

Abstract
Live cell imaging requires bright photostable dyes that can target intracellular organelles and proteins with high specificity in a no-wash protocol. Organic dyes possess the desired photochemical properties and can be covalently linked to various protein tags. The currently available fluorogenic dyes are in the green/yellow range where there is high cellular autofluorescence and the near-infrared (NIR) dyes need to be washed out. Protein-mediated activation of far-red fluorogenic dyes has the potential to address these challenges because the cell-permeant dye is small and nonfluorescent until bound to its activating protein, and this binding is rapid. In this study, three single chain variable fragment (scFv)-derived fluorogen activating proteins (FAPs), which activate far-red emitting fluorogens, were evaluated for targeting, brightness, and photostability in the cytosol, nucleus, mitochondria, peroxisomes, and endoplasmic reticulum with a cell-permeant malachite green analog in cultured mammalian cells. Efficient labeling was achieved within 20–30 min for each protein upon the addition of nM concentrations of dye, producing a signal that colocalized significantly with a linked mCerulean3 (mCer3) fluorescent protein and organelle specific dyes but showed divergent photostability and brightness properties dependent on the FAP. These FAPs and the ester of malachite green dye (MGe) can be used as specific, rapid, and wash-free labels for intracellular sites in live cells with far-red excitation and emission properties, useful in a variety of multicolor experiments.
Understanding of the dynamics of cellular proteins and organelles became possible with the discovery of fluorescent proteins (reviewed by S. J. Remington)1 and their application to live cell imaging2 as reporters and fusion proteins localized to intracellular organelles. Fluorescent proteins (FPs) are now available in a range of colors from blue to red and recently to the near-infrared;3 there are a set of photoconvertable, photoactivated, and phototoxic FPs. FPs can be split into two pieces to monitor protein–protein interactions; can be modified to act as Ca2+, pH, and redox sensors; and when unnatural amino acids are incorporated into the chromophore, function as specific hydrogen sulfide detectors.4 In spite of these considerable advances, these proteins have limitations. Photostability and brightness of FPs is typically lower than spectrally similar dye molecules, and chromophore maturation is a post-translational process that results in a long maturation time for most FPs. In addition, many proteins show photoactivation or photoconversion properties upon imaging that can result in complex effects in multicolor experiments.5 The cysteines of FPs can form unwanted disulfide bonds in oxidizing environments and suffer from misfolding so that cysteine-free versions have been developed.6
An alternative to genetically encoded fluorescent proteins is systems where proteins specifically attach organic dyes. Organic dyes are brighter than FPs, and the chemistry can be tailored for specific applications while preserving binding to the target protein. Such systems include SNAP/CLIP-tag,7,8 Halo-Tag,9,10 TMP-tag,11,12 LAP,13,14 Oligo-Asp tag,15 coiled-coil tag,16−18 and BL-tag.19−21 SNAP/CLIP and Halo tags now have far-red dye alternatives that function in the cell.22 A drawback of all of these methods is that washing is usually required to remove the unbound, intrinsically fluorescent dye.
Fluorogens are fluorescence generating dyes that are dark until constrained, bound or activated, ensuring that unreacted dye produces a minimal background signal. Fluorescence is produced only when the dye is bound or modified by the target, providing temporal control of the signal based on the addition of dye (e.g., before or after drug addition). Spatial control of the signal is achieved by fusing the activating protein to a specific protein or peptide that targets an organelle. Targeted fluorogenic signals have been demonstrated using FlAsH/ReAsH,23,24 SNAP-tag,22,25−27 and BL-tag28 systems; however each of these techniques requires long incubation times or washing. Recently, a method called PYP-tag exploited the environmental sensitivity of a coumarin-based fluorogen for the covalent labeling of cytosolic proteins within several minutes without washing.29 For actin or tubulin, live cell labeling can be achieved using SiR-actin or SiR-tubulin.30 For membrane proteins, SNAP-tag anchoring of the solvatochromic membrane dye Nile Red achieved fluorogenic labeling by proximal activation of the dye in the plasma membrane.31
Another promising method utilizes specifically selected Fluorogen Activating Proteins (FAPs) that bind and activate otherwise nonfluorescent dyes.32 FAPs are derived from a single chain variable fragment (scFv) scaffold and were selected to bind and activate fluorogenic dyes including various cyanine33 and triarylmethane dyes. Fluorogens derived from the malachite green chromophore are highly fluorogenic, emit in the far-red spectral region, and display low nonspecific activation in living cells, even when used in a cell-permeant form.32 With maximum excitation and emission wavelengths in the far-red (e.g., ex 636 nm/em 664 nm), phototoxicity and autofluorescence are minimal; the signal is spectrally distinct from FPs for multicolor experiments, and for future applications, longer wavelengths show decreased scatter and allow for improved tissue penetration.
Previously, these fluoromodules, combined with cell impermeant and cell-permeant dyes have been used to study the transport of membrane proteins to and from the cell surface and in secretory compartments, but applications within living cells have been limited to actin labeling.34,35 The scFv-derived FAPs may not fold properly in the reducing environment of the cytosol due to the presence of disulfide bonds within each variable domain, and the cell-permeant dye may partition within different subcellular organelles, preventing effective labeling in some cellular contexts. In this study, we compared three different FAPs: dL5**, a synthetic dimer of a light chain36 with a disulfide forming pair of cysteines in each monomer; dH6.2, a synthetic dimer derived from a heavy chain with the second cysteine changed to alanine in each monomer;34,35 and p13-CW, a classic heavy-light scFv with the second cysteine in each domain changed to an alanine.37 In order to characterize the cell permeability, binding, and activation of the cell-permeant malachite green ester (MGe) fluorogen, each FAP was expressed as an mCer3 fusion protein in five different subcellular compartments in mammalian cells; the cytosol, nucleus, mitochondria, peroxisomes, and endoplasmic reticulum. Targeting and colocalization with organelle specific dyes, photostability, and rate of fluorescence activation were measured by confocal microscopy while relative brightness and optimal dye concentrations were characterized by flow cytometry using no wash protocols and submicromolar dye concentrations. This study shows that these new genetically encoded FAPs can be targeted to the cytosol and to organelles within the cytosol and when localized can function to rapidly activate the highly fluorogenic far-red emitting MGe dye with no nonspecific activation of the dye. All organelles produced a photostable, nonphototoxic signal using a no-wash method.
Results and Discussion
The fluorescent signal arises from a FAP-fluorogen complex only when both components are present at the same site and in functional form (e.g., properly folded protein, unmodified dye). Because of this two-component nature, it is important to understand both the access of the dye to a compartment and the function of the FAP in a particular subcellular environment. To assess these properties, we prepared a series of FAP-mCer338 linked proteins targeted to subcellular sites (Figure 1). The mCer3 is intrinsically fluorescent and spectrally well resolved from the MG-binding FAP signal and was used to independently confirm transfection success and protein targeting. Differences in FAP function or dye accessibility would reduce the MGe labeling signal relative to the mCer3 signal. The images of these reporters and colocalization with cell compartment markers allowed us to clearly demonstrate that FAPs can be expressed and targeted and function intracellularly. In addition, the MGe fluorogen can enter each of these cytoplasmic organelles and be activated by binding to a properly folded FAP. The disulfide containing dL5** FAP and disulfide-free dH6.2 and p13-CW FAPs were evaluated in the reducing environment of the cytosol and nucleus and the more oxidizing environments of the endoplasmic reticulum, peroxisomes, and the mitochondrial matrix.
Figure 1.

Diagram showing the cloning of targeting elements, FAPs and mCer3. All components were cloned as modules, except SKL was added into the reverse primer.
Targeting and Localization in Transiently Transfected HEK 293 Cells
As shown in confocal images in Figure 2, MGe (red) colabeled with mCer3 (green) for all three FAPs. The dL5** protein was then colocalized with SYTO, MitoTracker, ERTracker, and BODIPY-AKL. These results indicate that each FAP was properly targeted and folded and then activated the fluorogen in subcellular compartments of varying local environments (Supporting Information Figure S1). In most compartments, a low concentration of the MGe (25 nM) was sufficient to label the FAPs for imaging, whereas 50 nM and 175 nM of the MGe were needed to produce adequate fluorescent signals for the dH6.2 and p13-CW FAPs, respectively, in the cytosol, perhaps the result of higher Kd’s of these FAPs. The heterogeneous expression levels observed in these images are a common result of transient transfections. The pBABE vector uses the Moloney Murine Leukemia Virus (MMLV) promoter to express proteins at a moderate level, chosen to reduce import saturation that sometimes results from overexpression. Under these conditions, each FAP was properly targeted and folded, and the MGe dye was able to penetrate the cellular compartment to achieve effective labeling at moderate dye concentrations. Colocalization with the compartment specific markers showed correct localization, quantitatively assessed using Pearson’s colocalization correlation analysis.39
Figure 2.
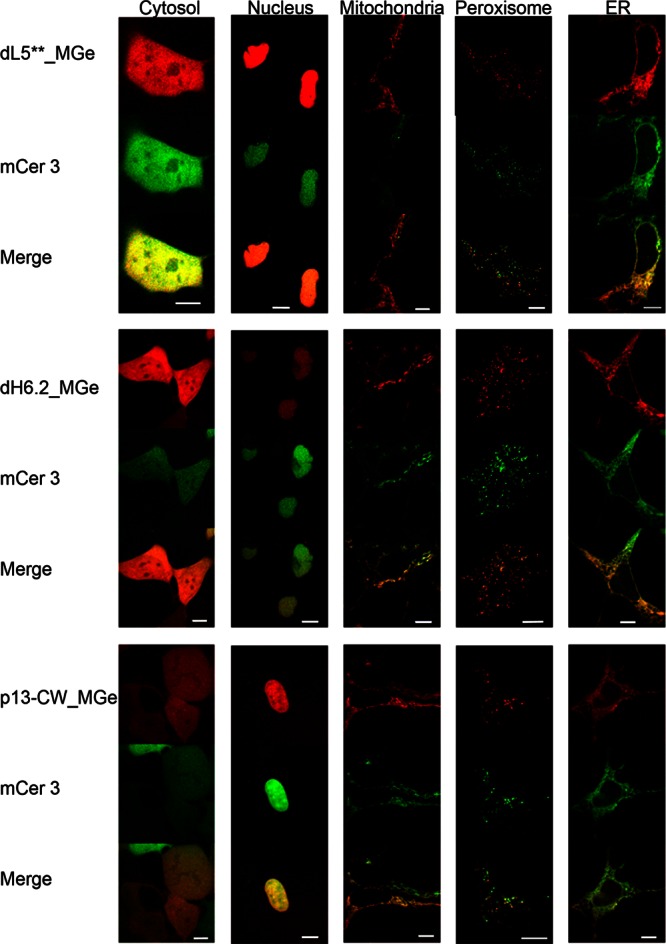
Confocal images showing the fluorescence pattern following expression of three different FAP-mCer3 tandems (side labels) targeted to five different cellular locations (top label). Signal from FAP_MGe (red) colocalizes with that of mCer3 (green), resulting in a yellow color in the merged images. Scale bars are 10 μm.
Expression from a plasmid encoding the fusion protein with no targeting sequence results in cytosolic synthesis and release of freely diffusing FAP-mCer3 protein in the cell. These proteins are not directed to an organelle, so they remain in the cytosol and diffuse through the 10–20 nm opening of the nuclear pore,40 resulting in a homogeneous fluorescent signal of the cytosol and nucleoplasm (cytosol in Figure 2). The FAPs employed here are 2–3 nm in diameter,36 and FPs are 2.5 × 4 nm.1 Therefore these easily diffuse in and out of the nucleus because they are below the size that is restricted by the nuclear pore (∼60–70 kDa).
The nuclear targeting sequence from the Mak16p protein,41 a protein involved in ribosome assembly, was fused to the N-terminus of the FAP-mCer3 protein. Although it is smaller than the commonly used SV40 nuclear localization signal (NLS), it efficiently targets to and retains the FAP-mCer3 proteins in the nucleus (Nucleus in Figure 2). Each FAP_MGe consistently showed a nuclear signal in both channels and colocalization with SYTO nuclear stain (Supporting Information Figure S1).
Peroxisome targeting was achieved by the addition to the C-terminus of an SKL tripeptide, the peroxisome transport signal, PTS1. Proteins synthesized on cytosolic ribosomes and folded in the cytosolic environment are transported through an import pore without unfolding.42 The fluorescence signal from these organelles (Peroxisome in Figure 2) is the brightest due to the concentration of all translated protein in the small volumes of the peroxisomes. BODIPY-AKL conjugates,43 a proven peroxisomal marker, colocalized with the mCer3 and FAP_MGe signal, confirming organelle identity (Supporting Information Figure S1).
Initial experiments targeting the mitochondria with a mitochondrial signal peptide produced mixed results. When amino acids 1–22 of COX IV44 were used, inconsistent localization was observed. Transient expression showed that the fusions were present in the mitochondria and could be labeled properly (Mitochondria in Figure 2), but selection to stable cell populations with puromycin or fluorescence activated cell sorting resulted in cytosolic localization. These results were observed in both the FAP_MGe and mCer3 channels, indicating that the protein transport into the mitochondria was perturbed and that it was not simply due to failure of the dye to penetrate the mitochondria. These observations could be the result of stable folding of the tandem protein prior to import, leader sequence blocking of import,45,46 puromycin blocking of import,47 misfolding upon import, intermembrane space oxidation/reduction,48 or collapse of the membrane potential.49 Previous studies have shown that when this signal peptide alone was used from 15 to 100 μM, it caused mitochondria to swell, decreased membrane potential, and released cytochrome c.50 The mitochondrial targeting sequence was then modified by the addition of the COX VIII signal sequence after the COX IV sequence, which resulted in consistent mitochondrial localization and no detectable residual cytoplasmic signal, even after selection to stably expressing cell populations (Supporting Information Figure S1).
Synthesis of endoplasmic reticulum (ER) proteins begins on free cytosolic ribosomes, revealing a nascent signal peptide that is rapidly recognized by the signal recognition particle (SRP), stalling further synthesis and delivering the ribosome to the SRP receptor for completion of synthesis and insertion into the oxidizing ER lumen through the translocon. The murine Ig-kappa leader sequence serves as the signal peptide in this study, and the ER retention signal tetrapeptide KDEL was added to the C-terminus of the proteins to keep the ER-soluble FAP-mCer3 resident in the early ER (ER in Figure 2). The FAPs and mCer colocalized with ER Tracker (Supporting Information Figure S1).
Fluorescent Brightness and Photostability
The photobleaching rate of the FAP_MGe and mCer3 signal was studied by time-lapse imaging over 300 sequential acquisitions. The remaining fluorescent intensity of the last image (f300) relative to the first image (f1) was calculated and expressed as a percentage of signal retained (Table 1). The mCer3 fluorescence was bleached consistently across all compartments, retaining a signal between 65 and 80% (mean ± SD = 73.13 ± 3.85) of the initial levels, and was insensitive to the presence of MGe bound to the associated FAP. In contrast, the FAPs, in the presence of excess dye, differed from each other and between compartments. The most generally photostable FAP was p13-CW (91.20 ± 2.78), then dL5** (73.20 ± 19.14), and the least was dH6.2 (40.60 + 16.71). No phototoxicity induced changes in cell morphology were observed in DIC images (Supporting Information Figure S2) at the end of the 300-frame timelapse (3 min).
Table 1. Percentage of Fluorescence Intensity Remaining in the 300th Image (f300) Compared to the First Image (f1) in a ∼3 min Timelapse (n: Number of Images Analyzed).
| FAP signal: MGe in bath | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cytosol | n | nucleus | n | mitochondria | n | peroxisome | n | ER | n | |
| dL5** | 89 ± 4 | 4 | 51 ± 2 | 3 | 63 ± 8 | 8 | 66 ± 2 | 3 | 97 ± 4 | 4 |
| dH6.2 | 23 ± 20 | 7 | 68 ± 11 | 8 | 41 ± 7 | 3 | 34 ± 10 | 3 | 37 ± 13 | 8 |
| p13-CW | 89 ± 4 | 4 | 90 ± 4 | 4 | 90 ± 2 | 4 | 96 ± 3 | 3 | 91 ± 9 | 4 |
| mCer3
signal: no MGe in bath | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cytosol | n | nucleus | n | mitochondria | n | peroxisome | n | ER | n | |
| dL5** | 64 ± 10 | 3 | 71 ± 3 | 4 | 70 ± 3 | 3 | 76 ± 11 | 3 | 72 ± 2 | 4 |
| dH6.2 | 75 ± 4 | 6 | 72 ± 3 | 6 | 77 ± 1 | 3 | 81 ± 12 | 4 | 73 ± 4 | 6 |
| p13-CW | 76 ± 4 | 3 | 71 ± 2 | 2 | 72 ± 3 | 3 | 75 ± 16 | 2 | 72 ± 6 | 4 |
After initial imaging experiments of transient transfections, cells were selected with puromycin, followed by fluorescence activated cell sorting for mCer3 positive cells. These cells were collected and expanded to produce the population for subsequent analysis. These stably expressing populations were analyzed for relative signals and dye loading rates by flow cytometry and time-lapse image analysis upon addition of the MGe fluorogen to the media. Figure 3 shows the flow-cytometry determined relative signal levels of the various organelle-targeted FAPs in the MGe channel (A) and the mCer3 channel (B), normalized to the highest signal in each panel, for each subcellular compartment. Across the compartments, the dL5** and dH6.2 are similarly bright, with the exception of the peroxisomes. The mCer3 signal levels are consistently high with the dL5** fusion, suggesting that there may be some expression defect with the other constructs, perhaps due to improper folding of the FAP and degradation of the fusion protein in different compartments. Table 2 shows the fold activation for the optimal staining conditions. Across many compartments, the dL5** shows both high-level expression and high fluorescence activation (∼100–900-fold compared to autofluorescence of cells), while expression level and relative brightness of the dH6.2 and the p13-CW constructs are more heterogeneous.
Figure 3.

Flow cytometric analysis of stably expressing FAP-mCer3 HEK cells in different organelles and untransfected HEK cells. (A) Bar graph representation showing the median fluorescence intensity (MFI) of dL5**, dH6.2, and p13-CW labeled with 200 nM MGe in different organelles. For each organelle, the MFI of FAP_MGe was normalized to the maximum fluorescence signal across the various clones. (B) Expression level of mCerulean3 (represented in median fluorescence intensity) was analyzed from unlabeled FAP expressing HEK cells. The MFI for mCer3 expression was normalized to the maximum in each organelle across the various clones. Cells with no FAP-mCer3 expression (untransfected HEK cells) labeled with 200 nM MGe (A) or without (B) showed no fluorescence signal in either channel. Error bars are SD of independent duplicate measurements on stable selected cell populations.
Table 2. Optimal Fold-activation Determined by Flow Cytometrya.
| cytoplasm | nucleus | peroxisomes | ER | |
|---|---|---|---|---|
| mCer3 | 44 (0) | 29 (0) | 55 (0) | 60 (0) |
| dL5** | 560 (500) | 720 (100) | 120 (100) | 920 (500) |
| dH6.2 | 760 (100) | 550 (100) | 300 (100) | 430 (500) |
| P13-CW | N.D. | 60 (500) | 79 (100) | 150 (500) |
Data show the fold activation relative to untransfected HEK293 cells labelled with the same MGe concentration (100, 200, or 500 nM, as indicated in parentheses). The optimal degree of activation was measured by comparing median fluorescent intensities of cell populations, in independent duplicate experiments, analyzing >20 000 cells per experiment.
The p13-CW FAP was photostable, but signal levels were inconsistent across multiple compartments compared to dL5**. In contrast, the dH6.2 was generally bright but considerably less photostable across multiple compartments. This property has proven useful for super-resolution imaging in previous studies35 but may not be ideal for conventional cellular microscopy. For general use within cells, the dL5** FAP appears to have the most consistent properties. In addition, this protein performs very well at the cell surface,36 providing a single FAP clone that can be used successfully across all cellular locations with a cell-permeant MG-based fluorogen. The origin of the low labeling level of the peroxisomes with the dL5** clone specifically may be an indication of sensitivity of the FAP to the particular conditions of this organelle and will be the subject of additional studies. Given the effective labeling of the dH6.2 peroxisomal construct, it cannot be explained by poor dye access in this compartment.
No-wash Labeling Dynamics
The rate of dye loading was analyzed by microscopy. A first evaluation of nonspecific activation and the impact of washing on the background fluorescence using nontransfected HEK293 cells showed that at 200 nM and 500 nM there was very low background activation on cells (<2X autofluorescence level), and washing showed no significant changes. At 1 μM, the dye displays some low-level nonspecific activation (4–5-fold over autofluorescence), weakly illuminating nuclei and dispersed puncta in the cytosol at levels far lower than the signal from FAP-expressing cells. This signal is decreased by washing (Supporting Information Figure S3). The dL5** clone was used to show the rapid dye activation at a variety of cellular locations when MGe dye is added to the media, even in the presence of serum. Figure 4 shows the activation kinetics when stably expressing cells are suddenly exposed to dye at a final concentration of 200 nM in the media. Under these conditions, the ER and mitochondria activate most quickly, reaching saturation within 10 min, while other organelles activate more slowly, requiring 20–30 min to reach a plateau. The half-time for activation of dye in all compartments was 15 min or less, and labeling patterns seen at the steady state were consistent with the imaged mCer3 patterns and properly colocalized. The addition of MGe therefore can be used to rapidly and specifically label intracellular targets without any required washing steps. The fact that the ER is the fastest labeling compartment, although it has multiple membranes to cross for labeling, suggests that there may be protein function differences in addition to dye access that control the activation rate in living cells. Analysis of the rate of loading of the nuclear targeted constructs for each FAP revealed that p13-CW and dL5** were activated at similar rates, while dH6.2 was activated about 2-fold more slowly (Supporting Information Figure S4). In contrast to the high affinity of dL5** reported at the cell surface, cytosolic FAPs reach saturation labeling above 200–500 nM concentrations (Supporting Information Figure S5), suggesting that functional binding or intracellular dye concentration is reduced in the cellular environment. The fact that ER labeling failed to reach saturation with a 200–500 nM dye concentration, in spite of the secretory environment for folding of the expressed FAP, suggests that a main limitation of the labeling here is the rate of dye penetration of the cellular membranes. This analysis is consistent with the rapid early dye labeling of FAP expressed in mitochondria, which may be driven by equilibrium accumulation of the cationic dye into the negative membrane potential of the mitochondria, followed by a slower accumulation phase that completes the mitochondrial FAP labeling. Importantly, although differences are seen, the loading half-time determined for all organelles with the MGe dye is in the range of 7–15 min, consistent with rapid fluorogenic labeling across all subcellular locations. Improvements to the dye structure may increase the rate of cell permeation and improve the effective labeling rates for FAPs in these cellular locations.
Figure 4.
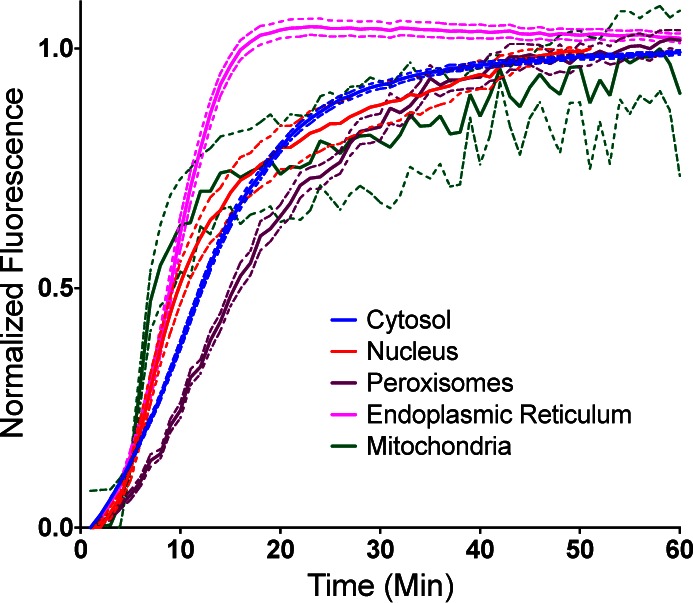
No wash time course of fluorescence signal following addition of 200 nM MGe dye to HEK cells expressing dL5**-mCer3 in the cytosol, nucleus, mitochondria, peroxisomes, and ER. The mean fluorescence intensity for each time point for all the cells in the field of view was calculated, normalized to the maximum fluorescence, and plotted with the bands representing the SEM of multiple fields of view (n = 20 fields for cytosol, nucleus, peroxisomes, endoplasmic reticulum; n = 3 fields for mitochondria).
In order to compare to the loading of the PYP-tag, the fastest reported intracellular fluorogenic label, we tested nuclear-targeted FAP loading rates in the presence of 1 μM dye, conditions identical to those examined in the work of Hori et al.29 The graph of intensity from the time-lapse imaging reveals rapid loading (Supporting Information Figure S6). The PYP-tag reportedly achieved a plateau in labeling after 6 min, while these results show that the MGe dye achieved its labeling plateau in 7 min, proving its value as a rapid, far-red alternative for fluorogenic labeling. A video showing the specific loading in 1 μM dye is included to show the specific and rapid intracellular labeling obtained (Supporting Information Video S1).
Conclusions
This study has presented a new labeling technology for cytoplasmic compartments that is no-wash, far-red, highly fluorogenic, photostable, and nonphototoxic and functions in all organelles. The cell-permeant MGe dye can cross the plasma membrane and label expressed FAPs in various intracellular compartments within minutes, when applied to the bath in nanomolar concentrations, where no significant background fluorescence or nonspecific activation is seen from the fluorogen alone. This rapid, general, and highly fluorogenic response paves the way for potential applications in complex tissues and model organisms where washing of an unbound label or high concentration application is not practical on an experimental time scale. Further, variations of the fluorogen have been shown to produce a variety of distinct spectral and sensing properties for a given FAP.33,51−55 Although these approaches have previously focused on applications at the cell surface, the availability of FAPs that function within cells suggests that cell-permeant versions of these varied fluorogens may be developed that deliver many of these functions to intracellular targets. Finally, fluorogenic labeling of organelles within cells using FAPs provides a far-red alternative to chemo- or osmotropic labeling with bath-applied dyes, one that is independent of organelle physiology, and which is compatible with independent multicolor experiments using other dyes and fluorescent proteins, as demonstrated here with mCer3, green organelle-tracker dyes, and the far-red FAP signal (Supporting Information Figure S1). The intracellular FAP-fluorogen approach outlined here is a versatile, effective fluorogenic labeling strategy.
Methods
Bacterial Strains and Vectors
The Escherichia coli (E. coli) bacterial strain MACH1-T1 (Invitrogen) was used as the host for cloning. The vector used was a derivative of the retroviral pBABE-puro plasmid (http://www.addgene.org/1764, Garry Nolan) with the SfiI site deleted.
DNA Constructions
The sequences of the oligonucleotides used are shown in Supporting Information Table S1. The pBABE vector was modified by introducing a new multiple cloning site in between the BamHI and SalI sites, and annealed oligonucleotides BamMCSfor and SalMCSrev were inserted, resulting in pBABEmod with BamHI, EcoRI, NdeI, AgeI, XhoI, and SalI sites (Figure 1).
The FAP, dL5**, with an amino terminal c-myc tag, EQKLISEEDL, and surrounded by SfiI sites, was amplified from pPNL6-dL5 NP138 with primers EcomycSfidL5F and NdeSfidL5R. For constructs without the c-myc tag, the primer EcoSfidL5F was used. Then the dH6.2 and p13-CW FAP were amplified using the primers, dH6SfiF and dH6SfiR, and CharlieSfiF and CharlieSfiR. PCR products were digested and ligated into a digested vector. The 2XG4S linker oligos, NdeG4SF and AgeG4SR, were annealed and ligated into a similarly digested vector. The mCer3 fluorescent protein38 was amplified using AgeCFPFor and XhoStCFPRev or XhoCFPRev, and the PCR product was digested with AgeI and XhoI prior to ligation.
For cytosolic expression, the Kozak and ATG were added by annealing oligos BamKozATGF and EcoKozATGR. To target the nucleus, a nuclear localization signal was formed by annealing oligos BamNLSfor and EcoNLSrev before ligation into the BamHI and EcoRI digested vector. The amino acid sequence that is coded for is N-terminal MNKNSAKRRKKGTSAKTKRPKV.41 The mitochondrial import sequence is derived from the human COX IV gene44 and codes for the first 22 amino acids MLATRVFSLVGKRAISTSVCVR. The COX VIII signal peptide sequence, MSVLTPLLLRGLTGSARRLPVPRAKIHSLPPEGKL, was added after the COX IV. For peroxisome import, the PTS1 recognition, SKL, was added to the C-terminus of the protein by using the CFP reverse primer, XhoStSKLCFPR. To target proteins into the ER, the murine Ig kappa-chain leader sequence from pDISPLAY (Invitrogen) was added into the BamHI and EcoRI sites by PCR amplifying with primers BamKappaF and EcoKappaR, and to retain the FAPs in the ER, KDEL was added to the C-terminus of the protein by annealing oligos XhoKDELSTOPF and XhoKDELSTOPF and inserting at the XhoI site.
Cell Lines for Tissue Culture
Untransfected HEK293 cells and HEK293 cells stably expressing constructs were cultured and maintained in a 100 mm dish in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (GIBCO, Life technologies). Every fourth day, cells were dissociated using trypsin, and the dissociated cells were plated at a density of 4 × 105 cells on 35 mm dishes for further experiments.
Transfection and Generation of Stable Cell Lines
HEK293 cells were transfected using MIRUS TransIT-LT1 Reagent (Mirus) and selected for puromycin resistance (2 ug mL–1) after 48 h. For the generation of stable lines, the puromycin resistant cells were harvested using trypsin (0.05% w/v), resuspended in PBS, and sorted using a Becton Dickinson FACS Vantage SE Flow Cytometer (Becton Dickinson) with excitation using the 405 nm laser, and emission filters were 450/50 nm. The gates were set above an untransfected control to retain those cells expressing mCer3. The 15 cell lines were transfected in three batches over three consecutive days and then sorted in three batches over three days also.
Imaging
Cells in 35 mm glass-bottom (∼0.17 μm thickness) dishes were imaged using an Andor Revolution XD system with a Spinning Disk Confocal microscope equipped with a Nikon 60x/1.49 NA TIRF oil immersion objective. Solid-state lasers of 405 and 640 nm were used to excite mCer3 and MGe, and emitted photons were transmitted through 525/50 and 685/70 nm filters, respectively, and detected on an Andor EMCCD camera (iXon X3 897 BV). Laser power (119 μW at the sample) of the 405 and 640 lines as well as exposure time (600 ms) and gain (300) of the camera were kept the same during all data acquisition. A sample with FAP-mCer3 expression in a cellular compartment was imaged first; then MGe (25–175 nM, see Results) was added in the dish for ≥20 min in the dark, and the sample was imaged again. The imaging included a sequential collection of confocal images and a DIC image, then a time-lapse of 300 image frames for either a FAP-mCer3 sample or FAP-mCer3_MGe sample, and at last a DIC image (to observe cell shape and size after exposure to laser illumination). The time interval for the time-lapse was set as fast as possible. Dishes and solutions were kept either in an incubator or a microscope stage incubator (Pathology Devices, Inc.) at 37 °C, 5% CO2, and humid conditions.
Colocalization of mCer3, MGe, and organelle marker dye signals were analyzed with Imaris software (Bitplane) for Pearson’s coefficient, and Costes’ automatic thresholding was utilized. Fluorescent intensity of images using time-lapse photography was measured with ImageJ.56 Multiple ROIs were manually drawn in cell regions for the mean and SD of pixel values. The percentage of remaining fluorescent intensity was also calculated (f300/f1 × 100%) using measured intensities of the first image (f1) and the last images (f300) using time-lapse photography. The mCer3 signal was measured without MGe in the media. Curve fit of exponential decay (y = y0 + Ae–x/t) on time-lapse data was performed with OriginPro 8 (OriginLab Corporation).
To study timing of MGe activation by FAPs, time-lapse imaging experiments were performed on the spinning disk confocal microscope using 405 and 640 nm lasers for excitation, coupled with a 447/60 nm emission filter for mCer3, and a 685/70 nm emission filter for MGe. Cells were incubated under physiological conditions as described above. A 37 °C stock of MGe dye twice the target concentration in culture media was mixed on the dish with an equal volume of media at 37 °C during imaging to achieve a well-mixed final solution in the concentration range of 10–500 nM MGe on the cells. Multiple positions (20) on one dish were selected for each condition. Time-lapse imaging was carried out at 1 min intervals over 90 min, imaging in each channel for every time point. Camera exposure time was 400 and 200 ms for 405 (laser power) and 640, respectively. All camera settings were kept consistent throughout the experiments. Intensity analysis of selected regions of interest was performed on the time-lapse data sets using ImageJ.
Flow Cytometry
The day prior to flow cytometry experiments, 4 × 105 HEK293 cells (stably expressing mCer3) were seeded into six well plates. Cells were incubated for 1 h with varying concentrations of MGe in the culture medium. After incubation, cells were washed three times with PBS and harvested by treating with trypsin (0.05% trypsin-EDTA). Cells were pelleted by gentle centrifugation and resuspended in PBS for the flow cytometric analysis on a Becton Dickinson FACS Vantage SE Flow Cytometer (Becton Dickinson). Violet Diode (405 nm) and Red HeNe (635 nm) lasers were used to excite mCer3 and FAP_MGe. Emission filters were 450/50 and 685/35 nm, respectively. A total of 20 000 events were collected for each sample, and the forward- and side-scatter properties were used to exclude the dead cells and debris from analysis. Median fluorescence intensity values were obtained by gating ∼80–85% of the total cell population on the basis of forward- and side-scatter characteristics using FlowJo software (TreeStar, Inc.). The same gate was applied to all the samples. Untransfected HEK293 cells without dye and with different dye concentrations were processed similarly and were used to normalize the background fluorescence signal in both channels. Histograms of data can be found in the Supporting Information, Figures S7 (mCer3-FAP cells) and S8 (untransfected cells). Graphs of analyzed data were generated using the GraphPad Prism software (GraphPad Software).
Acknowledgments
This work was supported in part by the National Institutes of Health, National Technology Center for Networks and Pathways Program U54GM103529 (M.P.B., C.A.T., H.T., S.A.) and R01EB017268 (M.P.B., C.A.T.) and the Human Frontiers Science Foundation grant RGP0063-2009 (R.V.).
Supporting Information Available
Primers used for cloning, colocalization data with organelle tracking dyes, cell morphology after photobleaching of FAP_MGe in organelles, graphs of nonspecific dye activation, graphs of dye-labeling rate for the various clones and graphs of concentration dependent activation in cells, and a graph and video of 1 μM MGe activation kinetics in the nucleus. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† These authors contributed similarly to this work.
The authors declare the following competing financial interest(s): M.B. is a founder of Sharp Edge Labs, which aims to exploit FAP-based labeling for applications in drug discovery.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Remington S. J. (2011) Green Fluorescent Protein: A Perspective. Protein Sci. 20, 1509–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenmann J.; Oswald F.; Nienhaus G. U. (2009) Fluorescent Proteins for Live Cell Imaging: Opportunities, Limitations, and Challenges. IUBMB Life 61, 1029–1042. [DOI] [PubMed] [Google Scholar]
- Shcherbakova D. M.; Verkhusha V. V. (2013) Near-Infrared Fluorescent Proteins for Multicolor in Vivo Imaging. Nat. Methods 10, 751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Chen Z. J.; Ren W.; Ai H. W. (2012) Reaction-Based Genetically Encoded Fluorescent Hydrogen Sulfide Sensors. J. Am. Chem. Soc. 134, 9589–9592. [DOI] [PubMed] [Google Scholar]
- Kremers G. J.; Hazelwood K. L.; Murphy C. S.; Davidson M. W.; Piston D. W. (2009) Photoconversion in Orange and Red Fluorescent Proteins. Nat. Methods 6, 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Arai S.; Takeuchi M.; Sakurai C.; Ebana H.; Higashi T.; Hashimoto H.; Hatsuzawa K.; Wada I. (2012) Development of Cysteine-Free Fluorescent Proteins for the Oxidative Environment. PLoS One 7, e37551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A.; Juillerat A.; Heinis C.; Correa I. R. Jr.; Kindermann M.; Beaufils F.; Johnsson K. (2008) An Engineered Protein Tag for Multiprotein Labeling in Living Cells. Chem. Biol. 15, 128–136. [DOI] [PubMed] [Google Scholar]
- Keppler A.; Gendreizig S.; Gronemeyer T.; Pick H.; Vogel H.; Johnsson K. (2003) A General Method for the Covalent Labeling of Fusion Proteins with Small Molecules in Vivo. Nat. Biotechnol. 21, 86–89. [DOI] [PubMed] [Google Scholar]
- Los G. V.; Wood K. (2007) The Halotag: A Novel Technology for Cell Imaging and Protein Analysis. Methods Mol. Biol. 356, 195–208. [DOI] [PubMed] [Google Scholar]
- Lee H. L.; Lord S. J.; Iwanaga S.; Zhan K.; Xie H.; Williams J. C.; Wang H.; Bowman G. R.; Goley E. D.; Shapiro L.; Twieg R. J.; Rao J.; Moerner W. E. (2010) Superresolution Imaging of Targeted Proteins in Fixed and Living Cells Using Photoactivatable Organic Fluorophores. J. Am. Chem. Soc. 132, 15099–15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Jing C.; Gallagher S. S.; Sheetz M. P.; Cornish V. W. (2012) Second-Generation Covalent Tmp-Tag for Live Cell Imaging. J. Am. Chem. Soc. 134, 13692–13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. W.; Cai Y.; Sheetz M. P.; Cornish V. W. (2005) In Vivo Protein Labeling with Trimethoprim Conjugates: A Flexible Chemical Tag. Nat. Methods 2, 255–257. [DOI] [PubMed] [Google Scholar]
- Yao J. Z.; Uttamapinant C.; Poloukhtine A.; Baskin J. M.; Codelli J. A.; Sletten E. M.; Bertozzi C. R.; Popik V. V.; Ting A. Y. (2012) Fluorophore Targeting to Cellular Proteins Via Enzyme-Mediated Azide Ligation and Strain-Promoted Cycloaddition. J. Am. Chem. Soc. 134, 3720–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttamapinant C.; White K. A.; Baruah H.; Thompson S.; Fernandez-Suarez M.; Puthenveetil S.; Ting A. Y. (2010) A Fluorophore Ligase for Site-Specific Protein Labeling inside Living Cells. Proc. Natl. Acad. Sci. U. S. A. 107, 10914–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka H.; Fujishima S. H.; Uchinomiya S. H.; Ojida A.; Hamachi I. (2010) Selective Covalent Labeling of Tag-Fused Gpcr Proteins on Live Cell Surface with a Synthetic Probe for Their Functional Analysis. J. Am. Chem. Soc. 132, 9301–9309. [DOI] [PubMed] [Google Scholar]
- Ono S.; Yano Y.; Matsuzaki K. (2012) Improvement of Probe Peptides for Coiled-Coil Labeling by Introducing Phosphoserines. Biopolymers 98, 234–238. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Egashira Y.; Kamiura R.; Kikuchi K. (2010) Noncovalent-Interaction-Promoted Ligation for Protein Labeling. Chembiochem 11, 646–648. [DOI] [PubMed] [Google Scholar]
- Tsutsumi H.; Nomura W.; Abe S.; Mino T.; Masuda A.; Ohashi N.; Tanaka T.; Ohba K.; Yamamoto N.; Akiyoshi K.; Tamamura H. (2009) Fluorogenically Active Leucine Zipper Peptides as Tag-Probe Pairs for Protein Imaging in Living Cells. Angew. Chem., Int. Ed. Engl. 48, 9164–9166. [DOI] [PubMed] [Google Scholar]
- Mizukami S.; Yamamoto T.; Yoshimura A.; Watanabe S.; Kikuchi K. (2011) Covalent Protein Labeling with a Lanthanide Complex and Its Application to Photoluminescence Lifetime-Based Multicolor Bioimaging. Angew. Chem., Int. Ed. Engl. 50, 8750–8752. [DOI] [PubMed] [Google Scholar]
- Watanabe S.; Mizukami S.; Akimoto Y.; Hori Y.; Kikuchi K. (2011) Intracellular Protein Labeling with Prodrug-Like Probes Using a Mutant Beta-Lactamase Tag. Chemistry 17, 8342–8349. [DOI] [PubMed] [Google Scholar]
- Watanabe S.; Mizukami S.; Hori Y.; Kikuchi K. (2010) Multicolor Protein Labeling in Living Cells Using Mutant Beta-Lactamase-Tag Technology. Bioconjugate Chem. 21, 2320–2326. [DOI] [PubMed] [Google Scholar]
- Lukinavicius G.; Umezawa K.; Olivier N.; Honigmann A.; Yang G.; Plass T.; Mueller V.; Reymond L.; Correa I. R. Jr.; Luo Z. G.; Schultz C.; Lemke E. A.; Heppenstall P.; Eggeling C.; Manley S.; Johnsson K. (2013) A near-Infrared Fluorophore for Live-Cell Super-Resolution Microscopy of Cellular Proteins. Nat. Chem. 5, 132–139. [DOI] [PubMed] [Google Scholar]
- Adams S. R.; Campbell R. E.; Gross L. A.; Martin B. R.; Walkup G. K.; Yao Y.; Llopis J.; Tsien R. Y. (2002) New Biarsenical Ligands and Tetracysteine Motifs for Protein Labeling in Vitro and in Vivo: Synthesis and Biological Applications. J. Am. Chem. Soc. 124, 6063–6076. [DOI] [PubMed] [Google Scholar]
- Griffin B. A.; Adams S. R.; Tsien R. Y. (1998) Specific Covalent Labeling of Recombinant Protein Molecules inside Live Cells. Science 281, 269–272. [DOI] [PubMed] [Google Scholar]
- Komatsu T.; Johnsson K.; Okuno H.; Bito H.; Inoue T.; Nagano T.; Urano Y. (2011) Real-Time Measurements of Protein Dynamics Using Fluorescence Activation-Coupled Protein Labeling Method. J. Am. Chem. Soc. 133, 6745–6751. [DOI] [PubMed] [Google Scholar]
- Zhang C. J.; Li L.; Chen G. Y.; Xu Q. H.; Yao S. Q. (2011) One- and Two-Photon Live Cell Imaging Using a Mutant Snap-Tag Protein and Its Fret Substrate Pairs. Org. Lett. 13, 4160–4163. [DOI] [PubMed] [Google Scholar]
- Sun X.; Zhang A.; Baker B.; Sun L.; Howard A.; Buswell J.; Maurel D.; Masharina A.; Johnsson K.; Noren C. J.; Xu M. Q.; Correa I. R. Jr. (2011) Development of Snap-Tag Fluorogenic Probes for Wash-Free Fluorescence Imaging. Chembiochem 12, 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizukami S.; Watanabe S.; Akimoto Y.; Kikuchi K. (2012) No-Wash Protein Labeling with Designed Fluorogenic Probes and Application to Real-Time Pulse-Chase Analysis. J. Am. Chem. Soc. 134, 1623–1629. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Norinobu T.; Sato M.; Arita K.; Shirakawa M.; Kikuchi K. (2013) Development of Fluorogenic Probes for Quick No-Wash Live-Cell Imaging of Intracellular Proteins. J. Am. Chem. Soc. 135, 12360–12365. [DOI] [PubMed] [Google Scholar]
- Lukinavicius G.; Reymond L.; D’Este E.; Masharina A.; Gottfert F.; Ta H.; Guther A.; Fournier M.; Rizzo S.; Waldmann H.; Blaukopf C.; Sommer C.; Gerlich D. W.; Arndt H. D.; Hell S. W.; Johnsson K. (2014) Fluorogenic Probes for Live-Cell Imaging of the Cytoskeleton. Nat. Methods 11, 731–733. [DOI] [PubMed] [Google Scholar]
- Prifti E.; Reymond L.; Umebayashi M.; Hovius R.; Riezman H.; Johnsson K. (2014) A Fluorogenic Probe for Snap-Tagged Plasma Membrane Proteins Based on the Solvatochromic Molecule Nile Red. ACS Chem. Biol. 9, 606–612. [DOI] [PubMed] [Google Scholar]
- Szent-Gyorgyi C.; Schmidt B. F.; Creeger Y.; Fisher G. W.; Zakel K. L.; Adler S.; Fitzpatrick J. A.; Woolford C. A.; Yan Q.; Vasilev K. V.; Berget P. B.; Bruchez M. P.; Jarvik J. W.; Waggoner A. (2008) Fluorogen-Activating Single-Chain Antibodies for Imaging Cell Surface Proteins. Nat. Biotechnol. 26, 235–240. [DOI] [PubMed] [Google Scholar]
- Ozhalici-Unal H.; Pow C. L.; Marks S. A.; Jesper L. D.; Silva G. L.; Shank N. I.; Jones E. W.; Burnette J. M. 3rd; Berget P. B.; Armitage B. A. (2008) A Rainbow of Fluoromodules: A Promiscuous Scfv Protein Binds to and Activates a Diverse Set of Fluorogenic Cyanine Dyes. J. Am. Chem. Soc. 130, 12620–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick J. A.; Yan Q.; Sieber J. J.; Dyba M.; Schwarz U.; Szent-Gyorgyi C.; Woolford C. A.; Berget P. B.; Waggoner A. S.; Bruchez M. P. (2009) Sted Nanoscopy in Living Cells Using Fluorogen Activating Proteins. Bioconjug Chem. 20, 1843–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q.; Schwartz S. L.; Maji S.; Huang F.; Szent-Gyorgyi C.; Lidke D. S.; Lidke K. A.; Bruchez M. P. (2014) Localization Microscopy Using Noncovalent Fluorogen Activation by Genetically Encoded Fluorogen-Activating Proteins. ChemPhysChem 15, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi C.; Stanfield R. L.; Andreko S.; Dempsey A.; Ahmed M.; Capek S.; Waggoner A. S.; Wilson I. A.; Bruchez M. P. (2013) Malachite Green Mediates Homodimerization of Antibody Vl Domains to Form a Fluorescent Ternary Complex with Singular Symmetric Interfaces. J. Mol. Biol. 425, 4595–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates B. P.; Peck M. A.; Berget P. B. (2013) Directed Evolution of a Fluorogen-Activating Single Chain Antibody for Function and Enhanced Brightness in the Cytoplasm. Mol. Biotechnol. 54, 829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwardt M. L.; Kremers G. J.; Kraft C. A.; Ray K.; Cranfill P. J.; Wilson K. A.; Day R. N.; Wachter R. M.; Davidson M. W.; Rizzo M. A. (2011) An Improved Cerulean Fluorescent Protein with Enhanced Brightness and Reduced Reversible Photoswitching. PLoS One 6, e17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn K. W.; Kamocka M. M.; McDonald J. H. (2011) A Practical Guide to Evaluating Colocalization in Biological Microscopy. Am. J. Physiol. Cell Physiol. 300, C723–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solmaz S. R.; Blobel G.; Melcak I. (2013) Ring Cycle for Dilating and Constricting the Nuclear Pore. Proc. Natl. Acad. Sci. U. S. A. 110, 5858–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timney B. L.; Tetenbaum-Novatt J.; Agate D. S.; Williams R.; Zhang W.; Chait B. T.; Rout M. P. (2006) Simple Kinetic Relationships and Nonspecific Competition Govern Nuclear Import Rates in Vivo. J. Cell Biol. 175, 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinecke M.; Cizmowski C.; Schliebs W.; Kruger V.; Beck S.; Wagner R.; Erdmann R. (2010) The Peroxisomal Importomer Constitutes a Large and Highly Dynamic Pore. Nat. Cell Biol. 12, 273–277. [DOI] [PubMed] [Google Scholar]
- Dansen T. B.; Pap E. H. W.; Wanders R. J.; Wirtz K. W. (2001) Targeted Fluorescent Probes in Peroxisome Function. Histochem. J. 33, 65–69. [DOI] [PubMed] [Google Scholar]
- Van Kuilenburg A. B.; Van Beeumen J. J.; Demol H.; Van den Bogert C.; Schouten I.; Muijsers A. O. (1992) Subunit Iv of Human Cytochrome C Oxidase, Polymorphism and a Putative Isoform. Biochim. Biophys. Acta 1119, 218–224. [DOI] [PubMed] [Google Scholar]
- Glaser S. M.; Cumsky M. G. (1990) A Synthetic Presequence Reversibly Inhibits Protein Import into Yeast Mitochondria. J. Biol. Chem. 265, 8808–8816. [PubMed] [Google Scholar]
- Glaser S. M.; Cumsky M. G. (1990) Localization of a Synthetic Presequence That Blocks Protein Import into Mitochondria. J. Biol. Chem. 265, 8817–8822. [PubMed] [Google Scholar]
- Price J.; Verner K. (1993) Puromycin Inhibits Protein Import into Mitochondria by Interfering with an Intramitochondrial Atp-Dependent Reaction. Biochim. Biophys. Acta 1150, 89–97. [DOI] [PubMed] [Google Scholar]
- Baker M. J.; Frazier A. E.; Gulbis J. M.; Ryan M. T. (2007) Mitochondrial Protein-Import Machinery: Correlating Structure with Function. Trends Cell Biol. 17, 456–464. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Beavis A. D. (1997) Effect of Leader Peptides on the Permeability of Mitochondria. J. Biol. Chem. 272, 13555–13561. [DOI] [PubMed] [Google Scholar]
- Kushnareva Y. E.; Polster B. M.; Sokolove P. M.; Kinnally K. W.; Fiskum G. (2001) Mitochondrial Precursor Signal Peptide Induces a Unique Permeability Transition and Release of Cytochrome C from Liver and Brain Mitochondria. Arch. Biochem. Biophys. 386, 251–260. [DOI] [PubMed] [Google Scholar]
- Fisher G. W.; Fuhrman M. H.; Adler S. A.; Szent-Gyorgyi C.; Waggoner A. S.; Jarvik J. W. (2014) Self-Checking Cell-Based Assays for Gpcr Desensitization and Resensitization. J. Biomol. Screen. 19, 1220–1226. [DOI] [PubMed] [Google Scholar]
- Grover A.; Schmidt B. F.; Salter R. D.; Watkins S. C.; Waggoner A. S.; Bruchez M. P. (2012) Genetically Encoded Ph Sensor for Tracking Surface Proteins through Endocytosis. Angew. Chem., Int. Ed. Engl. 51, 4838–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi C.; Schmidt B. F.; Fitzpatrick J. A.; Bruchez M. P. (2010) Fluorogenic Dendrons with Multiple Donor Chromophores as Bright Genetically Targeted and Activated Probes. J. Am. Chem. Soc. 132, 11103–11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shank N. I.; Pham H. H.; Waggoner A. S.; Armitage B. A. (2013) Twisted Cyanines: A Non-Planar Fluorogenic Dye with Superior Photostability and Its Use in a Protein-Based Fluoromodule. J. Am. Chem. Soc. 135, 242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shank N. I.; Zanotti K. J.; Lanni F.; Berget P. B.; Armitage B. A. (2009) Enhanced Photostability of Genetically Encodable Fluoromodules Based on Fluorogenic Cyanine Dyes and a Promiscuous Protein Partner. J. Am. Chem. Soc. 131, 12960–12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. (2012) NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.