

Abstract
Human microsomal epoxide hydrolase (mEH) is a biotransformation enzyme that metabolizes reactive epoxide intermediates to more water-soluble trans-dihydrodiol derivatives. We compared protein-coding sequences from six full-length human mEH DNA clones and assessed potential amino acid variation at seven positions. The prevalence of these variants was assessed in at least 37 unrelated individuals using polymerase chain reaction experiments. Only Tyr/His 113 (exon 3) and His/Arg 139 (exon 4) variants were observed. The genotype frequencies determined for residue 113 alleles indicate that this locus may not be in Hardy – Weinberg equilibrium, whereas frequencies observed for residue 139 alleles were similar to expected values. Nucleotide sequences coding for the variant amino acids were constructed in an mEH cDNA using site-directed mutagenesis, and each was expressed in vitro by transient transfection of COS-1 cells. Epoxide hydrolase mRNA level, catalytic activity, and immunoreactive protein were evaluated for each construct. The results of these analyses demonstrated relatively uniform levels of mEH RNA expression between the constructs. mEH enzymatic activity and immunoreactive protein were strongly correlated, indicating that mEH specific activity was similar for each variant. However, marked differences were noted in the relative amounts of immunoreactive protein and enzymatic activity resulting from the amino acid substitutions. These data suggest that common human mEH amino acid polymorphisms may alter enzymatic function, possibly by modifying protein stability.
Introduction
The ability of an organism to convert reactive chemicals to less harmful moieties is important for minimizing toxic effects which may result from such exposures. Xenobiotic-metabolizing microsomal epoxide hydrolase (mEH; E.C. 3.3.2.3) is one of four distinct epoxide hydrolases which are characterized by unique immunological properties, molecular weights, and substrate specificities. The gene encoding mEH (EPHX) is inducible by certain xenobiotic chemicals, and its protein product catalyzes the hydrolysis of arene and aliphatic epoxides to less reactive and more water soluble dihydrodiols by the trans addition of water. Although mEH may participate in the metabolism of endogenous steroids (1), the enzyme has been characterized most extensively with respect to xenobiotic chemical reactions (2), especially those processed in concert with the cytochrome P450 oxygenases which can generate reactive epoxides (3). Under certain circumstances, the chemical products resulting from cytochrome P450 and mEH interactive metabolism include highly reactive electrophiles (4). These activated chemical species may interact with cellular macromolecules (DNA, RNA, and proteins) and thus have the potential to participate in mutagenic, carcinogenic, or teratogenic processes. For example, evidence has been presented which suggests a relationship between low fetal mEH activity and increased potential for teratogenic outcome following maternal exposure to the anti-epileptic drug phenytoin (5).
mEH protein and nucleic acid sequences in humans, rats and rabbits (6 – 10) are evolutionarily conserved, with similarities between the relevant sequences exceeding 83 and 75%, respectively. This degree of structural relatedness between disparate mammalian species, in addition to the apparent ubiquitous organ expression of mEH (11), suggests an important functional role for this enzyme. Results from Southern blot hybridization experiments (6, 10, 12) and genomic sequencing (12; C.Hassett and C.J.Omiecinski, unpublished observations) indicate the existence of a single functional mEH gene per haploid genome in humans and rats. The human EPHX gene has been localized to the long arm of chromosome 1 (6). The absence of genetic complexity observed for mEH contrasts with other xenobiotic enzyme systems, for example the cytochrome P450s and glutathione transferases, and this should simplify the task of determining the functional significance of mEH genetic variation in human populations.
Currently, six distinct full-length human mEH DNA sequences have been cloned and are available for comparison. Four of these have been characterized in our laboratory, and two were reported by other investigators (6, 7). The respective protein sequences deduced from these DNAs demonstrate less than 2 % variation in human mEH amino acid content. Despite the similarity in mEH protein primary structure deduced from these cloned DNA sequences, there are striking inter-individual differences in mEH activity measured in human populations (13 – 15).
Due to the prominent role of this enzyme in xenobiotic metabolism, it is important to elucidate the molecular determinants that control mEH activity in humans, and ultimately whether variations in mEH expression correlate with sensitivity to chemically initiated toxicities or susceptibility to disease. In the study reported here, we examine unrelated individuals for the frequency of mEH protein variation at seven amino acid positions predicted to differ based on cloned DNA sequences. Allelic variants which were found to represent polymorphic loci (≥ 1 % frequency of occurrence) were expressed in vitro, and the effects of these amino acid variations on mEH RNA expression, protein, and enzymatic activity were evaluated.
Results
Full-length human mEH DNA sequences which have been cloned reveal variation at seven of 455 amino acid positions (Fig. 1). The variant positions are at amino acid residues 113 (exon 3), 139, 148 (exon 4), 348 (exon 8), 396, 406, and 420 (exon 9). Four of these variant amino acids (Asnl48, Ser348, Phe406, and Val420), are predicted from a single cDNA clone (7). Two variant amino acids are predicted from cloned genomic mEH DNA (exon 3, His 113 and exon 4, Argl39). The final variable amino acid was identified from the pHEH/33 cDNA which encodes Ile396 (exon 9). Overall, there is less than 2% variation in human mEH amino acid sequence when these six clones are compared to each other. Certain amino acids are most commonly found at the variant positions in these DNAs (i.e. Tyr113, His139, His148, Lys348, Thr396, Leu406, and Leu420). Based of the predominant frequency of occurrence, we tentatively designate this sequence as the most common human mEH allelic form, and refer to it as the ‘reference standard’ in subsequent discussion.
Figure 1.

Alignment of human mEH amino acid sequences predicted from six DNA clones. The locations of intron/exon boundaries are indicated by vertical arrows above the amino acids (shown in single-letter code) nearest to the respective junction. Sequences 1, 2, and 3 are from references 6, 7, and 8, respectively. Sequence 4 is compiled from the translated exons determined from the human gene. Sequences 5 and 6 are identified in the text as clones HEH/p33 and HEH/p53, respectively. Residues which are invariant between sequences are not displayed. Nonconservative amino acid substitutions are designated in bold face and with an asterisk below the variant amino acid.
Observed frequency of amino acid variants
A minimum of 37 individuals were genotyped for each of the seven human mEH variable amino acid positions. Because each individual contains a diploid complement of mEH genes, the genotype analysis reported here examines a minimum of 74 human chromosomes. In five of the seven variable amino acid positions predicted from the cloned DNA, there was no evidence for mEH variation in the analyzed DNA samples. Forty-nine persons assayed for variation at residues 148 and 348 were judged to be homozygous for His and Lys, respectively. Likewise, 45 individuals were homozygous for Thr at position 396. Finally, 37 persons were homozygous for Leu residues at both amino acid positions 406 and 420. For two of these amino acid positions, the conclusions derived from allele-specific oligomer hybridization were confirmed using diagnostic restriction enzyme digestion of the polymerase chain reaction (PCR) products. Each exon 4 PCR product digested with ApaLI indicated the presence of DNA coding for His148. None of these DNAs digested with HphI, which would be indicative of the Asn148 allele. Exon 8 PCR products were subjected to digestion with HinIII, and there were no products which showed evidence for the presence of the Ser348 allele.
In contrast to the absence of variation at these five positions, amino acid variants were confirmed at residues 113 and 139 (Table 1). A total of 105 individuals were evaluated for variation at residue 113. Thirty-eight individuals were classified as homozygous for Tyr (36.2%), eight were homozygous for His (7.6%), and 59 (56.2%) were heterozygotes. Figure 2 displays the specificity of allele-specific oligomers used to characterize residue 113 variation in human liver samples.
Table 1. Occurrence of epoxide hydrolase amino acid variants.
| Amino acid position | Amino acid genotyped Individuals (% total) | Total individuals examined | Total alleles examined | ||
|---|---|---|---|---|---|
| YY | YH | HH | |||
| 113 | 38 (36.2%) | 59 (56.2%) | 8 (7.6%) | 105 | 210 |
| HH | HR | RR | |||
| 139 | 64 (58.7%) | 40 (36.7%) | 5 (4.6%) | 109 | 218 |
The frequency of each of the two variable amino acid positions is shown, including the number of homozygous and heterozygous individuals. Amino acids are indicated by single letter designation
Figure 2.
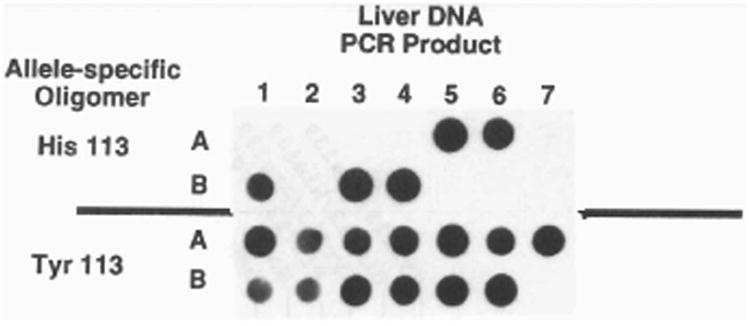
Exon 3 genotype analysis by dot blot hybridization. Thirteen human liver DNA samples were PCR amplified using primers flanking exon 3, denatured, and applied to a nylon membrane in duplicate. Nucleotides encoding amino acid 113 alleles were distinguished using oligomers with sequences diagnostic for either His113 (top) or Tyr113 (bottom). In this figure, five individuals are Tyr/His113 and eight are Tyr/Tyr113.
Cross-hybridization of allele-specific oligomers was encountered in efforts to genotype variation at amino acid 139. Despite repeated efforts to alter hybridization and wash temperature conditions, it was not possible to unambiguously assign this amino acid position by allele-specific oligomer hybridization. However, a diagnostic RsaI restriction site in the Arg139 allele permitted classification by RFLP analysis of PCR products. An example of this analysis, performed on lymphocyte DNA samples, is shown in Fig. 3. The presence of a common RsaI, site present in both alleles, enabled confirmation that RsaI digestion had occurred in all exon 4 products. Using this analysis, it was determined that of 109 individuals analyzed, 64 (58.7%) were homozygous for the His allele, five (4.6%) were homozygous for the Arg allele, and 40 (36.7%) were heterozygotes.
Figure 3.
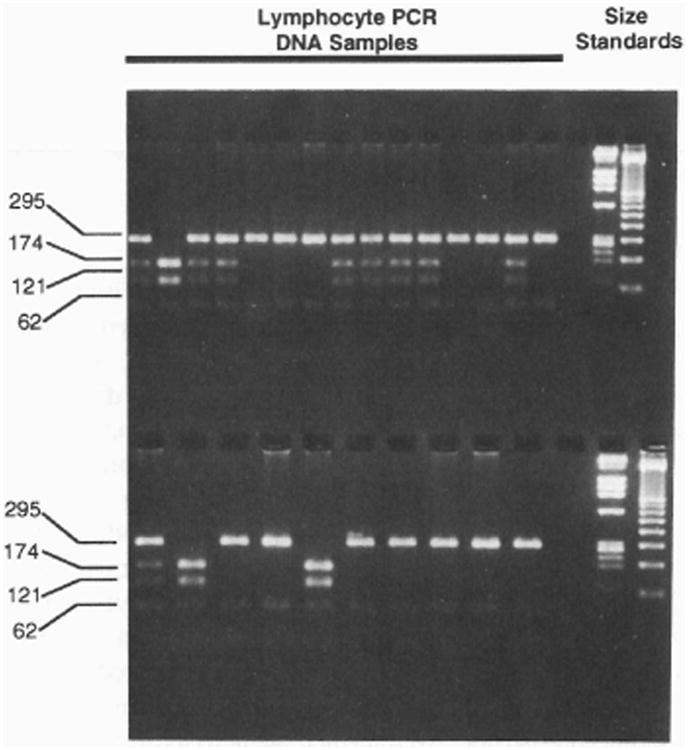
Exon 4 genotype determination by RFLP analysis. Lymphocyte DNA from 25 individuals was PCR amplified using primers flanking exon 4. DNA was digested with RsaI and size-separated on a 2% agarose gel. His 139 alleles are identified by two DNA bands (295 and 62 bp), whereas Arg139 alleles produce three bands (174, 121, and 62 bp) following digestion. Heterozygotes display a combination of all four DNA fragments.
The genotype frequencies observed from this analysis permit comparison to expected values, based on the Hardy–Weinberg equation (p2 + 2pq + q2 =1). For alleles at residue 113, the frequencies of observed/expected genotypes are as follows: Tyr/Tyr (0.362:0.413); Tyr/His (0.562:0.459); and His/His (0.076:0.127). Chi-squared analysis of observed and expected frequencies suggest that the alleles at amino acid 113 are not in Hardy–Weinberg equilibrium (p< 0.025). Statistical analysis of genotype frequency values for residue 139 alleles [His/His (0.587:0.594); His/Arg (0.367:0.353); and Arg/Arg (0.046:0.052)] indicate they are in Hardy-Weinberg equilibrium.
There were no individuals in our study population who were genotyped as homozygotes for both His113 and Arg139 alleles.
Site-directed mutagenesis
pHEH/33 was used as the template DNA for site-directed mutagenesis. This plasmid encodes Tyr113 and His139, the exon 3 and 4 amino acids most commonly identified by the PCR experiments. Mutant cDNAs were created using mutagenic oligomers, and three clones were identified which were identical to pHEH/33, except for nucleotides encoding amino acid 113 (Tyr to His), 139 (His to Arg), or 113/139 (both substitutions affected). The composition of each clone was confirmed by DNA sequence analyses.
When the site-directed mutagenesis studies were initiated, it was not yet appreciated that the Ile396 amino acid present in clone pHEH/33 was an infrequently occurring variant. Because we wished to identify the enzymatic effects of the 113 and 139 amino acid variants against a background of commonly occurring human mEH residues, it was necessary to substitute Thr396 for Ile396 in the mutant clones. Since the mutated sequences were subcloned in the expression vector pSG5, a strategy was devised whereby the Thr396 nucleotide sequence could be shuttled into the mutant constructs. The cDNA clone pHEH/53, which contains the nucleotide sequence encoding Thr396, was subcloned from the pSK vector into the pSG5 expression vector, and the following shuttle approach was utilized. Restriction enzyme digestion with EcoNI and BsaI permitted a fragment which encompassed the 3′ portion of the mEH sequence (including nucleotides coding for Ile396) and a portion of the vector DNA to be excised from each mutant construct. EcoNI/BsaI DNA containing the Thr396 nucleotide sequence isolated from pHEH/53 was ligated to DNA from each of the mutant constructs which contained the remainder of the mEH sequence, including mutant nucleotides, and the missing vector sequence. The nucleotide sequence for each construct was confirmed to encode Thr396, as well as the appropriate allelic nucleotides coding for amino acid residues 113 and/or 139.
In vitro analyses: mEH RNA expression
Total RNA was isolated from COS-1 cells transfected with each of four mEH constructs (i.e. Tyr113/His139, His113/His139, Tyr113/Arg139, or His113/Arg139). Also, RNA was isolated from cells subjected to transfection conditions, but in the absence of plasmid DNA in order to determine the background mEH synthesis in COS-1 cells. Northern-blotted RNA obtained from cells transfected with the mEH constructs hybridized to the mEH-specific oligomer, demonstrating relatively uniform intensity in each lane (Fig. 4A). RNA isolated from the mock-transfected COS-1 cells did not hybridize to the human mEH oligomer (Fig. 4A), demonstrating specificity of the probe, and suggesting that these cells have negligible or no transcription of an endogenous mEH gene (see below). When expression was normalized to 18S rRNA present in each lane (Fig. 4B), the amount of mEH RNA observed between the different constructs appeared essentially identical. This was verified using quantitative computer densitometry of slot blots (data not shown).
Figure 4.
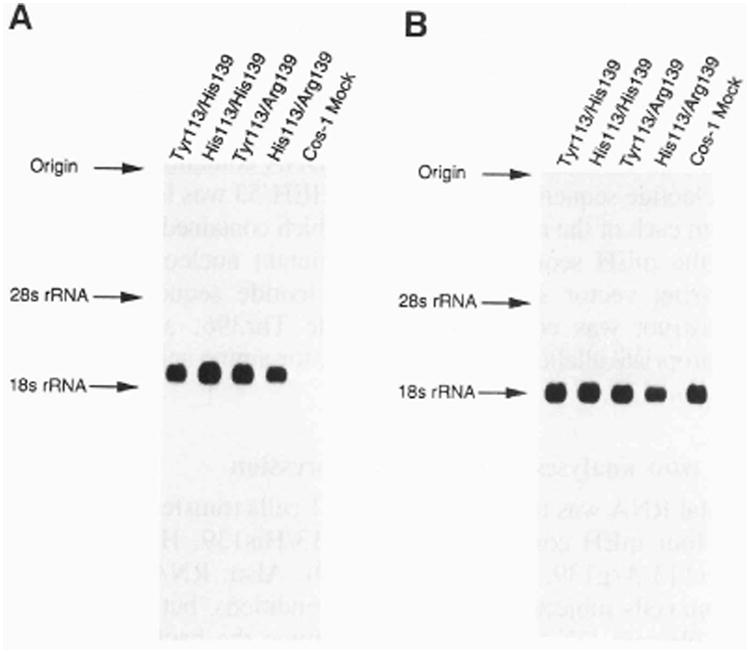
(A) Northern blot analysis of mEH RNA obtained from in vitro expressed allelic variants. Five μg of total RNA was size-separated in an agarose gel, transferred to GeneScreen Plus, and probed with an oligomer specific for the detection of mEH RNA. Clones containing the variant amino acid(s) used in the transfection experiment are designated above each lane by the allelic amino acid(s). COS-1 mock represents RNA isolated from COS-1 cells which were subjected to transfection conditions without a plasmid. The locations of 18S and 28S rRNA are indicated. (B) Northern analysis of 18S rRNA obtained from allelic variants expressed in vitro. The membrane used to generate (A) was dehybridized, and re-probed with an oligomer complementary to 18S rRNA.
mEH enzymatic activity
S9 protein isolated from COS-1 cells transfected with the cDNA constructs permitted determination of the mEH enzymatic activity associated with each of the variable amino acids. The amount of BP-4,5-diol product formed in this assay was dependent on the source of the S9 protein, with each construct yielding a different mEH activity profile. The results of a typical assay are presented in Table 2. The construct containing Tyr113 and His139 is the reference standard, and the end-product of 752.6 pmol of BP-4,5-diol/min per mg S9 protein is arbitrarily assigned 100% activity. When His113 is substituted for Tyr, a decrease to 462.5 pmol/min per mg protein was observed, a 39% loss in mEH activity. Replacing Arg139 for His resulted in an mEH activity increase of 125% (938.1 pmol/min per mg), relative to the reference construct. An intermediate activity level is observed when the mEH cDNA contains both His113 and Argl39 amino acids, 652 pmol/min per mg (87% of the reference activity). The relative profile differences in enzymatic activity among the respective mEH alleles have been reproduced in several separate transfection experiments. Enzymatic activity determined from S9 protein isolated from mock-transfected COS-1 cells was 6.3 pmol/min per mg protein (the detection limit of the assay is approximately 0.1 pmol of product/min per mg of protein). This suggests that COS-1 cells express low endogenous levels of mEH.
Table 2. In vitro expression analyses of mEH alleles in COS-1 cells.
| S9 activity1 | EH RNA (corr)2 | S9 Activity/EH RNA1,3 | EH protein4 | S9 activity/EH protein1,5 | |
|---|---|---|---|---|---|
| Tyr113/His139 | 752.6 (100%) | 1.0 | 752.6 (100%) | 1.0 | 752.6 (100%) |
| His113/Hisl39 | 462.5 (61%) | 1.04 | 444.7 (59%) | 0.56 | 825.9 (110%) |
| Tyr113/Arg139 | 938.1 (125%) | 1.01 | 928.8 (123%) | 1.54 | 609.2 (81%) |
| His113/Arg139 | 652.0 (87%) | 1.07 | 609.3 (81%) | 0.82 | 795.1 (106%) |
| Mock1 | 6.3 (0.8%) | 0.09 | 70.0 (9.3%) | 0.11 | 57.2 (7.6%) |
Numbers in parentheses represent percent changes relative to wild-type values.
Activity is expressed as pmol BP-4,5-diol formed per mg S9 protein per min of reaction. The limit of detection in this assay is approximately 0.1 pmol/min per mg protein.
mEH RNA levels were determined by computer densitometry analysis, and are corrected for levels of 18S rRNA. Values are expressed relative to wild-type controls (control = 1.0).
mEH activities expressed after normalization to respective mEH RNA levels.
Immunochemically detected levels of mEH protein were ascertained by computer densitometry and are expressed relative to wild-type controls (control = 1.0).
mEH activities expressed after normalization to respective immunoquantified mEH protein levels.
Human mEH immunoreactive protein
Western-blotted S9 protein obtained from COS-1 cells transiently transfected with each of the human mEH constructs reacted with polyclonal mEH-specific antibody. The extent of immunoreactivity was dependent on the specific mEH construct (Fig. 5). Furthermore, the amount of mEH protein (determined per unit of S9 protein extract) closely paralleled the mEH activity profile determined for each construct. S9 protein from each construct was applied to slot blots and immunoreactivity was analyzed by densitometric analysis. The construct containing Tyr113 and His139 yielded an optical density (OD) of 0.147, and was assigned a reference value of 100%. Immunoreactive protein from the mutant construct containing His113 had an OD of 0.083, indicating a 44% decrease of mEH-specific protein, relative to the reference. The construct containing Arg substituted for His139 demonstrated mEH protein levels 154% (OD=0.226) greater than reference construct. When both His113 and Arg139 were encoded by the mutant clone, the amount of mEH immunoreactive protein was 82% (OD=0.121) of the reference value. The correlation coefficient derived from comparing the mEH enzymatic activity for each construct with the corresponding level of mEH immunoreactive protein revealed a very high degree of association (r=0.98, p=0.02). Protein isolated from mock-transfected cells had an OD measure which was indistinguishable from background absorbance. These data are summarized in Table 2. The relative extent of immunoreactivity observed from S9 protein derived from the different mEH constructs was reproducible in multiple transfection experiments.
Figure 5.
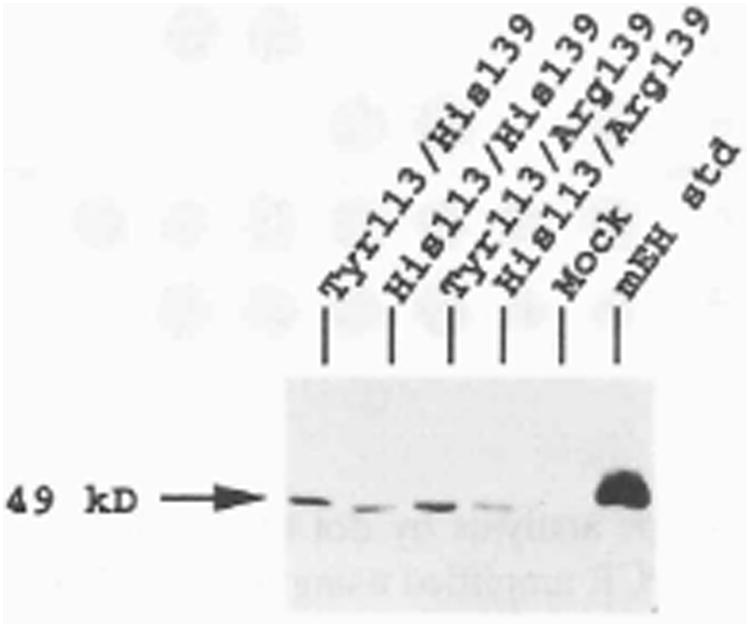
Western blot analysis of S9 protein. COS-1 cells were transfected with plasmids containing each of the mEH variant amino acids and 5 μg of S9 protein was electrophoretically separated, transferred to Immobilon-P, and reacted with antibody specific for mEH protein. The mEH standard is 100 ng of purified mEH protein. The size of the protein was estimated from molecular standards, run concurrently.
Discussion
In this report, we describe a PCR-based approach to estimate the frequency of mEH amino acid variation in humans. The human mEH protein contains 455 amino acids, and seven residue positions were identified as potentially polymorphic, based on six unique cloned human DNA sequences. We have examined a minimum of 74, and a maximum of 218 mEH alleles for polymorphism at these seven loci and confirm variation at only two residues, amino acids 113 and 139.
The reason that five of the allelic variants were not identified is unclear, although four of these variant residues were predicted from the sequence of a single cDNA clone (7). While no details are presented regarding the individual from whom this isolate was derived (16), it is possible that this particular cDNA contained four amino acids which occur infrequently in human mEH proteins. However, this conclusion seems somewhat unlikely, since two of the four allelic variants predicted from this clone represent nonconservative variation relative to other characterized human mEH DNA sequences. Other explanations for the presence of these residues include cloning and/or DNA sequencing artifacts.
The fifth predicted allelic variant not detected by our PCR screening strategy is Ile396. This amino acid is encoded by clone pHEH/33, isolated in our laboratory. The DNA sequence for this cDNA was confirmed multiple times. Therefore it appears likely Ile396 represents either an infrequently occurring variant or, possibly, a cloning artifact.
Considering occurrence in cloned mEH DNAs and frequency observed in PCR-genotype analyses, we tentatively identify human mEH amino acid sequences containing Tyr113 and His139 as the most frequently occurring alleles. Based on the populations examined in the study reported here, the variant amino acids His113 and Arg139 occur at frequencies >1% and represent classic genetic polymorphisms. The frequencies determined for these variant alleles also permits the prediction of Hardy–Weinberg equilibrium at each individual locus. Using these calculations, the His113 allele is observed to occur less frequently than would be expected. However, the amino acid 139 alleles (His or Arg) appear to be in Hardy–Weinberg equilibrium. Additional genotype data must be collected from a greater number of subjects in order to validate these conclusions. Also, it will be of interest to compare genotype frequencies for these variant alleles in ethnic populations other than predominantly Caucasian.
Because a substantial proportion of the population apparently possess mEH alleles which encode variant amino acids at residues 113 and 139, the functional significance of these changes on mEH activity and expression was undertaken. Direct determination of human in vivo expression patterns (i.e. the clinical phenotype) would be confounded by the diploid expression of mEH alleles, making it impossible to determine the effects of amino acid variation, except in the homozygous state. Therefore, this analysis was conducted using in vitro expression studies, where effects of distinct amino acid changes in the mEH enzyme could be studied, without the influence of multiple alleles.
Examination of mEH RNA expressed from cloned DNAs in vitro indicates there are no striking differences in the respective steady state RNA levels observed between the constructs (when cells are harvested for analysis 48 h post-transfection). Based on this observation, it is tentatively concluded that the nucleotide changes which accompany amino acid variation at residues 113 and 139 do not markedly influence RNA stability.
However, in vitro expression of cDNAs containing each of the common and variant amino acids at positions 113 and 139 suggests a functional significance for these residues with respect to mEH protein. Despite equivalent levels of expressed mRNA, substitution of His113 for the more commonly occurring Tyr113 residue decreased mEH activity approximately 40%, and this is accompanied by a similar reduction in the amount of immunoreactive mEH protein. In contrast, Arg139 substituted for His139 confers increased mEH activity when this variant is expressed in vitro. The increased mEH enzymatic activity is paralleled by similarly increased mEH-specific protein content. mEH protein containing both amino acid His113 and Arg139 demonstrate enzymatic activity and immunoreactive protein levels which approximate that observed for the construct containing Tyr113 and His139.
When the mEH enzymatic activities determined for each construct are normalized to the corresponding mEH immunoreactive protein contents, the data suggest that the specific activities of each amino acid variant are similar (Table 2). Considered together, these observations suggest that amino acid variation at residues 113 and 139 does not exert a primary influence on catalytic function, but rather may affect the stability of mEH protein. This hypothesis clearly needs to be explored further, with in vitro studies designed specifically to measure the respective translational efficiencies and protein turnover rates of the expressed constructs.
The research described in this report demonstrates that human mEH proteins contain allelic variation at residues 113 and 139. Furthermore, our analyses demonstrate that the variable amino acids occur in four allelic combinations. Cloned mEH sequences, derived from cDNA libraries, show that nucleotides encoding Tyr113 and His139 exist on the same mRNA strand. Genomic DNA, sequenced from a single DNA fragment, predicts the linkage of His113 and Arg139 amino acid residues. Lastly, from PCR-genotype analysis, we conclude that the allelic combinations Tyr113/Argl39 and His113/His139 exist, since there were several individuals identified with these homozygous genotypes. Our in vitro expression studies demonstrate that different combinations of amino acid residues at positions 113 and 139 in mEH proteins directly influence enzymatic activity, possibly by affecting protein stability. Therefore, it is reasonable to postulate that individuals with specific allelic combinations, especially in the homozygous state, may be at differential risk for the ability to metabolize reactive epoxides efficiently. Family studies, in which haplotypes and linkage can be determined, may reveal insightful segregation patterns of these amino acid combinations. Whether selective forces, or other explanations, are favoring particular amino acid combinations at residues 113 and 139 may indicate future direction for research.
Materials and Methods
Human mEH DNA sequences: amino acid variation
Three full-length human mEH cDNA clones have been reported (6–8). Each of these mEH clones is the product of a different cDNA library. Using a radiolabeled AvaI/NcoI mEH restriction fragment, a λgt11 library (Clontech, Palo Alto, CA) was screened for additional mEH cDNA clones. Phage DNAs were purified (17) from two clones (λB1B and λF1B), and these were subjected to limited digestion with EcoR1. The largest digestion fragments were subcloned in Bluescript plasmid vectors (Stratagene, La Jolla, CA), and re-named pHEH/33 and pHEH/53, respectively.
A human genomic library (a generous gift of T.Maniatis, Harvard Univ.) was screened in order to isolate the entire human mEH gene. Five overlapping or adjacent clones were identified which included nine exons, eight introns, and DNA flanking the 5′ and 3′ portions of the mEH gene. The exonic sequence derived from this genomic DNA represents the sixth distinct full-length sequence of human mEH.
Human DNA samples
DNA was obtained from archived tissue samples of unrelated, predominantly Caucasian (>85%) individuals (18, 19), and samples procured from either the Northwest Tissue Center or from the National Cancer Institute's National Human Tissue Network (Northwestern Division, Columbus OH). Procedures involving the use of human tissues were approved by the University of Washington human subjects review committee. DNA was isolated either from blood lymphocytes or liver tissues, essentially as described (20).
Oligonucleotides: PCR amplification and allele-specific hybridization
PCR reaction components included approximately 100 ng of genomic DNA, 50 mM KC1, 10 mM Tris HC1, pH 8.0, 1.0 mM MgCl2, and 1 μM each of the forward (sense) and reverse (antisense) amplification primers in a total volume of 50 μl. DNA was denatured at 95°C for 5 min followed by lowering of the reaction temperature to 85°C and addition of 1.5 U of Taq DNA polymerase (Promega, Madison, WI). Thirty cycles of amplification began with denaturation at 93°C, primer annealing at the empirically determined optimal temperature, and extension at 72°C, each step for 1 min. The primer annealing temperature for each exon was as follows: exon 3, 55°C; exon 4, 58°C; exon 8, 57°C; and exon 9, 52.5°C. Primers, at least 20 bases long and containing a GC ratio >50%, were designed to hybridize to intronic DNA flanking exons containing variable amino acids. Each oligomer flanks the corresponding exon by no more than 100 nucleotides, and usually less than 50 bases.
Allele-specific oligomers were 17–20 nucleotides long, with the mismatching base specifying the desired amino acid change centrally located. Oligomers were designed to hybridize exclusively to exonic sequence, with the exception of those detecting human mEH amino acid 348 variants. Because amino acid 348 is encoded by nucleotides beginning two bases from the 5′ intron/exon boundary of exon 8 (see Fig. 1), these allele-specific oligomers contain seven nucleotides of intron sequence at the 5′ terminus. Oligomers were synthesized with a Millipore Milligen/Biosearch Cyclone Plus DNA synthesizer. The nucleotide sequence of oligomers used for PCR amplification and allele-specific detection are presented in Table 3.
Table 3. PCR and allele-specific oligonucleotides.
| Primer | Direction | Sequence (5′–3′) |
|---|---|---|
| Exon 3 | ||
| Intron 2FP | Sense | CTTGAGCTCTGTCCTTCCCATCCC |
| Intron 3RP | Antisense | GACGGCCGTTCTCATGACATACATCC |
| Exon 3RP/Y113 | Antisense | AGTGAGGGTATCTGTTGAG |
| Exon 3RP/H113 | Antisense | AGTGAGGGTGTCTGTTGAG |
| Exon 4 | ||
| Intron 3FP | Sense | GGGGTACCAGAGCCTGACCGT |
| Intron 4RP | Antisense | AACACCGGGCCCACCCTTGGC |
| Exon 4RP/H139 | Antisense | TTCGGGGTATGGCCTGC |
| Exon 4RP/R139 | Antisense | TTCGGGGTACGGCCTGC |
| Exon 4RP/H148 | Antisense | GCTGATGGTGCACGGCTGGC |
| Exon 4RP/N148 | Antisense | GCTGATGGTGAACGGCTGGC |
| Exon 8 | ||
| Intron 7FP | Sense | CACACAACTGCAGGTGGCACTG |
| Intron 8RP | Antisense | GGCACCTGCAGGCAAGGCTA |
| Exon 8FP/K348 | Sense | TTCCCAGGAAGTTCTCCCTG |
| Exon 8FP/S348 | Sense | TTCCCAGAAGCTTCTCCCTG |
| Exon 9 | ||
| Intron 8FP | Sense | GTCGGCTCTTTCACTTCCAG |
| 3′ RP | Antisense | CGCTCATGTCACTGAGTTCC |
| Exon 9FP/T396 | Sense | TGTGCCCACTGGCTTCTC |
| Exon 9FP/I396 | Sense | TGTGCCCATTGGCTTCTC |
| Exon 9FP/L406 | Sense | GAGCTATTGCACACGCCTG |
| Exon 9FP/F406 | Sense | GAGCTATTCCACACGCCTG |
| Exon 9FP/L420 | Sense | GTACCCAAAGCTCATCTCC |
| Exon 9FP/V420 | Sense | GTACCCCAAAGTCATCTCC |
The two sequences displayed immediately below each exon are the PCR amplification primers. Allele-specific oligonucleotides, employed to distinguish the alternative alleles for each exon, are designated using the one letter amino acid abbreviation, followed by the corresponding residue number
Five μl of PCR-amplified DNA was denatured with 0.25 N NaOH for 5 min at room temperature and brought to a final volume of 100 μl in 0.125 N NaOH/0.125×SSC. Twenty μl of denatured DNA was applied to a dot blot manifold (Schleicher and Schuell, Keene, NH) and transferred on to a charged nylon membrane (GeneScreen Plus, NEN/Dupont, Boston, MA) as per the manufacturer's directions. Nucleic acid was cross-linked to the membrane using a UV Stratalinker 1800 (Stratagene, La Jolla, CA) followed by prehybridization in 5 × oligomer buffer, which contains 5×SSC, 1.0% (w/v) SDS, 25 mM Na phosphate, pH 6.5, 25 μg/ml polyadenylic acid, and 1 ×Dendhardt's solution (1 ×SSC: 1.5 M NaCl, 0.15 M NaCitrate; 1×Dendhardt's: 0.02% each of Ficoll, polyvinylpyrrolidone 360, bovine serum albumin). Allele-specific oligonucleotides were end-labeled as described (21), and added to the hybridization buffer at 5 × 105 dpm/ml. Hybridization was conducted overnight at the temperature specified in Table 4. Membranes were washed in 5 ×SSC/0.1% SDS for 20–60 min each, twice each at room temperature and the temperature specified in Table 4.
Table 4.
Determination of human mEH allele classification.
| (a) Allele-specific hybridization conditions | ||
|---|---|---|
| Amino acid position | Hybridization (T°C) | Wash (T°C) |
| Exon 3 | ||
| Tyr/His 113 | 52 | 52 |
| Exon 4 | ||
| His/Arg 139 | ND | ND |
| His/Asn 148 | 60 67.5 | |
| Exon 8 | ||
| Lys/Ser 348 | 54 | 54 |
| Exon 9 | ||
| Thr/Ile 396 | 57 | 57 |
| Leu/Phe 406 | 54 | 54 |
| Leu/Val 420 | 54 | 54 |
|
| ||
| (b) Allele-specific restriction enzymes | ||
| Allelic amino acid | Restriction enzyme sensitivity | |
| Exon 4 | ||
| Arg 139 | RsaI | |
| His 148 | ApaLI | |
| Asn 148 | HphI | |
| Exon 8 | ||
| Ser 348 | HindIII | |
Amino acid variants were genotyped in individuals by hybridization to allele-specific oligonucleotides under defined conditions (a), or, in some cases, by PCR-RFLP analysis of amplified exons (b). Optimal hybridization and wash conditions for allelic variants at residue 139 in exon 4 were not determined (ND)
Restriction digest of PCR products
Nucleotide sequences encoding several of the variable amino acids predicted a diagnostic restriction enzyme site in the PCR product (Table 4). In these instances, 5 μl of the PCR product was digested with 5–10 units of restriction enzyme in a total volume of 20 μl. The entire digestion mixture was subjected to electrophoresis in a 2% agarose gel, and digestion was evaluated on a UV transilluminator.
In vitro expression of cloned DNA
The human mEH cDNA p33 (containing Tyr113 and Hisl39) was mutated, using a solid-phase site-directed mutagenesis protocol, to create the appropriate nucleotide sequences encoding mEH amino acid variants. Reactions were conducted using the DoubleTake double-stranded mutagenesis kit, as per the manufacturer's instructions (Stratagene, La Jolla, CA). Two mutagenic primers were employed: 5′-CTCAACAGACACCCTCACT-3′ mutates the nucleic acid sequence for amino acid 113 from tyrosine to histidine; and 5′-GCAGGCCGTACCCCGAA-3′ mutates the nucleotide sequence encoding amino acid 139 from histidine to arginine (underlined nucleotides are mismatched relative to the template and specify the desired amino acid). The mutations were confirmed by DNA sequence analysis (22). The parent and mutated mEH DNA inserts were excised from the Bluescript vector and subcloned into the eucaryotic expression vector pSG5 (23), obtained commercially (Stratagene La Jolla, CA). Plasmids were prepared using a phenolless alkaline lysis protocol (24). The sequence of the mEH clones was reconfirmed after subcloning.
Cell culture and transfection
COS-1 monkey kidney cells (ATCC #CRL1650, Rockville, MD) were seeded at a density of 5 × 105 cells/ml in 75 cm2 tissue culture treated flasks. Cells were grown in DMEM/F12 (1:1) media supplemented with 15 mM Hepes pH 7.4 (Gibco-BRL, Grand Island, NY), 2 mM L-glutamine, 0.22% NaHCO3 (v/v), Pen-Strep (100 U/ml, 0.1 mg/ml) and 5% Nu-Serum (Collaborative Research Inc., Bedford, MA) to a density not greater than 40% before initiating transfection.
Transfection was mediated by the Lipofectin method (25), with modification. Cell culture medium was aspirated and cells were washed with OPTI-MEM (Gibco-BRL, Grand Island, NY). Cells were incubated with 5 ml of OPTI-MEM for 1 h prior to transfection. Each human mEH construct DNA (12.5 μg) was added to 3 ml of OPTI-MEM media, and 60 μl of Lipofectin (60 μg) was separately added to 3 ml of OPTI-MEM. The solutions were combined in each transfection flask, after aspiration of the pre-incubation medium. Flasks were swirled gently to disperse the coprecipitate and incubated at 37°C in 95% air/5% CO2 (v/v) for 6 h after which 14 ml of complete medium (minus Pen/Strep) plus 5% Nu-Serum was added to the transfection medium. This medium was replaced by 15 ml complete medium (plus Pen/Strep) 18 h after transfection, and the cells were cultured for a further 30 h. Each human mEH construct or mock-transfected control was done in triplicate flasks. Cells from each group were combined at the termination of the experiment and divided equally for analysis of RNA and protein.
Northern blot analyses
Ten ml of cell suspension obtained from each mEH construct transfection was centrifuged at 50 ×g for 10 min at room temperature. Five μg of total RNA was isolated (26), size-separated in a denaturing 6% formaldehyde/1.25% agarose gel and transferred to GeneScreen Plus using 10×SSC in a Bios transfer apparatus (Bios Corp., New Haven, CT). Also, 1, 2, or 5 μg of total RNA isolated from each tissue culture sample was applied to a nylon membrane using a slot blot apparatus (Schleicher and Schuell, Keene, NH), according to the manufacturer's directions. RNA was UV-crosslinked to membranes, dried at 80°C for 2 h, and prehybridized in 5 ×oligomer buffer at 52°C.
An antisense oligomer complementary to human mEH exon 3, with the sequence 5′-AGCTGTCCTCCAAAGGTGGG-3′, was end-labeled and added to the hybridization solution at 5 × 105 dpm/ml. Following overnight annealing at 52°C, the Northern and slot blot membranes were washed twice at room temperature and twice at 52°C (30 min/wash) in 5×SSC/0.1 %SDS and exposed to X-ray film.
Membranes were dehybridized in 0.1×SSC/1.0% SDS at 85°C. The blots were then pre-hybridized in 5 ×oligomer buffer at 52°C for at least 1 h. An oligomer complementary to human 18s rRNA (27), with the sequence 5′-CACCTCTAGCGGCGCAATAC-3′, was end-labeled and added to the hybridization solution at 5 × 103 dpm/ml. After overnight hybridization at 52°C, the membranes were washed and exposed to X-ray film.
Protein assessments
Transfected COS-1 cells were centrifuged as above, and cell pellets stored at −80°C until use. After thawing, 250 μl of solution A (0.15 M KC1, 0.05 M KH2PO4, pH 7.35) was added, and each sample was sonicated on ice for three 10 s bursts using a Cole Palmer 4710 ultrasonicator. Cell homogenates were centrifuged for 10 min at 9000 ×g at 4°C. A small amount of the supernatant (S9 fractions) was removed for protein determinations (Pierce BCA reagent system, Rockford, IL), and the remaining sample was stored at −80°C until use.
mEH enzymatic activity
The HPLC-based fluorometric assay, quantifying the mEH-mediated conversion of BP-4,5 oxide to the trans-BP-4,5-diol derivative was conducted essentially as described (15), except that the final dried extract was diluted with 1500 μl of acetone before 5 μl of the sample was injected. Enzymatic assays were performed using 100 μg of sample per reaction, and were conducted in either duplicate or triplicate. Two injections per each sample reaction were assessed. Blank reactions, which contained all of the reactants except the enzyme source, were included in all assays. The fluorescence intensity obtained from each injection was corrected for background fluorescence and averaged between values from the duplicate HPLC injections of each sample, and between values obtained from the replicate reactions. Fluorescence intensity, measured in arbitrary units, was converted to pmol of BP-4,5-diol product. All other materials used for mEH protein analysis were as described previously (15).
mEH antibody and immunoreactive protein
A synthetic peptide, corresponding to mEH amino acids 42 through 60, was used to generate polyclonal mEH antibody in rabbits. Purification methods and characterization of the antibody were reported previously (28).
S9 protein (5 μg) from transfected COS-1 cells, or purified mEH protein (100 ng), were electrophoretically separated on a 10% SDS-polyacrylamide gel according to established methods (20). Proteins were electroblotted on to an Immobilon-P membrane (Millipore Corp., Bedford, MA) using a SEMI-PHOR semi-dry transfer apparatus (Hoefer Scientific Instruments, San Francisco, CA). The membrane was soaked in TTBS +1 %B (Tris-buffered saline with 0.1% Tween 20 and 1 % BSA (w/v)) at room temperature for 1 h to inhibit non-specific antibody interaction with the membrane. The membrane was incubated with mEH-specific antibody at 0.3 μg/ml in TTBS+1%B for 1 h, followed by reaction with the secondary antibody, horseradish-peroxide conjugated goat anti-rabbit IgG (Pierce, Rockford, IL), at a 1:4000 dilution for 1 h. Each incubation step was followed by three washes with TTBS. Chemiluminescent visualization of the immunoreactive proteins was achieved with the ECL Western blotting detection system (Amersham Inc., Arlington Heights, IL) according to the manufacturer's protocol. In addition, S9 protein (0.5, 1.0 and 2.5 μg) obtained from each transfected COS-1 cell sample, or purified mEH protein (0.1, 0.5, 1.0, 2.0, 3.5 and 5.0 ng) was applied to an Immobilon-P membrane using a slot blot apparatus. Samples were absorbed to the membrane by gravity, after which it was briefly rinsed in TBS, reacted with antibody and ECL solutions, as described above.
Quantitation of mEH RNA and protein
The relative intensity of autoradiographic signals obtained from RNA or protein samples was quantified using Millipore Biolmage software (Ann Arbor, MI) and a Howtek Scanmaster 3+ densitometer, in conjunction with a Sun sparcstation II computer. Analyses were conducted on slot-blotted samples with autoradiographic exposures in the linear response range of the X-ray film. Membranes were exposed to either X-OMAT-AR X-ray film (Kodak, Rochester, NY) or Reflection autoradiography film (DuPont, Wilmington, DE).
Acknowledgments
This paper is dedicated to the memory of Neil M. Wilson, Ph.D. who began the characterization of human microsomal epoxide hydrolase in our laboratory. The authors thank Dr Arno Motulsky for helpful discussions, and Drs Harvey Checkoway and Carrie A. Redlich for assistance with sample procurement. The technical assistance of Mr Lothar Uher and Ms Kirsten Robinson for work characterizing the mEH gene is acknowledged. This research was supported by National Institute of Health grant ES-04978. Portions of this research were presented at die Society of Toxicology meeting, March 17, 1993
Abbreviations
- mEH
microsomal epoxide hydrolase
- PCR
polymerase chain reaction
- BP
(±)-Benzo[a]pyrene
- kb
kilobases
- RFLP
restriction fragment length polymorphism
References
- 1.Vogel-Bindel U, Bendey P, Oesch F. Eur J Biochem. 1982;126:425–431. [PubMed] [Google Scholar]
- 2.Guengerich FP. In: Reviews in Biochemical Toxicology. Hodgson E, Bend JR, Philpot RM, editors. Vol. 4. Elsevier Biomedical; New York: 1982. pp. 5–30. [Google Scholar]
- 3.Lu AYH, Miwa GT. Annu Rev Pharmacol Toxicol. 1980;20:513–531. doi: 10.1146/annurev.pa.20.040180.002501. [DOI] [PubMed] [Google Scholar]
- 4.Sims P, Grover PL, Swaisland A, Pal K, Hewer A. Nature. 1974;252:326–328. doi: 10.1038/252326a0. [DOI] [PubMed] [Google Scholar]
- 5.Buehler BA, Delimont D, van Waes M, Finnell RH. N Engl J Med. 1990;322:1567–1572. doi: 10.1056/NEJM199005313222204. [DOI] [PubMed] [Google Scholar]
- 6.Skoda RC, Demierre A, McBride OW, Gonzalez FJ, Meyer UA. J Biol Chem. 1988;263:1549–1554. [PubMed] [Google Scholar]
- 7.Jackson MR, Craft JA, Burchell B. Nucleic Acids Res. 1987;15:7188. doi: 10.1093/nar/15.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson NM, Omiecinski CJ. GenBank X07936 [Google Scholar]
- 9.Porter TD, Beck TW, Kasper CB. Arch Biochem Biophys. 1986;248:121–129. doi: 10.1016/0003-9861(86)90408-x. [DOI] [PubMed] [Google Scholar]
- 10.Hassett C, Turnblom SM, DeAngeles A, Omiecinski CJ. Arch Biochem Biophys. 1989;271:380–389. doi: 10.1016/0003-9861(89)90287-7. [DOI] [PubMed] [Google Scholar]
- 11.Oesch F, Glatt H, Schmassmann H. Biochem Pharmacol. 1977;26:603–607. doi: 10.1016/0006-2952(77)90032-6. [DOI] [PubMed] [Google Scholar]
- 12.Falany CN, McQuiddy P, Kasper CB. J Biol Chem. 1987;262:5924–5930. [PubMed] [Google Scholar]
- 13.Mertes I, Fleischmann R, Glatt HR, Oesch F. Carcinogenesis. 1985;6:219–223. doi: 10.1093/carcin/6.2.219. [DOI] [PubMed] [Google Scholar]
- 14.Glatt H, Oesch F. In: Genetic Variability in Responses to Chemical Exposure Banbury Report. Omenn GS, Gelboin HV, editors. Vol. 16. Cold Spring Harbor; 1984. pp. 189–203. [Google Scholar]
- 15.Omiecinski CJ, Aicher L, Holubkov R, Checkoway H. Pharmacogenetics. 1993;3:150–158. doi: 10.1097/00008571-199306000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Kwok SCM, Ledley FD, DiLella AG, Robson KJH, Woo SLC. Biochemistry. 1985;24:556–561. doi: 10.1021/bi00324a002. [DOI] [PubMed] [Google Scholar]
- 17.Kaslow DC. Nucleic Acids Res. 1986;14:6767. doi: 10.1093/nar/14.16.6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Checkoway H, Costa LG, Camp J, Coccini T, Daniell WE, Dills RL. Br J Ind Med. 1992;49:560–565. doi: 10.1136/oem.49.8.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Checkoway H, Costa LG, Woods JS, Castoldi AF, Lund BO, Swanson PD. J Neural Transm [P-D Sect] 1992;4:283–290. doi: 10.1007/BF02260077. [DOI] [PubMed] [Google Scholar]
- 20.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor; 1989. [Google Scholar]
- 21.Giachelli CM, Omiecinski CJ. J Biol Chem. 1986;261:1359–1363. [PubMed] [Google Scholar]
- 22.Sanger F, Nicklen S, Coulson AR. Proc Natl Acad Sci. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green S, Issemann I, Sheer E. Nucleic Acids Res. 1988;16:369. doi: 10.1093/nar/16.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S, Rasheed S. BioTechniques. 1990;9:676–679. [PubMed] [Google Scholar]
- 25.Feigner PL, Gadek TR, Holm M, Roman M, Chan HW, Wenz M, Northrop JP, Ringold GM, Danielson M. Proc Natl Acad Sci. 1987;84:7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 27.McCallum FS, Maden BEH. Biochem J. 1985;232:725–733. doi: 10.1042/bj2320725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farin FM, Omiecinski CJ. J Toxicol Environ Health. 1993;40:323–341. doi: 10.1080/15287399309531797. [DOI] [PubMed] [Google Scholar]