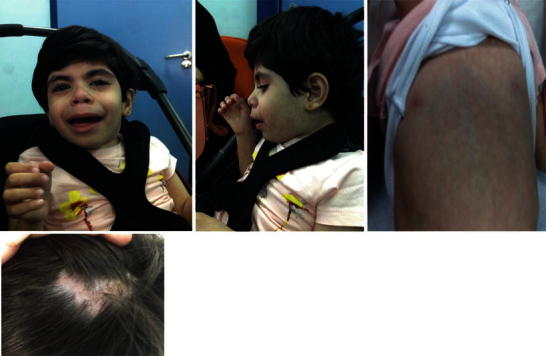
Photographs of the index case showing craniofacial dysmorphism (partial aplasia cutis congenita of the scalp, hypotelorism, broad base of nose, upturned nose, big mouth, prominent maxilla, large ears), inverted widely spaced nipples, abnormal distribution of fat at the buttocks, cutis marmorata, and broad thumbs
HPLC analysis of the transferrin isoforms showed a type 1 pattern (elevated diasialotransferrin (28%; reference value: 1.10 ± 0.72) and asialotransferrin (2%; reference value: not detectable). The activity of phosphomannomutase and lipid-linked oligosaccharides in fibroblasts were normal. The skeletal survey showed delayed bone age and mild skeletal dysplasia including mesomelic brachymelia with thickening of frontal and occipital bone, mild kyphosis of thoracolumbar spine, bilateral hip dislocation, round pelvis, brachycephaly, and shortening of greater sciatic notch. There are 11 ribs, symmetric with normal shape. The electroretinogram was normal. Brain MRI showed global cerebrum and cerebellar atrophy with delayed myelination.
The family history revealed three affected cousins (Fig. 2). Patients IV:8 and IV:13 have a similar presentation as patient IV:5 (Table 1). The only difference is that patient IV:8 has less severe developmental disability.
Fig. 2.
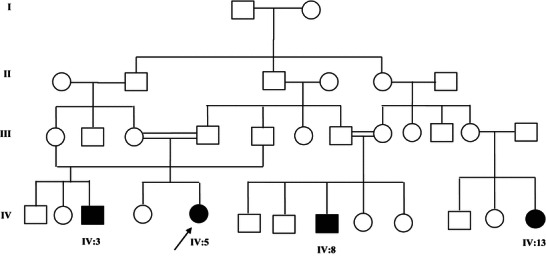
Pedigree of the family. The index case is indicated by an arrow
Patient IV:3 is a 25-day-old boy. He was diagnosed by fetal ultrasonography at 28 weeks of gestation with hydrops fetalis, which consisted of severe skin edema, pericardial effusion, and ascites. At 37 weeks of gestation, a cesarean section was performed because of breech presentation. Birth weight was 3,700 g (above 97th centile). Apgar scores were 7 at 1 min and 9 at 5 min. He had neck, feet, and skin edema. He had facial dysmorphism including a short nose, with a long philtrum, and a short neck. Echocardiography showed an atrial septal defect and mild dilatation of the right ventricle. Abdominal ultrasound was normal. Skeletal survey showed mild skeletal dysplasia. Cranial ultrasound showed wide subarachnoid spaces.
Discussion
So far only six patients with an ALG9-related phenotype have been reported in the literature (Frank et al. 2004; Tham et al. 2015; Vleugels et al. 2009; Weinstein et al. 2005). In this paper we report four more patients. Our patients’ phenotype, except patient IV:3, is similar to that of the patients described by Frank et al. (2004), Vleugels et al. (2009), and Weinstein et al. (2005). All of these patients, including our three, presented with a phenotype including infantile epileptic encephalopathy, progressive microcephaly, failure to thrive, and global developmental disability. Hepatomegaly, cardiac anomalies, mild skeletal dysplasia, and dysmorphism were observed in some patients. Hydrops fetalis was observed in only one patient. The three patients reported by Tham et al. had more severe phenotype (severe skeletal dysplasia, polycystic kidney, and multiple malformations) similar to that first reported by Gillessen-Kaesbach and Nishimura (Gillessen-Kaesbach et al. 1993; Nishimura et al. 1998). All of these patients died in utero. It is possible that the milder phenotypes of patients who were reported previously may be explained by a residual activity of the defective enzymes.
Our study confirms that skeletal dysplasia is a common symptom in ALG9-CDG syndrome, but this feature is also observed in other CDG including ALG3-CDG and ALG12-CDG (Lepais et al. 2015; Murali et al. 2014). Our study reports for the first time hydrops fetalis in ALG9-CDG. Also patients with PMM2-CDG and ALG8-CDG may present with hydrops fetalis (Höck et al. 2015; van de Kamp et al. 2007).
Although the number of patients is still small, there seems to be some phenotype-genotype correlation (Table 1). Patients with the most severe phenotype exhibit severe lethal skeletal dysplasia with visceral malformations. Their genotype is homozygous for a deleterious variant in the splice donor site of exon 10 in ALG9 (NG_009210.1:g. also denoted NM_024740.2:c.1173+2T>A). This variant results in skipping of exon 10 and leads to a shorter transcript. The other patients exhibit epilepsy and microcephaly, developmental disability, hepatomegaly, mild skeletal dysplasia, and cardiac anomalies. Interestingly, after an aggressive phase after 4–5 years with drug-resistant epilepsy and severe hypotonia, these patients become more stable and show some improvement in their milestones and epilepsy. Their genotype is a homozygous missense mutation.
In conclusion, the clinical and the biochemical findings described in these patients add to the spectrum of presentations of ALG9-CDG. Skeletal dysplasia, ranging in severity from mild to severe, appears to be a consistent feature. Other clinical features common to other forms of CDG are often associated including nonimmune hydrops fetalis.
Synopsis
The ALG9-CDG phenotype commonly includes skeletal dysplasia and may present with hydrops fetalis. A review of the literature suggests genotype-phenotype correlation.
Compliance with the Ethics Guidelines
Conflict of Interest
Sarah AlSubhi, Amal Hashem, Kalthoum Tlili, Saad AlShahwan, Dirk Lefeber, Fowzan S. Alkuraya, and Brahim Tabarki declare that they have no conflict of interest.
Details of the Contributions of Individual Authors
All authors (Sarah AlSubhi, Amal Hashem, Kalthoum Tlili, Saad AlShahwan, Dirk Lefeber, Fowzan S. Alkuraya, Brahim Tabarki) contributed equally in the planning, conducting, and reporting of the work described in the article.
Name of one author who serves as guarantor for the article, accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish: Brahim Tabarki
Details of Funding for All Research Studies
None.
“The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.”
Details of Ethics Approval
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki.
In the declaration of 1975, as revised in 2000, informed consent was obtained from the family for being included in the study.
A Patient Consent Statement
A consent form for the figures has been signed by the parents.
Footnotes
Competing interests: None declared
Contributor Information
Brahim Tabarki, Email: btabarki@hotmail.com.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Arndt T, Stanzel S, Sewell AC. Paediatric age-dependent serum transferrin isoform distribution studied by HPLC. Clin Lab. 2007;53:575–582. [PubMed] [Google Scholar]
- Frank CG, Grubenmann CE, Eyaid W, Berger EG, Aebi M, Hennet T. Identification and functional analysis of a defect in the human ALG9 gene: definition of congenital disorder of glycosylation type IL. Am J Hum Genet. 2004;75:146–150. doi: 10.1086/422367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillessen-Kaesbach G, Meinecke P, Garrett C, et al. New autosomal recessive lethal disorder with polycystic kidneys type Potter I, characteristic face, microcephaly, brachymelia, and congenital heart defects. Am J Med Genet. 1993;45:511–518. doi: 10.1002/ajmg.1320450422. [DOI] [PubMed] [Google Scholar]
- Höck M, Wegleiter K, Ralser E et al (2015) ALG8-CDG: novel patients and review of the literature. Orphanet J Rare Dis 10(1):73 [DOI] [PMC free article] [PubMed]
- Jaeken J. Congenital disorders of glycosylation. Handb Clin Neurol. 2013;113:1737–1743. doi: 10.1016/B978-0-444-59565-2.00044-7. [DOI] [PubMed] [Google Scholar]
- Lepais L, Cheillan D, Collardeau Frachon S et al (2015) ALG3-CDG: Report of two siblings with antenatal features carrying homozygous p.Gly96Arg mutation. Am J Med Genet A. doi:10.1002/ajmg.a.37232 [DOI] [PubMed]
- Murali C, Lu JT, Jain M, et al. Diagnosis of ALG12-CDG by exome sequencing in a case of severe skeletal dysplasia. Mol Genet Metab Rep. 2014;1:213–219. doi: 10.1016/j.ymgmr.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura G, Nakayama M, Fuke Y, et al. A lethal osteochondrodysplasia with mesomelic brachymelia, round pelvis, and congenital hepatic fibrosis: two siblings born to consanguineous parents. Pediatr Radiol. 1998;28:43–47. doi: 10.1007/s002470050289. [DOI] [PubMed] [Google Scholar]
- Saudi Mendeliome Group Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol. 2015;16(1):134. doi: 10.1186/s13059-015-0693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham E, Eklund EA, Hammarsjö A, et al. A novel phenotype in N-glycosylation disorders: Gillessen–Kaesbach–Nishimura skeletal dysplasia due to pathogenic variants in ALG9. Eur J Hum Genet. 2015 doi: 10.1038/ejhg.2015.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timal S, Hoischen A, Lehle L et al (2012) Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum Mol Genet 21:4151–4161 [DOI] [PubMed]
- van de Kamp JM, Lefeber DJ, Ruijter GJ et al (2007) Congenital disorder of glycosylation type Ia presenting with hydrops fetalis. J Med Genet 44:277–280 [DOI] [PMC free article] [PubMed]
- Vleugels W, Keldermans L, Jaeken J, et al. Quality control of glycoproteins bearing truncated glycans in an ALG9-defective (CDG-IL) patient. Glycobiology. 2009;19:910–917. doi: 10.1093/glycob/cwp067. [DOI] [PubMed] [Google Scholar]
- Weinstein M, Schollen E, Matthijs G, et al. CDG-IL: an infant with a novel mutation in the ALG9 gene and additional phenotypic features. Am J Med Genet. 2005;136A:194–197. doi: 10.1002/ajmg.a.30851. [DOI] [PubMed] [Google Scholar]