

Abstract
Synonymous mutations do not change the sequence of the polypeptide but they may still influence fitness. We investigated in Salmonella enterica how four synonymous mutations in the rpsT gene (encoding ribosomal protein S20) reduce fitness (i.e., growth rate) and the mechanisms by which this cost can be genetically compensated. The reduced growth rates of the synonymous mutants were correlated with reduced levels of the rpsT transcript and S20 protein. In an adaptive evolution experiment, these fitness impairments could be compensated by mutations that either caused up-regulation of S20 through increased gene dosage (due to duplications), increased transcription of the rpsT gene (due to an rpoD mutation or mutations in rpsT), or increased translation from the rpsT transcript (due to rpsT mutations). We suggest that the reduced levels of S20 in the synonymous mutants result in production of a defective subpopulation of 30S subunits lacking S20 that reduce protein synthesis and bacterial growth and that the compensatory mutations restore S20 levels and the number of functional ribosomes. Our results demonstrate how specific synonymous mutations can cause substantial fitness reductions and that many different types of intra- and extragenic compensatory mutations can efficiently restore fitness. Furthermore, this study highlights that also synonymous sites can be under strong selection, which may have implications for the use of dN/dS ratios as signature for selection.
Keywords: experimental evolution, synonymous mutations, bacterial fitness, compensatory mutations, Salmonella typhimurium, protein synthesis.
Introduction
The genetic code is redundant and since alternative codons can specify the same amino acid, one may assume that synonymous mutations do not cause any phenotype. However, sometimes synonymous mutations are associated with a phenotypic effect and a fitness cost (Carlini and Stephan 2003; Duan et al. 2003; Carlini 2004; Pagani et al. 2005; Kimchi-Sarfaty et al. 2007; Lind et al. 2010) and there are even examples of lethal synonymous mutations in viruses (Sanjuán et al. 2004; Carrasco et al. 2007). Synonymous mutations may act on several levels in the flow of genetic information, and explanations usually involve codon usage bias (Shields et al. 1988; Eyre-Walker and Bulmer 1993; Kudla et al. 2009; Chursov et al. 2013; Goodman et al. 2013). Two hypotheses for codon usage bias stand out as the most frequently discussed: mRNA secondary structure and stability, and translational efficiency with regard to speed and accuracy. These ideas are partly overlapping since the folding of the mRNA near the beginning of transcripts plays an important role during translation initiation (Eyre-Walker and Bulmer 1993; Kudla et al. 2009; Goodman et al. 2013; Brandis et al. 2016). Stem loops in this region can inhibit ribosome binding and consequently initiation of translation. In accordance with this, Gu et al. (2010) found a general trend toward lower folding stability near the translation initiation site in all three domains of life. Tuller et al. (2010) observed the same trend, although the low folding stability affected translation elongation rate rather than initiation. In addition, codon usage is important for mRNA structure and stability through the entire length of transcripts (Chamary and Hurst 2005; Shabalina et al. 2006; Chursov et al. 2013; Lind and Andersson 2013; Wan et al. 2014). Phenotypes of synonymous mutations that alter the secondary structure of the mRNA can be explained, for example, by differences in degradation patterns when RNase cleavage sites are more open for RNases to bind (Duan and Antezana 2003).
Apart from impairments in initiation of translation, synonymous mutations may affect both translational speed (Andersson and Kurland 1990; Cannarozzi et al. 2010) and accuracy (Kramer and Farabaugh 2007; Stoletzki and Eyre-Walker 2007). Drummond and Wilke (2008) proposed that misfolding of mistranslated peptides imparts a major evolutionary constraint resulting in codon usage bias through selection against codons that are often mistranslated. This is supported by a correlation between codon optimality and predicted structural importance of protein residues, with structurally important residues more often being encoded by optimal codons (Zhou et al. 2009). It is well known that tRNA isoacceptors and codon usage are co-evolved (Varenne et al. 1984; Ikemura 1985; Elf et al. 2003; Rocha 2004; Welch et al. 2009; Xu et al. 2013) and synonymous mutations could confer their effect by altering the balance between tRNA availability and codon concentration (Andersson and Kurland 1990). Accordingly, synonymous mutations could alter codon contexts and confer negative effects (Shields et al. 1988; Berg and Silva 1997), and it has been suggested that codons using the same tRNA are selected to be physically close on the mRNA (Cannarozzi et al. 2010), facilitating the re-use of the same tRNA-molecule before it has diffused away from the translating ribosome. Altered translational speed due to different codon usage could also influence protein misfolding or mRNA degradation caused by ribosome stalling at introduced rare codons (Lemm and Ross 2002; Baker and Mackie 2003; Deana and Blasco 2005; Kaberdin and Bläsi 2006; Lazrak et al. 2013), RNase cleavage sites being introduced or removed (Duan and Antezana 2003), and premature termination of transcription due to loss of transcription/translation coupling (Nudler and Gottesman 2002; Ciampi 2006). In addition, in eukaryotes selection for preservation of splicing regulatory sequences within exons puts selective constraints on synonymous codon usage, especially near intron–exon boundaries (Parmley and Hurst 2007).
Most studies of codon usage examine genome composition whereas experimental studies of fitness effects of synonymous mutations are scarce. We have used ribosomal protein S20 (encoded by the rpsT gene) as a model to examine fitness effects of synonymous mutations in Salmonella enterica and how these costs can be genetically ameliorated. S20 consists of 86 amino acids and binds to the 30S subunit of the ribosome where it has a local effect on the stability of the 16S rRNA (Ramaswamy and Woodson 2009). Transcription of rpsT is regulated negatively by DksA and ppGpp (Wirth et al. 1981; Lemke et al. 2011) and the transcripts are primarily degraded by RNase E (Mackie 2000; Baker and Mackie 2003). S20 is also known to self-regulate on a post-transcriptional level (Wirth et al. 1981, 1982; Parsons and Mackie 1983; Donly and Mackie 1988).
Mutants lacking S20 have severe defects in translation initiation due to impaired mRNA binding and docking of the ribosomal subunits (Götz et al. 1990; Rydén-Aulin et al. 1993; Tobin et al. 2010; Goodman et al. 2013). Here, we chose four deleterious synonymous S20 mutants in S. enterica that were constructed in a previous study and shown to have a substantial fitness cost (Lind et al. 2010), and asked why the synonymous mutations reduce fitness and how these costs can be genetically compensated. Our data suggest that the synonymous mutations in the rpsT gene reduce the levels of S20 leading to a subpopulation of initiation-defective ribosomes lacking S20. These problems were compensated by up-regulating S20 synthesis either by increasing gene copy number or by increasing rpsT transcript levels and/or translation.
Results
Synonymous Mutations Reduce rpsT mRNA and S20 Protein Levels
To determine why synonymous mutations reduced fitness and examine how S. enterica can compensate for the fitness reduction we used four synonymous single base pair substitutions in the rpsT gene that have previously been shown to confer significant fitness costs in conditions that support rapid growth (Lind et al. 2010). The synonymous mutations were located in stem-loop II (T36G and G48A) and IV (A150C and A150G) in the proposed secondary structure of the mRNA (Mackie 1992, 2013; fig. 1A). The relative growth rate in LB in early exponential phase was 86% for T36G, 82% for G48A, 58% for A150C, and 90% for A150G, as compared with an isogenic wild-type control (fig. 1B).
Fig. 1.

Four deleterious synonymous mutants in rpsT. (A) Structure of the rpsT P2 transcript (adapted from Mackie 1992). The structure of the E. coli rpsT mRNA was generated based on three methods of structural probing: Nuclease T1 and RNase CL3 cleavage pattern and dimethyl sulfate modification pattern (Mackie 1992). The four costly synonymous mutations are indicated in red. The roman numerals indicate the numbering of the stem-loops used throughout the text and black arrows indicate the three prominent RNase E cleavage sites in the E. coli rpsT transcript (Mackie 2013). The three primers used in the RT-qPCR are marked in grey (primer pair 1 = frw + rev1, primer pair 2 = frw + rev2). (B) Relative exponential growth rates of reconstructed synonymous mutants. Reported values represent the mean (±SD) of at least 12 independent experiments. The data for the growth measurements are listed in supplementyary table SI, Supplementary Material online. (C) Quantification of rpsT mRNA through RT-qPCR. Two primer pairs were used, as marked in (A) (supplementary table SII, Supplementary Material online). Filled bars represent primer pair 1, and striped bars primer pair 2. Reported values represent the mean (±SD) of at least ten replicates. *** indicate P < 0.001 (two-tailed Student’s t-test, equal variance) as calculated between mutant and wild-type. (D) Quantification of S20 through LC-MS/MS. Reported values represents the mean (±SD) of two independent experiments and is set relative to a wild-type control strain. (E) As in (D), but the average of all ribosomal proteins.
The levels of rpsT mRNA and S20 protein were reduced in the synonymous mutants, as demonstrated by reverse transcriptase quantitative PCR (RT-qPCR), and liquid chromatography tandem mass spectrometry (LC-MS/MS; fig. 1C and D) using tandem mass tagging (TMT). We used two sets of primers in the rpsT gene for the RT-qPCR (fig. 1A and supplementary table SII, Supplementary Material online). For the G48A and the A150C mutants, the relative quantities of both amplicons were lower than in wild-type, whereas for the T36G and A150G mutants, the product of only one primer pair differed (fig. 1C). The levels of S20 were lower in the reconstructed rpsT synonymous mutants than in the isogenic wild-type control, ranging from 49% (G48A) to 77% (A150G; fig. 1D). In general, the rpsT mRNA levels, S20 protein, and total ribosomal protein levels in the synonymous mutants followed the same pattern as the relative growth rates of the same mutants (fig. 1B–E), with the exception of the A150C mutant that had higher S20 levels than expected.
Compensatory Evolution
The fitness costs caused by the synonymous mutations were ameliorated in an evolution experiment. Eight independent lineages of each mutant and a wild-type control were serially passaged in LB medium for 200 generations (supplementary fig. S1, Supplementary Material online) and isolated clones from lineages that gained in fitness were whole genome sequenced (WGS). In a subset of WGS strains we found candidate compensatory mutations including large tandem duplications covering rpsT as well as mutations in rpoD and rpsT (table 1). None of these mutations were found in any of the evolved wild-type strains. Analyses of these mutations are presented in separate sections below.
Table 1.
Candidate Compensatory Mutations in a Subset of the Evolved Strains.
| rpsT allelea | Compensatory mutation | Designation in figures |
|---|---|---|
| wt | – | wt |
| G48A | rpsT(G-5T) | G48A G-5T |
| G48A | rpsT(G4A [Ala2Thr]) | G48A G4A |
| G48A | rpoD(A340C [Thr114Pro]) | rpoD |
| A150C | rpsT(G19A [Ala7Thr]) | A150C G19A |
| A150C | 0.2 Mbp duplb | 0.2 Mbp |
| A150C | 0.8 Mbp duplb | 0.8 Mbp |
| A150C | 1.4 Mbp duplb | 1.4 Mbp |
| A150C | 1.4 Mbp dupl partial Δb | 1.4 Mbp partial Δ |
aGrowth rates of reconstructed mutants are listed in supplementary table S1, Supplementary Material online.
bSee fig. 2A and supplementary table S1, Supplementary Material online for additional information about the evolved duplications.
Duplication of the rpsT Gene Can Suppress the Deleterious Effects of the Synonymous Mutations
Tandem duplications covering as much as one-third of the genome (0.2–1.4 Mbp) were found in four evolved populations with the A150C synonymous mutation (fig. 2A and table 1). The approximate endpoints are listed in supplementary table SI, Supplementary Material online. All duplications spanned rpsT and either partially restored the relative growth rates from ∼60% to ∼80% for the two larger duplications, or completely restored the growth rate for the smallest duplication (fig. 2B). In one of the A150C evolved populations, we additionally found a clone where 0.8 Mbp was deleted in one copy of the 1.4 Mbp duplication (fig. 2A), leading to an 18% increase in exponential growth rate (fig. 2B). DNA elements lost in the deletion included, for example, oriC and five rRNA-operons. The 0.2 and 1.4 Mbp duplications were each found in two populations. The levels of rpsT mRNA were increased to above that of the wild-type for all of the evolved strains with large duplications. The mass spectrometry failed to detect a corresponding increase in S20 protein levels (supplementary fig. S2, Supplementary Material online) as seen for other mutants measured for both rpsT mRNA levels and S20 levels (see ‘Discussion’ section).
Fig. 2.

Suppression by increased gene dosage. (A) Evolved duplications that suppress the cost of the synonymous mutation A150C. The graphs illustrate sequence read depth from Illumina reads assembled to a reference genome. Stretches with double coverage represent the duplicated regions (double amount of sequence reads assemble to the reference genome in these regions). The increased read depth marked in red corresponds to the Fels-1 prophage, indicating that this phage was replicating in the culture sent for WGS. Additional peaks in coverage illustrate short sequences present in multiple copies on the chromosome. rpsT is indicated with a red line and oriC with a yellow line. (B) Relative exponential growth rates of evolved strains with large duplications. Reported values represent the mean (±SD) of at least six independent experiments. (C) Quantification of rpsT mRNA through RT-qPCR in the four evolved duplication mutants using primer pair 1 as indicated in fig. 1A. Reported values are set relative to a wild-type control and represent the mean (±SD) of at least five replicates. See supplementary fig. S7, Supplementary Material online for values measured with a different set of primers. (D) Relative exponential growth rates of constructed duplications of rpsT (supplementary fig. S4, Supplementary Material online). Reported values represent the mean (±SD) of between seven and eight independent experiments. See supplementary fig. S5, Supplementary Material online for RT-qPCR on chromosomal DNA of the same strains. (E) Relative exponential growth rates of strains with rpsT complemented on a medium copy vector. Reported values represent the mean (±SD) of between 4 and 13 independent experiments. The raw data for the growth measurements is listed in supplementary table SI, Supplementary Material online.
To determine if the suppressing effect of the large evolved duplications (fig. 2A) was caused by an increased copy number of the rpsT gene, we constructed defined chromosomal duplications including only the synonymously mutated rpsT gene (supplementary figs. S4–S6, Supplementary Material online) and measured exponential growth rates (fig. 2D). For all four synonymous mutants, the growth rate increased to the levels of the isogenic wild-type with the same constructed duplication (∼96% of wild-type growth).
As an additional test, wild-type and synonymously mutated copies of the rpsT gene were cloned into the medium copy vector pJN3Kl (Näsvall et al. 2012) and were expressed under the strong PLlacO promoter in the synonymous mutants and in an isogenic wild-type control strain. For all synonymous mutants, the cloned rpsT alleles complemented the deleterious effect of the mutated rpsT (fig. 2E). The growth was restored to wild-type levels in all cases except for the most deleterious synonymous mutation (A150C) that only conferred partial complementation (92% of wild-type growth).
rpoD Compensatory Mutation
Within the genome of an evolved G48A synonymous mutant, an extragenic suppressor mutation was found in the rpoD gene, encoding the σ70 subunit of RNA polymerase (table 1). The mutation (A340C, Thr114Pro) was located in the conserved 1.2-domain that recognizes the -10 promoter element and regulates promoter escape (Bochkareva and Zenkin 2013). The compensatory mutation was introduced into a wild-type strain and into strains carrying the synonymous mutations (supplementary table SI, Supplementary Material online). Growth rates in early exponential phase were measured for the reconstructed mutants. When introduced into an isogenic wild-type the mutation conferred a 9% reduction in growth rate, whereas in the synonymous mutants it conferred sign epistasis such that the T36G, G48A, and A150G mutations in combination with the rpoD mutation had a higher growth rate than the isogenic wild-type strain with the same rpoD mutation (fig. 3A and supplementary table SI, Supplementary Material online). To test if the growth effects were also observed in a competition assay we measured the fitness of the rpoD mutation in the wild-type and G48A background by competition experiments. As expected, the competition assay confirmed the results from the growth rate measurements (fig. 3B). The relative rpsT mRNA and S20 protein levels were determined through RT-qPCR and LC-MS/MS in the same G48A rpoD and control strains (fig. 3C and D). The rpoD mutation increased the levels of both rpsT mRNA and S20 protein, suggesting an effect at the transcriptional level as expected for a subunit of RNA polymerase.
Fig. 3.

Compensatory rpoD mutation. (A) Relative exponential growth rates of reconstructed mutants carrying the candidate suppressor mutation rpoD (Thr114Pro). arpoD mutation introduced into a background where it was not selected. bStrain carrying an FRT scar sequence adjacent to the open reading frame of rpsT. All growth rates are set relative to isogenic wild-type controls (the wild-type strain carrying the FRT scar sequence is not shown in the figure). Reported values represent the mean (±SD). The raw data for all growth measurements are listed in supplementary table SI, Supplementary Material online. (B) Relative fitness in competitions. The reported values represent the average (± SD) of eight independent experiments. (C) Quantification of rpsT mRNA through RT-qPCR using primer pair 1 as indicated in fig. 1A. Reported values are set relative to an isogenic wild-type control and represent the mean (±SD) of at least five replicates. See supplementary fig. S7, Supplementary Material online for values measured with a different set of primers. (D) Quantification of S20 protein through LC-MS/MS. The values are the averages (±SD) of two biological replicates relative to a wild-type sample analyzed in the same 10-plex run.
Intragenic Compensatory Mutations and Directed Mutagenesis of rpsT
Three intragenic compensatory mutations near the 5′ end of the rpsT transcript were isolated during the compensatory evolution (fig. 4A and table 1). Among those, two mutations were changes of alanine GCU codons to threonine ACU codons (codons 2 and 7) in the first stem-loop of the transcript. These mutations completely restored the growth rate of the strains with the two synonymous mutations A150C and G48A, respectively (fig. 4B). The third compensatory mutation in rpsT, a mutation (G-5T) in the Shine–Dalgarno (SD) sequence partially restored the fitness of the G48A synonymous mutant.
Fig. 4.

Intragenic rpsT suppressor mutations. (A) Predicted secondary structure of the S. enterica rpsT P2 transcript and intragenic mutations compensating the cost of the synonymous mutations. The figure is adapted from the structure of the E. coli rpsT transcript (fig. 1; Mackie 1992), but altered to show the S. enterica sequence. The locations corresponding to the three prominent RNase E cleavage sites in the E. coli structure (Mackie 2013) are indicated, as well as the site for translational fusions. Letters in boxes indicate the four synonymous mutations studied and letters without boxes indicate mutations that compensate for the cost of the synonymous mutations. The mutations are color coded so that mutations in red compensate for the G48A mutation, turquoise for the A150C mutation and grey for the A150G mutation. aMutants found in the evolution experiment. All other mutants were generated through random mutagenesis. bThe C-19U and the A209G mutations were found together in the same mutant, cas were the A8C and the A245G mutations. (B) Growth rates of reconstructed mutants. All values are set relative to an isogenic wild-type and represent the mean (±SD) of at least four independent experiments. The values are also listed in supplementary table SI, Supplementary Material online. (C) Scatter plot of growth rate measurements for the compensatory mutations found in (A) plotted against transcript position. +1 is the first base pair in the UUG start codon. In the cases where the mutant was not re-constructed, growth rate measurements from the original mutant were used (see supplementary table SI, Supplementary Material online). Roman numbers refers to the stem loops found in (A). (D) Quantification of rpsT mRNA through RT-qPCR of reconstructed mutants using primer pair 1 as indicated in fig. 1A. Reported values are set relative to an isogenic wild-type control and represent the mean (±SD) of at least five replicates. See supplementary fig. S7, Supplementary Material online for values measured with a different set of primers. (E) Quantification of S20 protein through LC-MS/MS. The values are the averages (±SD) of two biological replicates relative to a wild-type sample analyzed in the same 10-plex set. (F) Scatter plot of the data in (D) and (E). Data for the T36G and A140G mutants are also included.
To investigate if other substitutions in the same gene could suppress the cost conferred by the synonymous mutations, we randomly mutagenized the rpsT 5′-untranslated region and open reading frame (originating from promoter P2) of the synonymous mutants and selected for improved growth rate. For the G48A, A150C, and A150G synonymous mutations; 13, 35, and 10 different rpsT mutations, respectively, that reduced the fitness cost were found (fig. 4A and supplementary table SI, Supplementary Material online). No mutations ameliorating the cost of the T36G mutant were found during the screening. In total, we identified 56 different strains with single compensatory mutations and two with double mutations (supplementary table SI, Supplementary Material online). The compensatory mutations clustered in the SD sequence and in the proposed stem loop structures I, II, IV, and V of the transcript (fig. 4A). In addition, a χ2 test did not support a uniform distribution (P < 2.2 × 10−16) and a Gaussian Kernel Density Estimation (Parzen 1962) indicated four clusters separated by stem loop III (supplementary fig. S3, Supplementary Material online). Two positions were frequently mutated and suppressor mutations in these positions were found for all three synonymous mutants: the A at position 18 (changed to a T or a C) and the T at position 195 (changed to any other nucleotide). At nine positions, we found mutations that compensated for more than one of the synonymous mutations, indicating that these compensations were not specific for any of the synonymous mutants. Compensated mutants with mutations in the beginning of the transcript had higher growth rate than those with mutations in the second half of the transcript (P < 0.0001, two-tailed Student’s t-test, equal variance; fig. 4C).
Nineteen intragenic suppressor mutants from the random mutagenesis, as well as the three evolved intragenic suppressor mutants, were reconstructed and the growth rates in exponential phase were determined (fig. 4B). All grew faster than the parental synonymous mutants. Seven of the reconstructed mutants (including the three evolved mutants) were also subjected to rpsT mRNA quantifications through RT-qPCR (fig. 4D). In all cases, the intragenic rpsT suppressor mutants had higher mRNA levels than the synonymous mutants alone, and in some cases the mRNA levels exceeded the levels of the wild-type control strain. S20 protein levels were increased to near wild-type levels, or higher, in strains with the three evolved internal compensatory mutations (fig. 4E). The rpsT mRNA levels correlated with the S20 protein levels (fig. 4F).
Effects of rpsT Mutations on rpsT Gene Expression
To further determine how the different rpsT mutations affected gene expression of the rpsT gene, translational fusions of rpsT to a yellow fluorescent protein (yfp) reporter gene were generated in the wild-type, the parental synonymous mutants and in 19 intragenically compensated mutants (fig. 5A and B and supplementary fig. S8 and supplementary table SI, Supplementary Material online). YFP fluorescence levels were measured to determine the intracellular concentration of S20-YFP fusion protein. Changes in YFP levels reflected the combined effects on transcript levels and translation that the different rpsT alleles have. The effects on transcript levels include direct effects such as transcription initiation and altered degradation by RNase cleavage, as well as downstream stabilizing effects of translating ribosomes.
Fig. 5.

rpsT expression measured by fusions of the rpsT gene to the reporter gene yfp. (A) Translational fusions of rpsT to yfp. aindicates different alleles of the rpsT gene. (B) Chromosomal location of the fusion described in (A). The construct was created through insertion duplication as described in supplementary fig. S6, Supplementary Material online, resulting in the rpsT::yfp fusion located next to the original rpsT gene with the same mutations as present in the fusion. We named this fusion rpsT*::yfp-(rpsT*). (C) Chromosomal location of fusions moved to a neutral position close to the terminus (cobA). A wild-type copy of rpsT is present in its native position. We named this fusion rpsT*::yfp_(rpsTwt). (D) Fluorescence of rpsT*::yfp-(rpsT*) translational fusions in relation to exponential growth rates of reconstructed rpsT alleles in the same rpsT mutants. All values are set relative to isogenic wild-type controls and represent the mean (±SD) of at least three independent experiments. (E) As in (D) but with rpsT*::yfp_(rpsTwt) fusions. (F) Correlation between the relative fluorescence of translational fusions rpsT*::yfp-(rpsT*) and rpsT*::yfp_(rpsTwt). The wild-type fusions have not been included in the calculations for the correlation coefficient.
The rpsT::yfp fusions were generated by forcing the duplication of part of rpsT by insertion of an yfp-kan (kanamycin resistance) cassette, in strains carrying the various rpsT alleles (figs. 4A and 5A). The rpsT locus in the resulting strains then contained a truncated rpsT fused to yfp, as well as a complete rpsT copy. Both the rpsT::yfp construction and the complete rpsT copy carried the same mutations (fig. 5B) and we refer to these fusions as rpsT*::yfp-(rpsT*) in text and figures. The phenotypes of the rpsT mutants could be expected to result in global effects on translation, which would further exacerbate the effect of the synonymous mutations on rpsT expression. Thus, we also wanted to test the effect of the mutations on expression from the rpsT transcript in the presence of fully functional ribosomes. We therefore transferred the rpsT::yfp-kan part of the constructs to the cobA locus in strains carrying the wild-type rpsT in its native locus (fig. 5C). We refer to these fusions as rpsT*::yfp_(rpsTwt). The expression of the fusions was measured in both of these contexts (fig. 5D and–E and supplementary fig. S8, Supplementary Material online).
When no wild-type copy of rpsT was present, the expression of the fusion proteins was more severely affected than when a wild-type copy was present (fig. 5D–F). This indicates that reduced levels of S20 cause a global effect on translation that further reduces S20 expression. Accordingly, the exponential growth rate of the rpsT*::yfp_(rpsTwt) fusions was identical to the growth of our wild-type strain without fusions (supplementary fig. S9, Supplementary Material online).
Five of the rpsT*::yfp_(rpsTwt) translational fusions with compensatory mutations showed higher expression levels than wild-type (fig. 5E and supplementary fig. S8B, Supplementary Material online). These compensatory mutations all localized in the beginning of the transcript. Expression of the rpsT*::yfp-(rpsT*) translational fusions correlated strongly with the corresponding rpsT*::yfp_(rpsTwt) fusions (fig. 5F). In addition, the general expression patterns among the synonymous mutants (measured by the fusions) strongly reflect that seen for growth (fig. 1B), where A150C has the strongest effects followed by G48A, T36G, and lastly A150G.
Do the rpsT Mutations Confer Their Effect on Transcript Level or Translation?
Translating ribosomes are known to protect the mRNA from degradation (Lemm and Ross 2002; Baker and Mackie 2003; Deana and Belasco 2005; Kaberdin and Bläsi 2006; Lazrak et al. 2013). This further complicates studies of mechanistic effects of mutations since a mutation that appears to primarily affect the levels of mRNA can be a downstream effect of an altered degradation pattern due to a change in ribosomal protection of the transcript, making expression analyses difficult to interpret. In order to circumvent this complication in the study of the mechanistic effects of the rpsT mutations we exchanged the GGAGT SD sequence of rpsT to CCTCC in 12 of the rpsT*::yfp_(rpsTwt) translational fusions and thereby effectively eliminated translation initiation from the transcript (fig. 6A and supplementary fig. S10, Supplementary Material online). These mutants were analyzed by quantitative RT-PCR, using one primer pair that bridges rpsT and yfp and one that only amplifies a stretch of the yfp-part of the transcript (fig. 6A, supplementary table SII, Supplementary Material online). The two primer pairs gave very similar results (fig. 6B and supplementary fig. S11, Supplementary Material online). Removing the SD (and translation) resulted in a ∼17-fold decrease of transcript levels (supplementary fig. S11, Supplementary Material online), reflecting the importance of translating ribosomes in stabilizing mRNA (“ribosomal protection”).
Fig. 6.
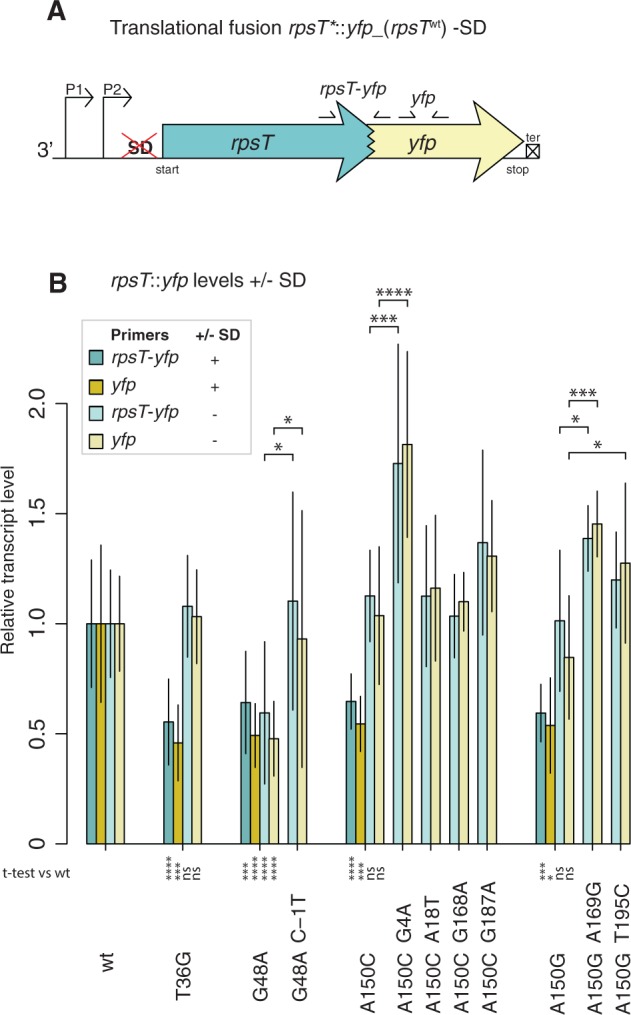
Transcript levels of rpsT*::yfp_(rpsTwt) +/- rpsT -SD (A) Constructed rpsT*::yfp_(rpsTwt) translational fusions lacking the SD of rpsT -SD. (B) Quantifications of the rpsT::yfp transcript through RT-qPCR, in the presence and absence of rpsT SD. Reported values are first related to two reference genes and then normalized to the transcript level in an isogenic wild-type control and represent the mean (±SD) of at least four replicates. Two primer pairs were used as indicated in (A). *indicates P < 0.05, ***P < 0.001, and ****P < 0.0001 (two-tailed Student’s t-test, equal variance) as calculated between mutant and wild-type if not annotated otherwise. For values that are only normalized to the reference genes see supplementary fig. S11, Supplementary Material online.
With an intact SD, all the synonymous mutants had lower levels of the transcript as compared with an isogenic wild-type control (fig. 6B), as seen earlier both with the YFP measurements from the same strains and with RT-qPCR measurements on the synonymous mutants with no fusions (supplementary fig. S8B, Supplementary Material online and fig. 1C). When eliminating translation, the G48A mutant still had reduced rpsT mRNA levels compared with an isogenic wild-type (fig. 6B), indicating that this mutation at least partially confers its deleterious effects by lowering the amount of rpsT mRNA independently from any effect on translation. With this method we cannot separate any combination effect on transcript levels and translation from effects on transcript level only. For the T36G, A150C, and A150G -SD mutants, the mRNA levels were not significantly different from wild-type (fig. 6B), suggesting that these mutations confer their effect on a translational level since the effect is no longer detectable when the transcript is not translated. Similarly, we saw a trend for the C-1T, G4A, A169G, and T195C compensatory mutations in the -SD background to have higher rpsT mRNA levels than their corresponding synonymous mutant alone (fig. 6B), indicating that parts of their compensating effect was on the transcript level, whereas the rpsT mRNA levels for the A18T, A168G, and A187G compensatory mutation did not differ from the corresponding synonymous mutant, indicating that these compensatory mutations act on a translational level rather than on the transcript level.
Discussion
We investigated the cause for the fitness costs of four different synonymous base pair substitutions in the gene rpsT and how these costs can be compensated. With regard to the fitness costs of synonymous mutations, we found that mRNA and protein levels generally co-varied with the relative growth rates of the mutants (fig. 1B–D and supplementary fig. S8, Supplementary Material online), suggesting that a reduction in S20 levels in the mutants is the primary cause of the deleterious effects (fig. 7). More specifically, the initial dysfunction of the synonymous mutants was indicated to be at the level of translation for the T36G, A150C, and A150G mutants, and at least partially on the transcript level for the G48A mutant, as revealed when measuring the transcript levels of the rpsT translational fusions deleted of the SD sequence (fig. 6).
Fig. 7.
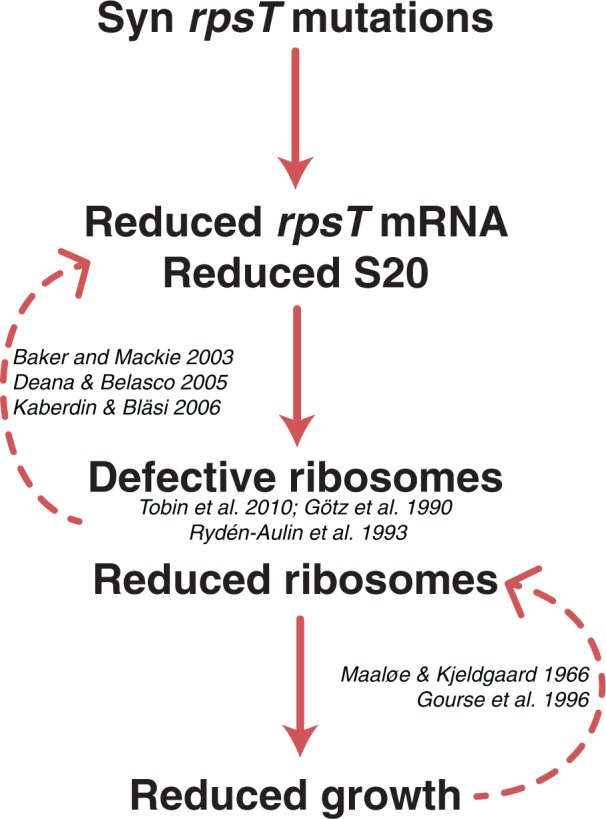
Model describing the cost of the rpsT synonymous mutants. The synonymous rpsT mutations result in reduced levels of S20 protein, generating a subpopulation of 30S subunits lacking S20. Ribosomes lacking S20 are defective in translation initiation (Götz et al. 1990; Rydén-Aulin et al. 1993; Tobin et al. 2010). Inefficient translation initiation on the rpsT mRNA alters the coverage by ribosomes on the transcript, leaving it more accessible to nucleases (Baker and Mackie 2003; Deana and Belasco 2005; Kaberdin and Bläsi 2006), thus further reducing S20 expression. Global effects on translation initiation leads to reduced growth rate, which may reduce ribosome synthesis through growth rate dependent regulation (Maaløe and Kjeldgaard 1966; Gourse et al. 1996).
Deleting the ribosomal binding sites caused ∼17-fold decrease in mRNA levels (supplementary fig. S11, Supplementary Material online), which is probably due to the loss of the protecting effect of mRNA by translating ribosomes. It is likely that the synonymous mutants have a similar but not as severe effect on ribosomal protection (fig. 7). In accordance with this idea, the synonymous mutants showed less reduction in expression of the translational fusions when a wild-type copy of the rpsT gene was present in the genome compared with when a mutant copy was present (fig. 5D–F). Further supporting the importance of ribosomal protection on rpsT is a study by Baker and Mackie (2003) in which they found that stabilization of the Escherichia coli rpsT mRNA is dependent on efficient translation initiation and transit of ribosomes past the third RNase E cleavage site.
A reduction in S20 level is likely to cause a global impairment of translation both by decreasing the total amount of ribosomes (figs. 1E and 7) and by increasing the fraction of ribosomes lacking S20. S. enterica mutant ribosomes that lack S20 (due to deletion of the rpsT gene) are defective in translation initiation due to a reduction of the rate of mRNA binding and rate of formation of 70S initiation complexes (Tobin et al. 2010). Similar conclusions have been drawn from studies on S20 deficient E. coli strains (Götz et al. 1990; Rydén-Aulin et al. 1993). Thus, 30S initiation complexes lacking S20 are unable to dock to the 50S subunit, blocking translation initiation. As a consequence, also fully functional ribosomes containing S20 would have reduced access to the same mRNA, resulting in further reduction of translation and concomitant decrease in fitness (fig. 7). However, it should be noted that we cannot exclude that the reduction in S20 levels confer other pleiotropic effects that reduce fitness. Ribosome synthesis is under growth rate dependent regulation and therefore a reduction in growth rate is expected to result in reduced ribosome synthesis, which could have unpredictable effects on the fitness of rpsT mutants.
To select for mutations that compensate for the cost of the synonymous mutations, we evolved independent lineages of each mutant strain until the growth rates were restored. Large duplications including rpsT as well as mutations in rpoD and rpsT could compensate for the fitness costs of the synonymous mutations (table 1). Regarding the duplications (fig. 2A), subsequent plasmid complementation and construction of duplications that only included the rpsT gene and its promoters confirmed that the increased gene copy number of rpsT compensate for the synonymous mutations (fig. 2D and E). Our protein quantification through mass spectrometry did not indicate any increase in S20 in the strains with large duplications including the rpsT gene (supplementary fig. S2, Supplementary Material online). We see no obvious explanation for this, but it might be due to a problem with normalization of the protein levels in strains with large duplications. The rpoD mutation caused increased rpsT expression (fig. 3C and D and supplementary fig. S7, Supplementary Material online), and we propose that the compensatory effect caused by the rpoD mutation is due to increased transcription of the rpsT gene.
In the evolution experiment and the targeted mutagenesis, we found 61 mutations that could compensate the cost of the three synonymous rpsT mutations (fig. 4A and C, supplementary table SI, Supplementary Material online). They caused up-regulation of both rpsT transcript and S20 protein levels (fig. 4D–F), and fully or partially restored exponential growth rates (fig. 4B and C). The most extreme case was seen for the G19A compensatory mutation in the A150C background, which resulted in a 72% increase in exponential growth rate. We observed a high degree of overlap (20%) in the positions for compensatory mutations when comparing the three synonymous mutants (fig. 4A), and in six cases the suppressing base pair substitutions were identical for the synonymous mutants. This finding indicated that a substantial proportion of the compensatory mutations acted as general suppressors of the synonymous mutations, and that there are many possibilities for compensation by up-regulation of this gene. The compensatory mutations caused both synonymous and nonsynonymous changes, suggesting that at least some of the compensatory mutations were at the level of mRNA rather than protein.
The compensatory mutations clustered in the beginning of the transcript and in stem loop structures I, II, VI, and V (fig. 4A and supplementary fig. S3, Supplementary Material online), with many large effect mutations in and before stem loop I (fig. 4C). S20 synthesis is subject to feedback auto-regulation, such that excess S20 inhibits translation initiation on the rpsT mRNA to balance S20 expression with rRNA synthesis (Parsons and Mackie 1983). The rpsT leader sequence and the UUG start codon in stem loop I have previously been reported to be critical for auto-regulation as well as translation efficiency (Parsons et al. 1988; Mackie 2013). With an intact auto-regulation mechanism S20 levels would therefore be expected not to exceed the wild-type levels. Five of the compensatory rpsT mutations expressed more S20-YFP fusion protein than the wild-type (supplementary fig. S8B, Supplementary Material online). These mutations cluster in stem loop I and in the translation–initiation region, and may have compensated the reduced expression of S20 by disabling the negative feedback loop. Other mutations in stem loop I may have affected translation initiation without disabling the feedback regulation. We see no obvious explanation for the effects of the compensatory mutations in stem loops II, IV, and V.
What are the general implications of these results? First, they demonstrate how compensatory mutations can serve as a driving force for evolution, by rapidly and efficiently restoring fitness to defective mutants. In this study, we characterized 61 intragenic and five extragenic mutations that in many cases could restore fitness to near wild-type levels. This observation appears general, as similar results have been obtained in many other systems. For example, several studies show that viruses with de-optimized codons that reduce fitness can adapt by compensatory mutations after multiple passages in cell cultures. In some cases, the gain in fitness was slow as predicted but in other cases it was relatively rapid (Bull et al. 2012; Martrus et al. 2013). One outstanding finding in eukaryotic viruses is a strong selection against the dinucleotides CpG and UpA, which are targets for innate immunity (Burns et al. 2009; Atkinson et al. 2014; Tulloch et al. 2014). This again points toward multiple conflicting selective pressures within genes that affect codon bias. Similar to what we report here, mutations that reverse the phenotypic effects of codon-deoptimization in viruses generally do not revert the original synonymous changes, and often fall outside the altered genomic regions (Bull et al. 2012; Martrus et al. 2013; Bull 2015). Furthermore, many studies of compensatory evolution of drug-resistant bacterial and viral mutants show that fitness-restoring compensatory mutations are pervasive and common (Andersson and Hughes 2010; zur Wiesch et al. 2010; Hughes and Andersson 2015). Overall, these reports highlight the fact that compensatory mutations that occur somewhere else in the gene or at a different locus in the genome are generally more common than reversion of a specific nucleotide change, increasing the likelihood that fitness restoration occurs by compensation rather than true reversion.
Second, it should be noted that the costs of synonymous mutations varies extensively depending on their location and type. The strength of selection (s) for optimal codon usage has been estimated to be between 10−4–10−9 per codon and generation (Bulmer 1991; Hartl et al. 1994), and recent experiments give s-values in the range of 10−5–10−4 per codon and generation in the highly expressed bacterial tufA gene carrying multiple de-optimizing synonymous mutations (Brandis and Hughes, 2016). However, several studies have reported substantial effects on fitness (s ≥ 10−2) or protein expression from single synonymous mutations (Sanjuán et al. 2004; Carrasco et al. 2007; Kudla et al. 2009; Lind et al. 2010). The effects caused by these synonymous mutations are several orders of magnitude larger than estimated or experimentally determined s values against nonoptimal codons (Brandis and Hughes, 2016, Bulmer 1991; Hartl et al. 1994). Thus, the substantial deleterious effects from large-effect mutations can probably not be explained by effects on rate or accuracy of translating a single codon. It should be noted that we chose the most costly synonymous mutations in rpsT for this study. Thus, in the case of rpsT the probable explanation for the large deleterious effects is that the mutations trap the cells in a vicious circle, where less S20 protein leads to accumulation of dysfunctional ribosomes, causing less translation and more degradation of the rpsT mRNA, further reducing S20 levels and so on (fig. 7). Similar vicious circles are perhaps likely to be found in other highly expressed genes involved in the transcription or translation machinery where reduced expression of the mutated gene could lead to further reduction in its own expression. However, even in the presence of a wild-type rpsT gene, our gene fusions reported lower expression when they contained the four synonymous mutations than when they contained the wild-type sequence (fig. 5E and supplementary fig. S8, Supplementary Material online), indicating that the primary defect that got the cells into the vicious circle in the first place is significant even when the vicious circle is broken. Similarly, some synonymous mutations in the genes encoding GFP and TEM-1 β-lactamase cause reduced expression of the corresponding proteins (Kudla et al. 2009; Firnberg et al. 2014), hinting toward a general phenomenon of deleterious “silent” mutations. Thus, this study and others highlight the presence of several overlapping codes within genes, with gene-specific trade-offs in selection for translation rate and accuracy, protein folding, transcription rate and processivity, mRNA stability, processing, and turnover. This may have implications for the use of dN/dS ratios as signatures for selection as, in contrast to what is commonly assumed, synonymous sites can be under strong selection.
Materials and Methods
Strains and Media
Salmonella enterica subsp. enterica serovar Typhimurium str. LT2 (designated S. enterica in the text), or derivatives thereof, was used in all experiments, except for the cloning where the E. coli cloning strain DH5α was used. Phage P22 HT int (Schmieger 1972) was used for transductions. LB (5 g yeast extract, 10 g Tryptone, and 10 g NaCl L−1) was used as liquid medium and LA (LB with 15g L−1 agar) as solid medium. When needed, antibiotics in the following concentrations were supplemented to the media: 100 mg L−1 ampicillin, 12.5 mg L−1 chloramphenicol, 50 mg L−1 kanamycin, and 7.5 mg L−1 tetracycline. To select for loss of the cat-sacB cassette, LA without NaCl, supplemented with 50 g L−1 sucrose was used. When growing cells to exponential phase to prepare samples for LC-MS/MS, FACS analysis or mRNA extractions for RT-qPCR LB was supplemented with 2 g L−1 glucose in order to allow for exponential growth at higher OD. In addition, LB without NaCl was used when growing cultures for making electro-competent cells.
Strain Construction
All strains are listed in supplementary table SI, Supplementary Material online and primers used for the constructions are listed in supplementary table SII, Supplementary Material online. All new constructs were verified with PCR and sequencing and all constructs made through λ red recombineering (Datsenko and Wanner 2000; Yu et al. 2000) were moved to other strains through P22 transduction before use. The latter was to minimize the risk of unwanted mutations that could have formed during expression of the λ red system from the pSIM5-Tet plasmid (Koskiniemi et al. 2011). For many constructs, multiple independently constructed strains were made in parallel (supplementary table SI, Supplementary Material online). If, during analysis, the independent strains deviated from each other, the strains were reconstructed. This happened at two instances.
Construction of Strains for Compensatory Evolution
The synonymous mutants used in the evolution experiment are derivatives of strains described in Lind et al. (2010). Prior to the compensatory evolution experiment, the kanamycin resistance marker (FRT-kanR-FRT from pKD4) adjacent to rpsT was removed from these strains by expression of FLP recombinase from plasmid pCP20 (Cherepanov and Wackernagel 1995).
Reconstruction of Synonymous Mutants
Whole genome sequencing of evolved strains reveled that some of the synonymous mutants carried additional mutations that were accidentally brought in during strain construction and were therefore present before the evolution. Those were, for example, an SNP in the ribF gene, neighboring rpsT, and a deletion of 1 bp in the FRT scar sequence in the A150C mutant (supplementary table SIII, Supplementary Material online). The synonymous mutants were therefore reconstructed in a mutation free background. To remove mutations nearby rpsT (co-transduces at high frequency) we first introduced a chloramphenicol resistance marker (cat) from pEL3C16(FRT) (lab collection) upstream of rpsT in the wild-type strain through λ red recombineering (Datsenko and Wanner 2000; Yu et al. 2000). In a second λ red recombineering step, the synonymously mutated rpsT and the downstream kanR was moved from the strains constructed by Lind et al. (2010), into the strain with cat upstream of rpsT. To find strains that recombined to reconstruct synonymously mutated rpsT-kanR we screened for mutants resistant to kanamycin and sensitive to chloramphenicol. Thereafter the kanR was removed by expressing the FLP recombinase from plasmid pCP20 (Cherepanov and Wackernagel 1995).
To lose the FRT scar mutation in the A150C mutant and to reconstruct the three evolved rpsT alleles, the FRT-kanR-FRT from pKD4 (Datsenko and Wanner 2000) was re-introduced downstream rpsT through λ red recombineering (Datsenko and Wanner 2000; Yu et al. 2000). The constructs were moved with P22 transduction into the wild-type DA6192 strain, where after the kanR was removed as described above, leaving an intact FRT scar. Reconstruction of four of the synonymous mutants reveled that the unintentional mutations conferred a detectable decrease in fitness in the A150C and A150G mutants (7.5% and 6.3%, respectively; supplementary table SIII, Supplementary Material online). The evolved duplications were not reconstructed and thereby carried a deletion of 1 bp in the FRT scar sequence.
Reconstruction of Compensated Mutants
To move the evolved rpoD mutation into the reconstructed synonymous mutants, FRT-cat-FRT from pEL3C16(FRT) (lab collection) was first inserted ∼5 kb away from the mutation in rpoD (yielding ∼50% co-transduction frequencies), by means of λ red recombineering (Datsenko and Wanner 2000; Yu et al. 2000). P22 transduction and selection for chloramphenicol resistance was used to introduce the mutation in the synonymous mutants. The rpoD gene was sequenced in several transductants to find clones that had inherited the mutated allele of rpoD, as well as those that only inherited the cat-cassette. The latter strains were used as controls in the further experiments. These reconstructed strains were later used for growth rate measurements, competitions and mass spectrometry analysis, as indicated in supplementary table SI, Supplementary Material online.
Construction of Scar Free Strains
The FRT scar sequence adjacent to the rpsT open reading frame was found to confer a 6% growth defect in our wild-type strain (supplementary table SIII, Supplementary Material online). We therefore constructed scar-free synonymous mutants in which we introduced the majority of the compensatory mutations. This was performed by first exchanging the FRT scars in the synonymous mutants, and in the three evolved rpsT alleles, with a cat-sacB cassette (GenBank: KM018298), using λ red recombineering (Datsenko and Wanner 2000; Yu et al. 2000). Then, a 70-bp oligo (purified by size) spanning the site the cat-sacB insertion was allowed to recombine to exchange cat-sacB (Ellis et al. 2001) followed by selection for sucrose resistance thus generating cat-sacB- strains. To make sure no other mutations were introduced during the recombination, P22 lysates were grown on both the cat-sacB strain and the cat-sacB- strains. The cat-sacB was transduced into our wild-type strain (DA6192). There after the cat-sacB- lysates were transduced into the reconstructed cat-sacB strain, followed by selection for sucrose resistance. The compensatory mutation in rpoD was later introduced into these scar-free strains in the same manner as described above. The reconstructed scar-free strains were used for growth rate measurements, quantitative PCR, directed mutagenesis, plasmid complementation, rpsT duplications, and translational fusions with rpsT. The FRT scar sequence was found to be deleterious in the wild-type control strain (−5%), while in the A150C mutant it conferred a benefit (5%). In the other synonymous mutants, the FRT scar had no detectable effect on growth rate (supplementary table SIII, Supplementary Material online).
Construction of Strains for Competitions
Chromosomal copies of the fluorescent protein genes bfp (mtagbfp2, blue; Subach et al. 2011; Gullberg et al. 2014) and yfp (syfp2, yellow; Kremers et al. 2006; Gullberg et al. 2014) were moved with P22 transduction into strains used in the competitions. The two fluorescent protein genes were localized within the galK-locus of Salmonella.
Construction of Plasmids
To construct rpsT-complementation vectors we amplified the rpsT wild-type sequence and synonymously mutated rpsT together with the rpsT RBS sequence (13 bp upstream the TTG start codon). The primers used were designed to contain the EcoRI restriction site on the forward primer and PstI on the reverse primer. The rpsT sequences were cloned under the PLlacO promoter in the cloning vector pJN3Kl-<mRFP1> (Näsvall et al. 2012).
Constructed Duplications
Chromosomal insertion–duplications were constructed to form the rpsT-duplications (supplementary fig. S4, Supplementary Material online and fig. 2D) and the rpsT translational fusions (fig. 5A–C). A schematic representation of the construction procedure is found in supplementary fig. S6, Supplementary Material online and primers used in the construction are found in supplementary table SII, Supplementary Material online.
To construct the rpsT-duplications (supplementary fig. S4, Supplementary Material online) a bla resistance marker flanked by transcriptional terminators was amplified from pEL3A17 (lab collection) and used as the selectable marker. The two known promoters of rpsT (Mackie and Parsons 1983) as well as the rpsT terminator were included in the duplicated region. Quantitative Real-time PCR runs on strains with these constructed duplications showed no signs of further amplifications of rpsT (supplementary fig. S5, Supplementary Material online).
For the translational fusions of rpsT to the yfp reporter gene (rpsT-yfp-kanR-rpsT; fig. 5A–C and supplementary fig. S8, Supplementary Material online), the yfp and kanR were PCR amplified from a strain carrying the yfp gene (syfp2; Kremers et al. 2006; Gullberg et al. 2014) upstream of a kanR cassette that originates from pKD4 (Datsenko and Wanner 2000). For the rpsT translational fusion, the forward primer was instead designed so that the rpsT open reading frame merged in frame with the open reading frame of yfp, excluding the yfp start codon. The amplified fragments were recombined into the reconstructed rpsT-mutant strains through λ red recombineering (Datsenko and Wanner 2000; Yu et al. 2000).
A subset of the translational fusions were moved into the cobA locus of an otherwise wild-type strain (harboring a wild-type copy of the rpsT gene in its native position) through λ red recombineering (Datsenko and Wanner 2000; Yu et al. 2000).
Construction of rpsT -SD Strains
For a subset of the rpsT translational fusions located in cobA we wanted to eliminate the SD sequence of the fusions so that the transcript could no longer be translated. To achieve this, we first introduced a cat-sacB cassette in the position for the expected rpsT SD (GGAGT) and re-constructed this strain by P22 transduction into our wild-type strain. We then exchanged the cat-sacB cassette through λ red recombineering (Ellis et al. 2001) by an oligo designed to cover 39 bp on each side of the expected SD and that had the SD exchanged to CCTCC. For re-construction of the genetically engineered strains, we moved the -SD constructs into the re-constructed cat-sacB strain and selected for sucrose resistance. All primers used are found in supplementary table SII, Supplementary Material online. No YFP expression was detected from the transcripts, neither in exponentially growing cultures (supplementary fig. S10, Supplementary Material online), nor in stationary cultures (data not shown).
Analysis of Translational Fusions
Overnight cultures were diluted 100-fold in LB supplemented with 2 g L−1 glucose and appropriate antibiotic and were grown to early exponential phase (the strict majority of OD600 were between 0.18 and 0.35) and outliers did not affect fluorescence showing that all cultures of the same strain were in the same growth phase. 15 µL culture was removed and mixed in phosphate buffered saline (PBS; 2.7 mM KCl, 1.4 mM KH2PO4, 137 mM NaCl, 4.3 mM Na2H2PO4, pH 7.3), supplemented with 30 mg L−1 tetracycline (used to stop translation) and the fluorescent proteins were allowed to mature for at least 45 min before quantification. The fluorescence of 105 cells was measured using a fluorescence-activated cell sorter (BD FACS Aria) and set relative to the fluorescence from isogenic wild-type controls.
Directed Mutagenesis
The synonymously mutated rpsT genes were exchanged by mutagenized copies of the same gene where after mutants with increased growth rates were selected. First, mutagenic PCRs were performed in 50 µL reactions with 1.5 mM MgCl2, 1× taq buffer (MgCl2-free), 200 µM dNTPs, 0.3 µM of each primer, 5 U Taq AmpliTaq Gold DNA Polymerase, 0.1 ng/µL DNA template, and 2–10 µL mutagenic buffer containing: 4 mM dTTP, 4 mM dCTP, 27.5 mM MgCl2, and 2.5 mM MnCl2 (Rasila et al. 2009). The primers were placed so that the entire transcript (excluding the terminator) from the rpsT P1 promoter would be mutagenized. The templates for the mutagenic PCR consisted of PCR products of synonymously mutated rpsT amplified under nonmutagenic conditions. To amplify the template, the same primers were used as those used later in the error prone PCR.
λ red recombineering (Datsenko and Wanner 2000; Yu et al. 2000) was used to exchange the synonymously mutated rpsT along with the cat-sacB cassette in the strains having a cat-sacB cassette adjacent to rpsT with the mutagenized rpsT alleles. To lower the risk for genetically identical clones generated during recovery, the cells were aliquoted into ten tubes containing 1 mL LB, where after they were allowed to recover overnight at 37 °C. The following day the cultures were diluted 100-fold and plated on sucrose selective plates. Faster growing colonies were picked and re-streaked on the same selective media. To confirm that the mutants were growing faster than the parental, synonymous mutants, growth rate measurements were performed (described below), and rpsT was sequenced. In total, 13 different rpsT alleles were picked that suppressed the cost of the G48A mutation, 37 were picked for A150C and ten for A150G. No suppressor mutations were found for T36G in the single experiment we performed. This could be due to, for example, spontaneous sucrose resistant mutants occurring in the overnight culture for this strain. Since we found much overlap in the suppressor mutations isolated for the three other synonymous mutants we decided to not repeat the experiment for the T36G mutation. A selection of 19 of those were reconstructed in a clean background by P22 transduction into strains with the cat-sacB cassette and counter selected for sucrose resistance. These strains were subjected for further studies including growth rate measurements, RT-qPCR, and translational fusions.
Compensatory Evolution
To select for mutations that compensate for the cost of the synonymous mutations, the synonymous mutants were serially passaged in LB at 37°C in a shaking incubator (supplementary fig. S1, Supplementary Material online). Initially, eight independent overnight cultures for each synonymous mutant were started from independent colonies. Each day for 20 days, the lineages were serially passaged by 1,000-fold dilution in 1 mL batch cultures, allowing for ∼200 generations of growth. At days 10 and 20, growth rate measurements (as described below) were conducted and every fifth cycle the strains were frozen in 10% DMSO. In parallel, eight independent wild-type lineages were evolved under the same conditions as the synonymous mutants. This was done to control for growth medium adaptations.
Whole Genome Sequencing and Sequence Analyses
Lineages that conferred increased growth after the serial passage were subjected to whole genome sequencing (BGI, Beijing, China) in order to find candidate compensatory mutations. Independent clones were isolated from the evolved populations and grown overnight in 5 mL LB where after genomic DNA was prepared using Qiagen Genomic Tip 100/G. The obtained Illumina reads were assembled toward a reference genome using CLC Genomics Workbench (CLC Bio, Aarhus, Denmark). SNPs and indels were detected using the probabilistic variant detection tool included in CLC Genomics Workbench. Duplications were found as regions of increased read depth by visual scanning of the assembled reads along the reference genome, and duplication junctions were found as hybrid reads at the borders of the regions with increased coverage. The raw Illumina sequencing datasets have been deposited in the National Center for Biotechnology Information Sequence Read Archive (www.ncbi.nlm.nih.gov/Traces/sra/) with accession number SRP068642.
Growth Rate Measurements
Growth rates in early exponential phase were obtained by measuring optical density at 600 nm every 4 min by using a Bioscreen C Reader (Oy Growth Curves) at 37°C with shaking. Overnight cultures from separate clones of, on average 7 (median 5) independent replicates (supplementary table SI, Supplementary Material online) of each mutant were diluted 1500- or 2000-fold in LB and 300 µL was loaded in wells of a 100-well honeycomb plate. The growth rates were calculated in the OD600 interval 0.02–0.1. The growth was normalized to the growth of isogenic wild-type controls included in each experiment. If several parallel experiments with the same mutants were run, the means were calculated after normalization to the wild-type controls within the same run.
Competitions
Competition experiments to measure relative fitness were performed using the genetically tagged strains with chromosomal copies of bfp and yfp described above. Overnight cultures were grown and the wild-type strain tagged with one of the two fluorescent markers was mixed 1:1 with a mutant carrying the other marker. The following day the competing strains were passaged through 1,000-fold dilution into fresh medium and were allowed to grow for another 24 h. Before the passage and after the last 24-h incubation, the ratio mutant to wild-type was measured by counting 105 cells using a fluorescence-activated cell sorter (BD FACS Aria). Eight independent competitions for each mutant were made where four were between a mutant carrying an yfp marker and a wild-type carrying bfp, and four with strains carrying the opposite markers. Selection coefficients were determined using the regression model s = [ln(R(t)/R(0))]/[t] (Dykhuizen 1990), where R is the ratio of mutant to wild-type and t is number of generations. Since the relative cost of the fluorescent markers varied with genetic background, the average fitness between the four + four replicates were used to account for the cost of the markers. The standard deviation was calculated within the two sets of dye swaps and the average SD between the two sets was used as a measurement for variation.
Reverse Transcriptase Quantitative PCR (RT-qPCR) and Quantitative PCR (qPCR)
For the RT-qPCR overnight cultures were diluted 100-fold in LB supplemented with 2 g L−1 glucose and were allowed to grow to early exponential phase (OD600 0.19–0.28). 500 µL culture was removed and total RNA was prepared, using RNeasy Mini Kit (Qiagen), according to the manufacturer. The RNA was DNase treated, using the Turbo DNA-free kit (Ambion) following the manufacturer’s instructions, and was visually inspected on a 2% agarose gel. 500 ng RNA (quantified using the Qubit RNA BR Assay Kit, according to the manufacturer) was synthesized into cDNA using the High Capacity Reverse Transcription kit (Applied Biosystems), following the manufacturer’s instructions. RT-qPCRs based on the high affinity of SYBR Green dye for double-stranded DNA was used for measuring relative mRNA levels, using PerfeCTa SYBR Green SuperMix (Quanta Biosciences, Gaithersburg, MD), according to the manufacturer. The result was recorded as the CT value, which is inversely related to the presence of the transcript studied. The levels of two rpsT transcript fragments (fig. 1A, supplementary table SII, Supplementary Material online) were calculated relative to the geometrical mean of the two reference genes used (cysG and hcaT), and normalized to the levels of the same two fragments in wild-type cells. Eight biological replicates were made for the wild-type, four for the synonymous mutants, and two for the compensated synonymous mutants. In addition, quantitative PCRs were run at least once in a 1-, 10-, and 100-fold dilution series for all samples, generating a standard curve of three dilutions for each biological replicate. The same variation in [rpsT mRNA] was seen between the biological replicates as between the different dilutions of the same biological replicate, whereby the biological and technical repeats were treated evenly in calculations. In the few cases where one of the three dilutions clearly deviated from the straight line the reaction was excluded.
For rpsT DNA copy number determination, template DNA was prepared from cultures grown over night in LB at 37°C. 100 µL culture was removed and incubated at 95°C for 8 min where after the cell debris was pelleted by centrifugation for 5 min at 13,000 rpm. The supernatant was diluted at 10-, 100-, and 1,000-fold and used as template in qPCR, as described above.
Mass Spectrometry Analysis
Relative quantification of peptides was performed by the Proteomics Core Facility at Sahlgrenska Academy, Gothenburg University. In brief, 200 µL of overnight cultures were diluted to 25 mL LB supplemented with 0.2% glucose and were grown to early exponential phase. Thereafter cells were pelleted, washed three times in PBS, and frozen for further sample preparation. The samples were homogenized in 200 µL lysis buffer (50 mM Triethylammoinium bicarbonate [TEAB; Fluka, Sigma Aldrich, St. Louis, MO] and 8 M Urea using a FastPrep-24 instrument [MP Biomedicals, OH, USA]). The sample buffer was adjusted to 2% sodium dodecyl sulfate, put on a shaker for 30 min and total protein concentration was determined with Pierce BCA Protein Assay (Thermo Scientific). The same amount of total protein was analyzed for each sample. The proteins were trypsin digested using the filter-aided sample preparation method (Wiśniewski et al. 2009) and peptides subjected to chemical labeling using 10-plex isobaric mass tagging reagent TMT according to the manufacturer’s instructions (Thermo Scientific).
The labeled samples were mixed per TMT set, fractionated by strong cation exchange and desalted before LC-MS/MS analysis on a Q Exactive hybrid instrument (Thermo Fisher Scientific) interfaced with an Easy-nLC autosampler (Thermo Fisher Scientific). Peptides were separated on-line with a 0.075-mm inner diameter C18 reversed phase column using 0.2% formic acid and an acetonitrile gradient. Ions were injected in data-dependent positive ion mode and MS/MS spectra were acquired using higher energy collision dissociation in a stepped format at 25%, 35%, and 45% from m/z 110 for the ten most abundant precursors at a resolution of 35,000. Dynamic exclusion during 30 s after selection for MS/MS was enabled to allow for detection of as many precursors as possible.
Data analysis for relative quantification and identification was performed with Proteome Discoverer version 1.4 (Thermo Fisher Scientific). A database search was performed against Uniprot S. enterica subsp. enterica serovar Typhimurium str. LT2 reference proteome with some modifications, MS peptide tolerance of 5 ppm and MS/MS tolerance for identification of 50 millimass units (mmu). Tryptic peptides were accepted with zero missed cleavages and the threshold in the software was set to 1% False Discovery Rate by searching against a reversed database. The relative quantification was calculated for fragment ions within 3 mmu and co-isolation of <10% was allowed. Each 10-plex set included biological duplicates of one wild-type and four mutants. The protein ratio in each sample was calculated toward one of the wild-type controls within the same 10-plex, only peptides unique for a given protein were considered for relative quantification and ratios normalized using the protein median. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al. 2016) partner repository with the dataset identifier PXD003495.
Supplementary Material
Supplementary material, table SI–SIII, and figures S1–S11 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We would like to thank Erik Gullberg and Pikkei Yuen for helping with statistical analyses, and Erik Lundin for the unpublished plasmids pEL3A17 and pEL3C16(FRT). We are also grateful to Hervé Nicoloff, Gerrit Brandis, Linus Sandegren, Gerhart Wagner, Jon Jerlström-Hultquist, Lisa Albrecht and Cédric Romilly for insightful discussions and helpful comments on the manuscript. The Proteomics Core Facility at Sahlgrenska Academy, Gothenburg University, performed the analysis for protein quantification. This study was supported by grants from the Swedish Research Council to DIA.
References
- Andersson DI, Hughes D. 2010. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol. 8:260–271. [DOI] [PubMed] [Google Scholar]
- Andersson SG, Kurland CG. 1990. Codon preferences in free-living microorganisms. Microbiol Rev. 54:198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson NJ, Witteveldt J, Evans DJ, Simmonds P. 2014. The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication. Nucleic Acids Res. 42:4527–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KE, Mackie GA. 2003. Ectopic RNase E sites promote bypass of 5’-end-dependent mRNA decay in Escherichia coli. Mol Microbiol. 47:75–88. [DOI] [PubMed] [Google Scholar]
- Berg OG, Silva PJ. 1997. Codon bias in Escherichia coli: the influence of codon context on mutation and selection. Nucleic Acids Res. 25:1397–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkareva A, Zenkin N. 2013. The σ70 region 1.2 regulates promoter escape by unwinding DNA downstream of the transcription start site. Nucleic Acids Res. 41:4565–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandis G, Bergman JM, Hughes D. 2016. Autoregulation of the tufB gene in Salmonella. Molecular Microbiol. DOI:10.1111/mmi.13364. [DOI] [PubMed] [Google Scholar]
- Brandis G, Hughes D. 2016. The Selective Advantage of Synonymous Codon Usage Bias in Salmonella. PLoS Genet. DOI:10.1371/journal.pgen.1005926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ. 2015. Evolutionary reversion of live viral vaccines: can genetic engineering subdue it? Virus Evol. 1:vev005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ, Molineux IJ, Wilke CO. 2012. Slow fitness recovery in a codon-modified viral genome. Mol Biol Evol. 29:2997–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer M. 1991. The selection-mutation-drift theory of synonymous codon usage. Genetics 129:897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CC, Campagnoli R, Shaw J, Vincent A, Jorba J, Kew O. 2009. Genetic inactivation of poliovirus infectivity by increasing the frequencies of CpG and UpA dinucleotides within and across synonymous capsid region codons. J Virol. 83:9957–9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannarozzi G, Cannarrozzi G, Schraudolph NN, Faty M, von Rohr P, Friberg MT, Roth AC, Gonnet P, Gonnet G, Barral Y. 2010. A role for codon order in translation dynamics. Cell 141:355–367. [DOI] [PubMed] [Google Scholar]
- Carlini DB. 2004. Experimental reduction of codon bias in the Drosophila alcohol dehydrogenase gene results in decreased ethanol tolerance of adult flies. J Evol Biol. 17:779–785. [DOI] [PubMed] [Google Scholar]
- Carlini DB, Stephan W. 2003. In vivo introduction of unpreferred synonymous codons into the Drosophila Adh gene results in reduced levels of ADH protein. Genetics 163:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco P, de la Iglesia F, Elena SF. 2007. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco Etch virus. J Virol. 81:12979–12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary JV, Hurst LD. 2005. Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol. 6:R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. [DOI] [PubMed] [Google Scholar]
- Chursov A, Frishman D, Shneider A. 2013. Conservation of mRNA secondary structures may filter out mutations in Escherichia coli evolution. Nucleic Acids Res. 41:7854–7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampi MS. 2006. Rho-dependent terminators and transcription termination. Microbiology 152:2515–2528. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deana A, Belasco JG. 2005. Lost in translation: the influence of ribosomes on bacterial mRNA decay. Genes Dev. 19:2526–2533. [DOI] [PubMed] [Google Scholar]
- Donly BC, Mackie GA. 1988. Affinities of ribosomal protein S20 and C-terminal deletion mutants for 16S rRNA and S20 mRNA. Nucleic Acids Res. 16:997–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Antezana MA. 2003. Mammalian mutation pressure, synonymous codon choice, and mRNA degradation. J Mol Evol. 57:694–701. [DOI] [PubMed] [Google Scholar]
- Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, Gejman PV. 2003. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet. 12:205–216. [DOI] [PubMed] [Google Scholar]
- Dykhuizen D. 1990. Experimental studies of natural selection in bacteria. Annu Rev Ecol Syst. 21:373–398. [Google Scholar]
- Elf J, Nilsson D, Tenson T, Ehrenberg M. 2003. Selective charging of tRNA isoacceptors explains patterns of codon usage. Science 300:1718–1722. [DOI] [PubMed] [Google Scholar]
- Ellis HM, Yu D, DiTizio T, Court DL. 2001. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc Natl Acad Sci USA. 98:6742–6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A, Bulmer M. 1993. Reduced synonymous substitution rate at the start of enterobacterial genes. Nucleic Acids Res. 21:4599–4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firnberg E, Labonte JW, Gray JJ, Ostermeier M. 2014. A comprehensive, high-resolution map of a gene’s fitness landscape. Mol Biol Evol. 31:1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman DB, Church GM, Kosuri S. 2013. Causes and effects of N-terminal codon bias in bacterial genes. Science 342:475–479. [DOI] [PubMed] [Google Scholar]
- Götz F, Dabbs ER, Gualerzi CO. 1990. Escherichia coli 30S mutants lacking protein S20 are defective in translation initiation. Biochim Biophys Acta 1050:93–97. [DOI] [PubMed] [Google Scholar]
- Gourse RL, Gaal T, Bartlett MS, Appleman Ja, Ross W. 1996. rRNA transcription and growth rate-dependent regulation of ribosome synthesis in Escherichia coli. Annu Rev Microbiol. 50:645–677. [DOI] [PubMed] [Google Scholar]
- Gu W, Zhou T, Wilke CO. 2010. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput Biol. 6:e1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullberg E, Albrecht LM, Karlsson C, Sandegren L, Andersson DI. 2014. Selection of a multidrug resistance plasmid by sublethal levels of antibiotics and heavy metals. MBIO 5:e01918–e01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Moriyama EN, Sawyer SA. 1994. Selection intensity for codon bias. Genetics 138:227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes D, Andersson DI. 2015. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet. 16:459–471. [DOI] [PubMed] [Google Scholar]
- Ikemura T. 1985. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 2:13–34. [DOI] [PubMed] [Google Scholar]
- Kaberdin VR, Bläsi U. 2006. Translation initiation and the fate of bacterial mRNAs. FEMS Microbiol Rev. 30:967–979. [DOI] [PubMed] [Google Scholar]
- Kimchi-Sarfaty C, Oh JM, Kim I-W, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM. 2007. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315:525–528. [DOI] [PubMed] [Google Scholar]
- Koskiniemi S, Pränting M, Gullberg E, Näsvall J, Andersson DI. 2011. Activation of cryptic aminoglycoside resistance in Salmonella enterica. Mol Microbiol. 80:1464–1478. [DOI] [PubMed] [Google Scholar]
- Kramer EB, Farabaugh PJ. 2007. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 13:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers G-J, Goedhart J, van Munster EB, Gadella TWJ. 2006. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Förster radius. Biochemistry 45:6570–6580. [DOI] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Science 324:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazrak A, Fu L, Bali V, Bartoszewski R, Rab A, Havasi V, Keiles S, Kappes J, Kumar R, Lefkowitz E, et al. 2013. The silent codon change I507-ATC->ATT contributes to the severity of the ΔF508 CFTR channel dysfunction. FASEB J. 27:4630–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke JJ, Sanchez-Vazquez P, Burgos HL, Hedberg G, Ross W, Gourse RL. 2011. Direct regulation of Escherichia coli ribosomal protein promoters by the transcription factors ppGpp and DksA. Proc Natl Acad Sci USA. 108:5712–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemm I, Ross J. 2002. Regulation of c-myc mRNA decay by translational pausing in a coding region instability determinant. Mol Cell Biol. 22:3959–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind PA, Andersson DI. 2013. Fitness costs of synonymous mutations in the rpsT gene can be compensated by restoring mRNA base pairing. PLoS One 8:e63373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind PA, Berg OG, Andersson DI. 2010. Mutational robustness of ribosomal protein genes. Science 330:825–827. [DOI] [PubMed] [Google Scholar]
- Maaløe O, Kjeldgaard NO. 1966. Control of macromolecular synthesis; a study of DNA, RNA, and protein synthesis in bacteria. New York: W. A. Banjamin. [Google Scholar]
- Mackie GA. 1992. Secondary structure of the mRNA for ribosomal protein S20. Implications for cleavage by ribonuclease E. J Biol Chem. 267:1054–1061. [PubMed] [Google Scholar]
- Mackie GA. 2000. Stabilization of circular rpsT mRNA demonstrates the 5’-end dependence of RNase E action in vivo. J Biol Chem. 275:25069–25072. [DOI] [PubMed] [Google Scholar]
- Mackie GA. 2013. Determinants in the rpsT mRNAs recognized by the 5’-sensor domain of RNase E. Mol Microbiol. 89:388–402. [DOI] [PubMed] [Google Scholar]
- Mackie GA, Parsons GD. 1983. Tandem promoters in the gene for ribosomal protein S20. J Biol Chem. 258:7840–7846. [PubMed] [Google Scholar]
- Martrus G, Nevot M, Andres C, Clotet B, Martinez MA. 2013. Changes in codon-pair bias of human immunodeficiency virus type 1 have profound effects on virus replication in cell culture. Retrovirology 10:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näsvall J, Sun L, Roth JR, Andersson DI. 2012. Real-time evolution of new genes by innovation, amplification, and divergence. Science 338:384–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudler E, Gottesman ME. 2002. Transcription termination and anti-termination in E. coli. Genes Cells 7:755–768. [DOI] [PubMed] [Google Scholar]
- Pagani F, Raponi M, Baralle FE. 2005. Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. Proc Natl Acad Sci USA. 102:6368–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmley JL, Hurst LD. 2007. Exonic splicing regulatory elements skew synonymous codon usage near intron-exon boundaries in mammals. Mol Biol Evol. 24:1600–1603. [DOI] [PubMed] [Google Scholar]
- Parsons GD, Donly BC, Mackie GA. 1988. Mutations in the leader sequence and initiation codon of the gene for ribosomal protein S20 (rpsT) affect both translational efficiency and autoregulation. J Bacteriol. 170:2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons GD, Mackie GA. 1983. Expression of the gene for ribosomal protein S20: effects of gene dosage. J Bacteriol. 154:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parzen E. 1962. On estimation of a probability density function and mode. Ann Math Stat. 33:1065–1076. [Google Scholar]
- Ramaswamy P, Woodson SA. 2009. Global stabilization of rRNA structure by ribosomal proteins S4, S17, and S20. J Mol Biol. 392:666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasila TS, Pajunen MI, Savilahti H. 2009. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Anal Biochem. 388:71–80. [DOI] [PubMed] [Google Scholar]
- Rocha EPC. 2004. Codon usage bias from tRNA’s point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 14:2279–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydén-Aulin M, Shaoping Z, Kylsten P, Isaksson LA. 1993. Ribosome activity and modification of 16S RNA are influenced by deletion of ribosomal protein S20. Mol Microbiol. 7:983–992. [DOI] [PubMed] [Google Scholar]
- Sanjuán R, Moya A, Elena SF. 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc Natl Acad Sci USA. 101:8396–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieger H. 1972. Phage P22-mutants with increased or decreased transduction abilities. MGG Mol Gen Genet. 119:75–88. [DOI] [PubMed] [Google Scholar]
- Shabalina SA, Ogurtsov AY, Spiridonov NA. 2006. A periodic pattern of mRNA secondary structure created by the genetic code. Nucleic Acids Res. 34:2428–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields DC, Sharp PM, Higgins DG, Wright F. 1988. “Silent” sites in Drosophila genes are not neutral: evidence of selection among synonymous codons. Mol Biol Evol. 5:704–716. [DOI] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. 2007. Synonymous codon usage in Escherichia coli: selection for translational accuracy. Mol Biol Evol. 24:374–381. [DOI] [PubMed] [Google Scholar]
- Subach OM, Cranfill PJ, Davidson MW, Verkhusha VV. 2011. An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS One 6:e28674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin C, Mandava CS, Ehrenberg M, Andersson DI, Sanyal S. 2010. Ribosomes lacking protein S20 are defective in mRNA binding and subunit association. J Mol Biol. 397:767–776. [DOI] [PubMed] [Google Scholar]
- Tuller T, Waldman YY, Kupiec M, Ruppin E. 2010. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci USA. 107:3645–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulloch F, Atkinson NJ, Evans DJ, Ryan MD, Simmonds P. 2014. RNA virus attenuation by codon pair deoptimisation is an artefact of increases in CpG/UpA dinucleotide frequencies. Elife 3:e04531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varenne S, Buc J, Lloubes R, Lazdunski C. 1984. Translation is a non-uniform process. J Mol Biol. 180:549–576. [DOI] [PubMed] [Google Scholar]
- Vizcaíno JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. 2016. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44:D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, et al. 2014. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505:706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C. 2009. Design parameters to control synthetic gene expression in Escherichia coli. PLoS One 4:e7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth R, Kohles V, Böck A. 1981. Factors modulating transcription and translation in vitro of ribosomal protein S20 and isoleucyl-tRNA synthetase from Escherichia coli. Eur J Biochem. 114:429–437. [DOI] [PubMed] [Google Scholar]
- Wirth R, Littlechild J, Böck A. 1982. Ribosomal protein S20 purified under mild conditions almost completely inhibits its own translation. Mol Gen Genet. MGG 188:164–166. [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat Methods. 6:359–362. [DOI] [PubMed] [Google Scholar]
- Xu C, Dong J, Tong C, Gong X, Wen Q, Zhuge Q. 2013. Analysis of synonymous codon usage patterns in seven different citrus species. Evol Bioinform. Online 9:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins Na, Copeland NG, Court DL. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 97:5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Weems M, Wilke CO. 2009. Translationally optimal codons associate with structurally sensitive sites in proteins. Mol Biol Evol. 26:1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Wiesch PS, Engelstädter J, Bonhoeffer S. 2010. Compensation of fitness costs and reversibility of antibiotic resistance mutations. Antimicrob Agents Chemother. 54:2085–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.