

Abstract
High levels of microRNA-155 (miR-155) are associated with poor outcome in acute myeloid leukemia (AML). In AML, miR-155 is regulated by NF-κB, the activity of which is, in part, controlled by the NEDD8-dependent ubiquitin ligases. We demonstrate that MLN4924, an inhibitor of NEDD8-activating enzyme presently being evaluated in clinical trials, decreases binding of NF-κB to the miR-155 promoter and downregulates miR-155 in AML cells. This results in the upregulation of the miR-155 targets SHIP1, an inhibitor of the PI3K/Akt pathway, and PU.1, a transcription factor important for myeloid differentiation, leading to monocytic differentiation and apoptosis. Consistent with these results, overexpression of miR-155 diminishes MLN4924-induced antileukemic effects. In vivo, MLN4924 reduces miR-155 expression and prolongs the survival of mice engrafted with leukemic cells. Our study demonstrates the potential of miR-155 as a novel therapeutic target in AML via pharmacologic interference with NF-κB-dependent regulatory mechanisms. We show the targeting of this oncogenic microRNA with MLN4924, a compound presently being evaluatedin clinical trials in AML. As high miR-155 levels have been consistently associated with aggressive clinical phenotypes, our work opens new avenues for microRNA-targeting therapeutic approaches to leukemia and cancer patients.
INTRODUCTION
Acute myeloid leukemia (AML) is a malignant disease of the hematopoietic system characterized by maturation arrest and hyperproliferation of clonal myeloid precursors. Standard treatment for AML generally consists of cytarabine/anthracycline-based chemotherapy; however, only a minority of patients are cured with this approach.1 More intensive treatments, including allogeneic stem cell transplantation, albeit more effective, often results in increased toxicity and treatment-related mortality. Thus, novel therapies are urgently needed.
microRNA-155 (miR-155) is a noncoding RNA frequently deregulated in cancer and leukemia and its upregulation has been associated with more aggressive disease2 and chemoresistance.3 In AML, high expression of miR-155 independently predicts poor outcome in cytogenetically normal patients4 and is associated with high-risk FLT3 internal tandem duplication (ITD) mutations.5 Ectopic expression of miR-155 in hematopoietic progenitors can induce either a myeloproliferative disorder,6 or an aggressive B-cell leukemia in mice,7 supporting a leukemogenic role of miR-155. Furthermore, miR-155 overexpression in human CD34+ progenitors perturbs normal myeloid and erythroid differentiation.8 Among the downstream targets of miR-155, relevant to normal and clinical hematopoiesis, are the Src homology-2 domain-containing inositol 5-phosphatase 1 (SHIP1) tumor suppressor9 and PU.1,10,11 a transcription factor critical for myelomonocytic differentiation.12 miR-155-mediated downregula-tion of these targets may partly explain the leukemogeneic role of this oncomiR.
In Epstein–Barr virus-transformed B cells, miR-155 expression is regulated by NF-κB,13 a transcription factor that is constitutively active in AML.14 Recently, Gerloff et al.11 reported that miR-155 transcription in AML is also regulated by NF-κB. The transcriptional activity of NF-κB is primarily regulated by physical interaction with inhibitory IκB proteins (IκBα and IκBβ), which prevents its nuclear translocation. IκB complex stability is regulated by signal-induced phosphorylation at residues Ser32 and Ser36, which results in its degradation and release of NF-κB complex into the nucleus, where it can transactivate its target genes.15–17 Degradation of the IκBα subunit of IκB is mediated by the ubiquitin/proteasome system. Importantly, activity of the E3 ligase responsible for IκBα ubiquitination is controlled by neddylation.18
The neddylation cascade begins with activation of the small ubiquitin-like molecule, NEDD8, by the NEDD8-activating enzyme (NAE). Subsequently, NEDD8 is conjugated onto Cullin-RING E3 ligases (CRLs), a class of ubiquitin ligases, which require neddylation for their activity. Neddylated CRLs polyubiquitinate their substrates, resulting in proteasomal degradation.19 In addition to IκBα, CRL substrates include a number of other tumor suppressors, such as the cyclin-dependent kinase inhibitors p27 (ref. 20) and p21,21 as well as CDT1 (DNA replication factor 1),22 NRF2 (Nuclear factor (erythroid-derived)23 and CHK1 (Check point kinase 1).24
Neddylation can be selectively blocked by MLN4924.25 In the presence of enzymatically active NAE, MLN4924 forms a stable MLN4924–NEDD8 adduct preventing NAE from further activation of NEDD8.25 This terminates the pathway at an early step. In preclinical studies, MLN4924 has shown promising activity against diffuse large B-cell lymphoma and AML, correlating with inhibition of NF-κB activity.26,27 These data suggest that the inhibition of NF-κB is a key component of this compound’s activity in AML; however, the effects of MLN4924 on key downstream targets of NF-κB remain poorly understood.
Given the role of miR-155 in promoting leukemogenesis and its regulation by NF-κB, which is partially controlled by neddylation, we hypothesized that MLN4924 would decrease miR-155 levels in AML cells and result in antileukemic effects. Importantly, MLN4924 is already being used in clinical trials for AML,28 allowing the rapid translation of positive results to the clinic. Indeed, we found that MLN4924 decreased miR-155 expression through decreasing NF-κB binding to the miR-155 promoter. In turn, it restored expression of the miR-155 targets, SHIP1, PU.1 and the macrophage colony-stimulating factor receptor. MLN4924 treatment also triggered monocytic differentiation of human AML cells and prolonged survival of mice xenografted with human leukemic cells. Furthermore, the forced expression of miR-155 partially abrogated the effects of MLN4924, demonstrating that the inhibition of this miR is a critical component of its activity in AML.
MATERIALS AND METHODS
Cell culture and treatment
AML cell lines MV4-11 (CRL-9591), THP-1 (TIB-202) Kasumi-1 (CRL-2724) and HEK-293T (CRL-11268) were from American Type Culture Collection (ATCC). MOLM-13 cells (ACC 554) were from German Collection of Microorganisms and Cell Cultures (DSMZ, Germany). CG-SH cells,29 PU.1-null cell line (503) and its stable transfectant with PU.1-estrogen receptor α fusion (503/PUER)30 were described. All AML lines were cultured in RPMI-1640, whereas HEK-293T cells were grown in DMEM, both media supplemented with 10% fetal bovine serum and 1% Antibiotic–Antimycotic (Gibco Inc., Grand Island, NY, USA). Cells were treated with MLN4924 (Millennium Pharmaceuticals Inc., Cambridge, MA, USA), or vehicle control (0.01% DMSO (dimethyl sulfoxide)) at concentrations indicated.
Primary AML samples
Blasts from AML patients were obtained from The Ohio State University (OSU) Leukemia Tissue Bank. All patient samples were selected based on high miR-155 expression and were FLT3-ITD+. All patients provided written informed consent in accordance with the Declaration of Helsinki under an Institutional Review Board approved protocol and according to OSU institutional guidelines for tissue collection and the use in research. Primary blasts were cultured in StemSpan SFEM medium (Stem Cell Technologies, Vancouver, BC, Canada), supplemented with 20% fetal bovine serum and StemSpan CC100 cytokine cocktail (Stem Cell Technologies). Blasts were treated with MLN4924 (Millennium Pharma-ceuticals Inc.) at concentrations specified in the figure legends, or vehicle control (0.01% DMSO).
Mice
Female NSG (NOD/SCIDγ; Non-Obese Diabetic/Severe Combined Immuno-deficient γ) mice were from Jackson Laboratories (Bar Harbor, ME, USA). The mice used were between 4 and 6 weeks of age. Animal studies followed the rules and regulations of the Institutional Animal Care and Use Committee at the Ohio State University.
MV4-11 xenograft mouse model
Spleen cells (3 × 105) from MV4-11-transplanted NSG mice were intravenously injected into secondary NSG recipient mice via tail vein. One week later, the engrafted mice received intraperitoneal treatments of 180 mg/kg every other day, consisting of a total of nine injections. Mice in the control group were treated similarly with the vehicle alone (20% 2-hydroxypropyl-betacyclodextrin). Peripheral blood was collected 24 and 48 h post first injection to measure the expression of pri-miR-155 and mature miR-155. Mononuclear cells, spleen, sternum, liver and kidney were collected from mice 21 days after the first injection and analyzed for white blood cell count and hematoxylin–eosin staining of tissues. For the survival study, the leukemic mice were treated with MLN4924 180 mg/kg, or vehicle two weeks after MV4-11 cell transplantation every other day for 21 days and followed longitudinally for disease progression and survival.
Statistical analysis
Data were represented as mean ± s.d. and analyzed using the two-tailed Student's t-test. The mean and s.d. were calculated and displayed in bar graphs as the height and the corresponding error bar, respectively. Mouse survival data were analyzed using the Kaplan–Meier and long-rank test methods (Graph Pad Prism, GraphPad Software, La Jolla, CA, USA). A P<0.05 was considered statistically significant (*P<0.05, **P<0.01 and ***P<0.001).
Other methods
For standard techniques including real-time PCR, chromatin immunoprecipitation, western blotting, flow cytometry, clonogenic assay, electro-phoretic mobility shift assay (EMSA), plasmid constructions, transient transfections and luciferase assay cell proliferation viability and apoptosis assays, morphological cell examination and synthetic miR treatment, see Supplementary Methods.
RESULTS
Antileukemic effects of inhibiting miR-155 expression in AML cells
High expression of miR-155 is associated with FLT3-ITD mutations and correlates with poor prognosis in AML.4,5,31 MOLM-13 and MV4-11 AML cells harbor FLT3-ITD and express relatively high levels of miR-155 (Supplementary Figures S1A and B). Using transferrin-conjugated nanoparticles (Tf-NP),32 we delivered antagomiR-155, or a scramble control to these cells. The transferrin (Tf) receptor is generally expressed on AML cells, thereby facilitating Tf-NP uptake via Tf receptor-mediated endocytosis. Forty-eight hours after Tf-NP treatment, MV4-11 cells receiving antagomiR-155 had a fivefold decrease in mature miR-155 levels compared with cells receiving a scramble control (Figure 1a). This was paralleled by an increase in SHIP1 protein (a direct miR-155 target) and cleaved caspase-3 (Figure 1b), indicating the onset of apoptosis. AntagomiR-155 treatment also resulted in a threefold reduction in the number of colonies relative to control in MV4-11 cells treated with Tf-NPs, plated in methylcellulose and scored 7 days later (Figure 1c). Similar results were obtained in primary blasts expressing high levels of miR-155 (Figure 1d and Supplementary Figure S2) and treated with Tf-NPs containing antagomiR-155 or a scramble control (Figures 1e–g). These results demonstrate that decreasing miR-155 levels blocks leukemia growth.
Figure 1.
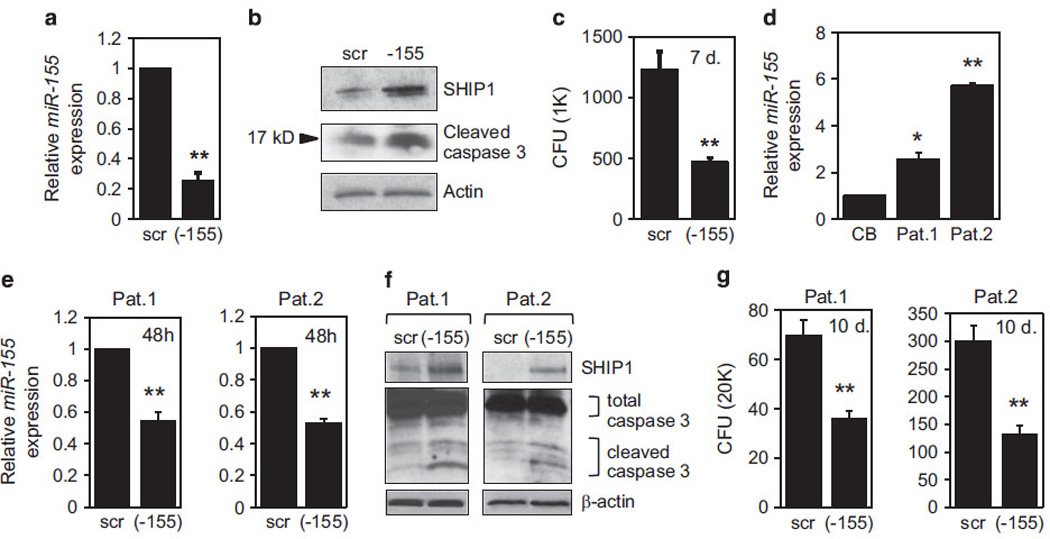
Antileukemic effects of miR-155 inhibition. (a) Nanoparticles loaded with scramble oligo (scr), or antagomiR-155 (-155), were delivered to MV4-11 cells and mature miR-155 expression was determined 48 h later by TaqMan assay. The expression in scramble control was set to 1. (b) Nanoparticle-treated MV4-11 cells were analyzed by western blot for SHIP1 (top) and cleaved caspase-3 levels (middle). The same blot was stained with anti-actin antibody for loading control (bottom). (c) MV4-11 cells were treated with nanoparticles, 1000 cells were plated in methycellulose and colonies (CFU) were counted 7 days later. (d) TaqMan assay of miR-155 expression in CD34+/CD38+ cells purified from two AML patient samples (Pat. 1 and Pat. 2) compared with CD34+/CD38+ cells from normal cord blood (CB), which was set to 1. (e) AML patient blasts were treated with scramble oligo (scr) or antagomiR-155 (-155) loaded nanoparticles and mature miR-155 expression was measured 48 h later by TaqMan. Scramble controls were set to 1. (f) Whole-cell extracts from AML blasts treated with nanoparticles for 48 h were analyzed for the expression of SHIP1 (top) and the presence of cleaved caspase-3 (middle). Staining for anti-actin (bottom) served as a loading control. (g) Following nanoparticle treatment, primary AML cells were analyzed for clonogenic activity by plating 20 000 cells in methylcellulose. Colonies (CFU) were scored 10 days later.
Treatment with MLN4924 downregulates miR-155 expression in FLT3-ITD AML
Given the potential prognostic11 and mechanistic role of miR-155 in differentiation block and aberrant blast proliferation in AML, we sought for clinically available compounds that could interfere with the expression of this oncomiR. We first confirmed that the miR-155 promoter is positively regulated by the NF-κB transcription factor.11,13 Using EMSA with an oligonucleotide containing the canonical NF-κB site, we found that NF-κB constitutively binds to this element in nuclear extracts from MV4-11 cells (Supplementary Figure S3A, lane 2). Co-incubation with antibodies against the p65 and p50 subunits of NF-κB resulted in supershifted complexes, confirming NF-κB binding in this assay (Supplementary Figure S3A, lanes 3 and 4).
A neddylation inhibitor with antineoplastic activity, MLN4924, has been developed33 and is being tested in clinical trials.28,33,34 One of the downstream effects of MLN4924 is the inhibition of NF-κB activity.26,27 Consistent with a previous report,27 we showed that DNA binding of NF-κB was strongly inhibited by treatment of MV4-11 cells with 500 nm MLN4924 (Supplementary Figure S3A, lane 5), which had no effect on the levels of p65/NF-κB (Supplementary Figure S3B). We also observed that the treatment of cell lines (Supplementary Figure S4A) and primary blasts carrying FLT3-ITD mutations (Supplementary Figure S4B) with MLN4924 in the nanomolar range (100–1000 nm), resulted in a time- and concentration-dependent decrease in cell proliferation (Supplementary Figures S4A and B; Supplementary Table S2) and induction of apoptosis (Supplementary Figure S4C).
Thus, we hypothesized that MLN4924 decreases the expression of miR-155 in AML through the inhibition of NFκB. To test this, MV4-11 cells were cultured in 100–500 nm MLN4924 and miR-155 levels were determined by quantitative real-time reverse transcription-PCR. MLN4924 treatment resulted in a significant downregulation of miR-155 expression specifically in FLT3-ITD cells in a dose- (Figures 2a and b; MOLM-13 and MV4-11 cells) and time-dependent (Figure 2b) manner. In FLT3-ITD MV4-11 cells miR-155 levels were decreased by ~ 50% after treatment with 300–500 nm MLN4924 for 12 h (P<0.01, Figures 2b and c). MLN4924 also decreased miR-155 expression in three primary AML samples (Figure 2d) with relatively high levels of miR-155 (Supplementary Figure S2).
Figure 2.
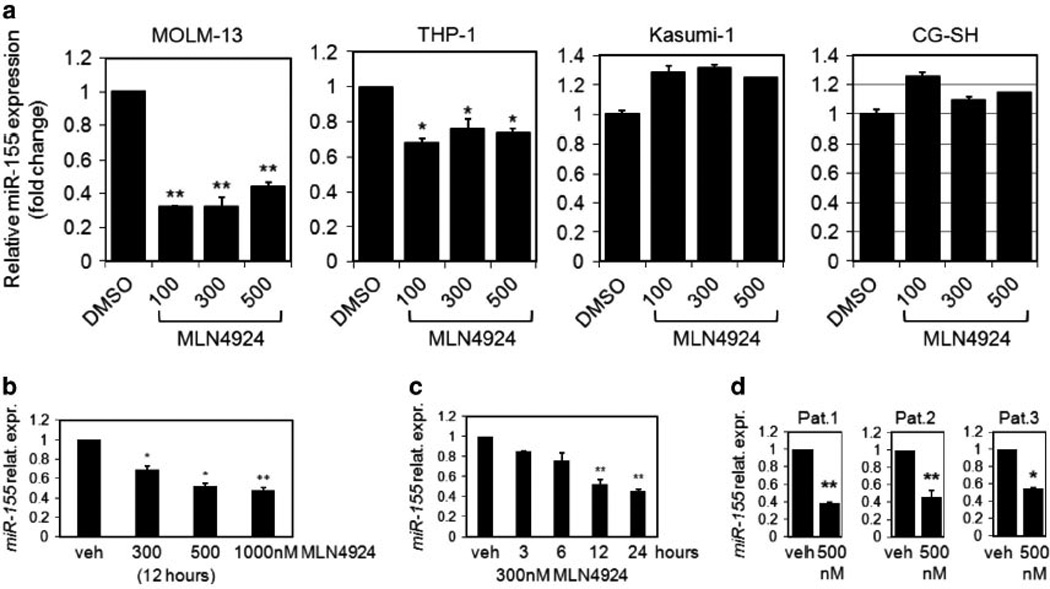
MLN4924 treatment reduces miR-155 expression in FLT3-ITD AML cell lines and primary patient blasts. (a) AML cell lines: FLT3-ITD carrying MOLM-13 and wt FLT3 expressing THP-1, Kasumi-1 and CG-SH were cultured in the presence of indicated concentrations of MLN4924, or vehicle control (DMSO) for 12 h and the expression of miR-155 was determined by the TaqMan assay and normalized to U44 levels. (b) Mature miR-155 expression was analyzed by the TaqMan method in MV4-11 cells, following 12 h of treatment with MLN4924 at the indicated concentrations. The results were normalized to U44. On the basis of triplicate readings of an average of three separate experiments, the average relative fold change was calculated in comparison with the vehicle controls, which were set to 1. (c) Time course of MV4-11 treatment with 300 nm MLN4924. (d) TaqMan assay of miR-155 expression in FLT3-ITD AML primary patient blasts treated with MLN4924 for 12 h. Results are shown as relative fold change after normalization with U44 and 2 Ct, based on triplicate readings of an average of three readings in the same experiments.
MLN4924 disrupts binding of NF-κB to the miR-155 promoter
From a mechanistic standpoint, we confirmed that treatment with MLN4924 caused extensive inhibition of NAE (measured by the levels of neddylated cullins; Supplementary Figure S5A), and cytoplasmic accumulation of the known CRL substrate, pSer32-I-κBα (Supplementary Figure S5A). Accumulation of the active I-κBα (phosphorylated on Ser32) is expected to retain the active p65/p50 heterodimer of NF-κB in the cytoplasm, preventing its nuclear translocation, and thus diminishing NF-κB-dependent transcription. Indeed, we observed a decrease in nuclear localization of p65 in MV4-11 cells treated with MLN4924 (Supplementary Figure S5B), which correlated with a decrease in NF-κB transcriptional activity (Supplementary Figure 5C), similar to that achieved with siRNAs against the p65/NF-κB.
Two NF-κB-binding sites on the miR-155 promoter were previously identified in Epstein–Barr virus-positive B cells.13 To verify that NF-κB also binds to the miR-155 promoter in AML cells, we used EMSA with nuclear extracts from MV4-11 cells. We detected strong binding of the NF-κB complex to both NF-κB sites in vitro (κB1 and κB2; Figure 3a, lanes 2 and 12), which was significantly decreased after 12 h of treatment with 300 or 500 nm MLN4924 (Figure 3a, lanes 3, 4, 13 and 14). NF-κB binding to the miR-155 promoter was diminished by the addition of cold oligonucleotides containing the canonical NF-κB site (IgκB site; Figure 3a, lanes 5 and 15), but not by the ones containing an unrelated C/EBP-binding site (Figure 3a, lanes 6 and 16). In addition, supershifting complexes were observed in the presence of anti-p50 and anti-p65 antibodies, but not with normal IgG, or c-Rel antibody (Figure 3a, compare lanes 8, 9, 18, 19 to 7, 10, 17 and 20). Similarly, chromatin immunoprecipitation experiments showed that MLN4924 dramatically inhibited enrichment of NF-κB on the miR-155 promoter in intact cells (Figure 3b). Treatment of MV4-11 cells with 300 nm MLN4924 for 12 h led to over twofold inhibition of NF-κB enrichment on both binding sites of the miR-155 promoter (Figure 3b; P <0.01).
Figure 3.
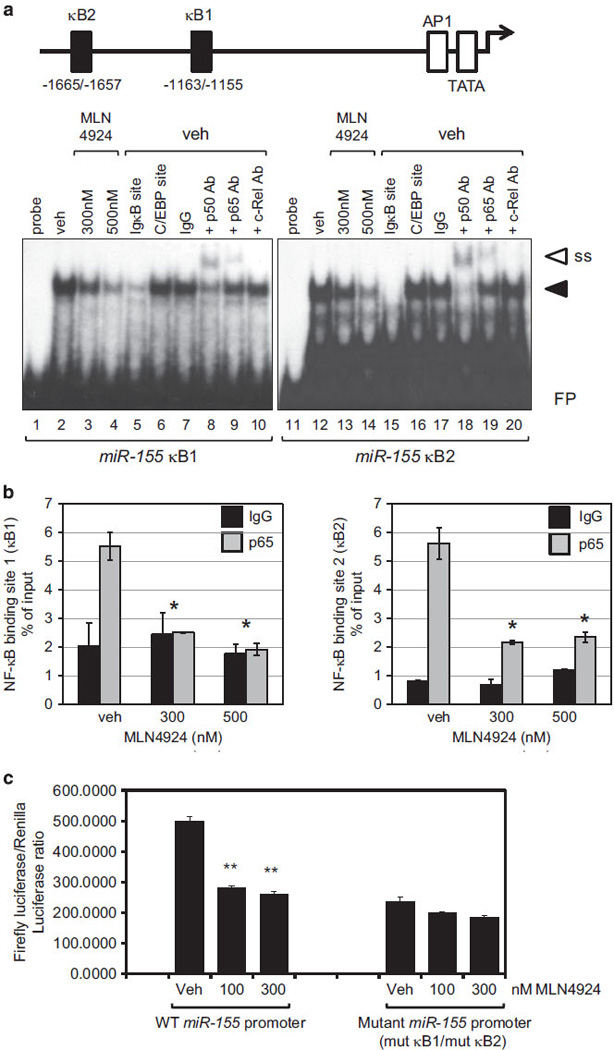
MLN4924 inhibits binding of NF-κB to the miR-155 gene promoter. (a) Top: diagram of the miR-155 promoter showing known NF-κB-binding sites: κ1 (nts − 1163/ − 1155) and κ2 (nts − 1665/ − 1657). Lower panel: nuclear extracts from MV4-11 cells treated with vehicle control (DMSO), 300 or 500 nm MLN4924 (as indicated above the lanes) for 12 h were used in EMSA with radiolabeled probes containing NF-κB-binding sites from human miR-155 promoter (κ1 and κ2). NF-κB/DNA complexes are indicated by a black arrowhead. Specificity of binding is shown by using antibodies against the NF-κB subunits, p65 and p50, to supershift the DNA–protein complexes (marked with white arrowhead; SS) and competition with unlabeled oligos containing either the Igκ gene-derived NF-κB site (IgκB), or unrelated C/EBP-binding site. Free probe (FP) is indicated. (b) Chromatin immunoprecipitation using MV4-11 cells treated with 300 or 500 nm MLN4924 for 12 h. DMSO treatment served as a control (veh). Relative percent enrichment over input was measured by real-time PCR with specific primers covering the miR-155 promoter region containing the two NF-κB-binding sites (κ1 on the left and κ2 on the right). An average of three measurements is shown. Triplicate readings were averaged and error bars indicate s.d. (c) 293T cells were stimulated with 10ng/ml PMA for 2 h to activate NF-κB and then treated with either DMSO (veh), or MLN4924 (100 and 300 nm) for an additional 6 h, followed by transient transfection with firefly luciferase vectors driven by the miR-155 promoter, either wild type (WT) or with both NF-κB sites mutated (mut κB1/mut κB2) together with TK-promoter-driven Renilla luciferase vector. Cell lysates were analyzed for luciferase activity.
To determine whether MLN4924-induced reduction in NF-κB binding to the miR-155 promoter results in decreased NF-κB transcriptional activity, we mutagenized both NF-κB sites in the miR-155 promoter. As tested in EMSA, both mutated oligonucleotides lost their capacity to compete for NF-κB binding (Supplementary Figure S6, lanes 4 and 8). Next, we needed a system to measure the NF-κB-dependent transcriptional activity in response to MLN4924. Owing to low transfection efficiency of MV4-11 cells (30% using optimized nucleofection protocol; Lonza), we chose to transfect miR-155 promoter-luciferase constructs into 293T cells treated with phorbol myristate acetate (PMA), a known activator of the NF-κB pathway. As shown by EMSA (Supplementary Figure S7, lanes 3 and 9), the exposure of 293T cells to 10 ng/ml of PMA for 2 h led to robust NF-κB DNA binding. Similar to the effect of MLN4924 in MV4-11 cells, addition of MLN4924 to PMA-stimulated 293T cells strongly diminished NF-κB DNA binding to the miR-155 promoter (Supplementary Figure S7, lanes 6 and 12), and miR-155 promoter-driven luciferase activity (Figure 3c). In contrast, a miR-155 promoter with both NF-κB sites mutated was less effective in driving luciferase activity and unaffected by MLN4924 treatment (Figure 3c).
These findings demonstrate that MLN4924 decreases binding of NF-κB to the miR-155 promoter and NF-κB-dependent transcriptional activity, leading to the downregulation of miR-155 expression.
MLN4924 treatment results in upregulation of the miR-155 target, SHIP1 and inhibition of the PI3K/AKT pathway in AML cells
The tumor suppressor SHIP1 is a direct target of miR-155, and its knockdown in hematopoietic cells, similar to the over-expression of miR-155, can result in a myeloproliferative phenotype, or a preleukemic pre-B-cell proliferation followed by B-cell malignancy.6,7,9,35 Thus, we hypothesized that MLN4924-induced decrease in miR-155 expression might also result in derepression of SHIP1 expression. To test this, levels of SHIP1 mRNA and protein were assessed in MLN4924-treated AML cells. Upon 24 h of treatment with concentrations of MLN4924 near the IC50, SHIP1 mRNA increased ninefold in MV4-11 cells (P <0.01), and 2–2.5-fold in samples from two patients with high levels of miR-155 (P <0.05; Figure 4a). SHIP1 protein was also upregulated in all samples, as shown by the western blot (Figure 4a).
Figure 4.
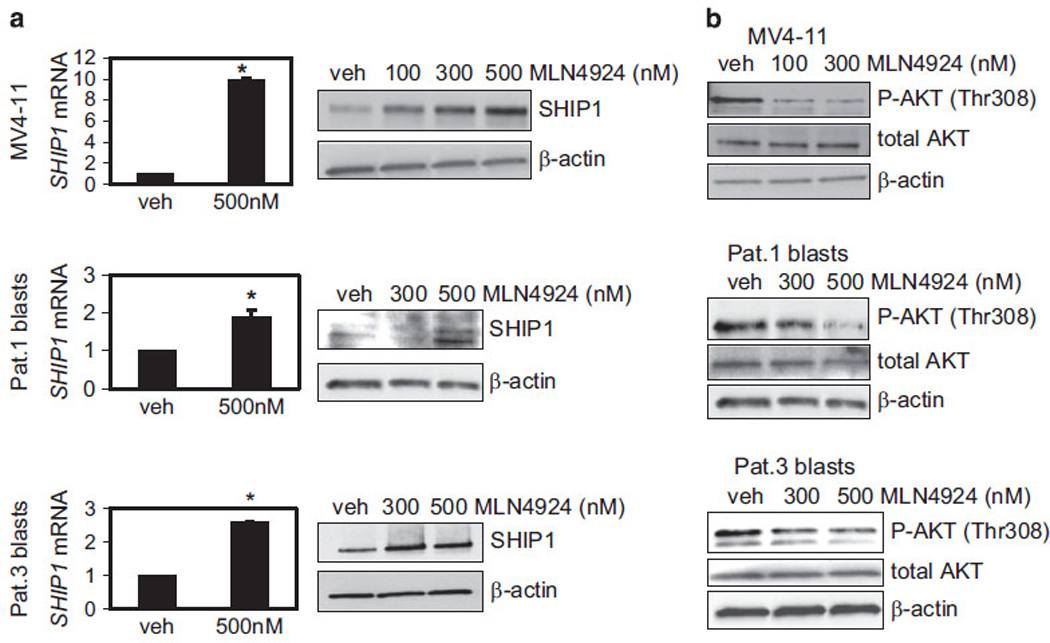
MLN4924 treatment restores the expression of the SHIP1 tumor suppressor and inhibits the PI3K/AKT pathway in MV4-11 cells and primary AML patient blasts. (a) MV4-11 cells and two patient blast samples were cultured in the presence of 500 nm MLN4924 for 24 h. The same samples were also similarly treated with vehicle control (DMSO; veh). SHIP1 mRNA was tested by real-time PCR (left) and the results are shown as relative fold change after normalization with 18S and 2 Ct calculations. Triplicate measurements were averaged and error bars show s.d. (P <0.01 for MV4-11 and AML patients P <0.05). Protein expression was evaluated by western blot (right). β-actin served as a loading control. (b) Phospho-AKT (Thr308, P-AKT) and total AKT protein expression in MV4-11 and two primary AML samples following treatment with 100, 300 or 500 nm MLN4924, or vehicle control for 24 h is shown by western blot. Staining with β-actin antibody was used as a loading control.
SHIP1 functions by hydrolyzing the 5′ phosphate of phosphatidylinositol (3,4,5)-triphosphate to generate phosphatidylinositol (3,4)-bisphosphate. This blocks the PI3K-mediated membrane localization of Pleckstrin homology domain-containing signaling molecules, such as AKT and PLCγ, which have critical regulatory roles in diverse cellular processes. Overexpression of miR-155 has been shown to activate the AKT pathway via the downregulation of SHIP1 in natural killer cell lymphoma/leukemia.36 Thus, we postulated that MLN4924-induced upregulation of SHIP1 would result in the downregulation of the PI3K/AKT pathway. As expected, AML cells treated with MLN4924 showed decreased levels of active phospho-AKT (Thr308) in both MV4-11 cells and primary AML patient blasts (Figure 4b) treated with MLN4924, as assessed by western blot.
In summary, decreased miR-155 expression by MLN4924 resulted in the reactivation of SHIP1 tumor suppressor expression and inhibition of active AKT.
MLN4924 induces monocytic differentiation of AML cells
It has been demonstrated that the introduction of miRNA-155 into CD34+ hematopoietic progenitor cells greatly reduced myeloid and erythroid colony formation, suggesting that miR-155 may inhibit myeloid differentiation.8 In addition, miR-155 has been reported to directly target PU.1,10,11,37 C/EBPβ7 and the M-CSF receptor,38 key players in myelomonocytic differentiation. As MLN4924 decreases miR-155 expression in AML cells (Figure 2), we hypothesized that treatment with this compound might induce cell differentiation. To avoid rapid apoptosis, we tested MV4-11 cells using relatively low concentrations of MLN4924. Cells were cultured in the presence of 20, 50 or 100 nm MLN4924, or 0.01% DMSO and monitored daily for morphological changes by Wright–Giemsa staining of cytospins. On day 6, MV4-11 cells acquired monocytic morphology at every tested concentration of MLN4924 (Figure 5a) and a decreased clonogenic ability (Figure 5b). Furthermore, cell surface staining and flow cytometric analyses indicated a dose-responsive increase in monocytic markers, CD14+ and CD115+ (macrophage colony-stimulating factor receptor; Figure 5c). We also observed an increase in myeloid maturation markers, CD11b+ and CD11c+, and a modest decrease in CD33 expression (Supplementary Figure S8). Finally, treatment with MLN4924 strongly inhibited the expression of c-myc protein (Figure 5d), a critical event for allowing early myeloid precursors to enter a differentiation pathway.39 We confirmed these findings in primary cells. Blasts from patient 1 were treated with 20, 50 and 100 nm MLN4924 for up to 6 days. Wright–Giemsa-stained cytospins revealed an increase in cells with monocyte/macrophage morphology even at concentrations as low as 20 nm (Figure 5e). Phenotypically, 50 and 100 nm concentrations led to a decrease in the CD33+ population, as well as an increase in CD14+, CD115+, CD11b+, CD11c+ and CD36+ populations (Figure 5f; Supplementary Table S3). MLN4924-induced differentiation of AML cells prompted us to investigate the expression of PU.1, a transcription factor indispensable for myeloid differentiation12 and a direct target of miR-155.10 Indeed, exposure to 100 nm MLN4924 for as little as 24 h resulted in a 22-fold increase in PU.1 protein expression in MV4-11 cells (Figure 5g; top panel) and a threefold increase in PU.1 protein expression in patient blasts (Figure 5g; bottom panel).
Figure 5.
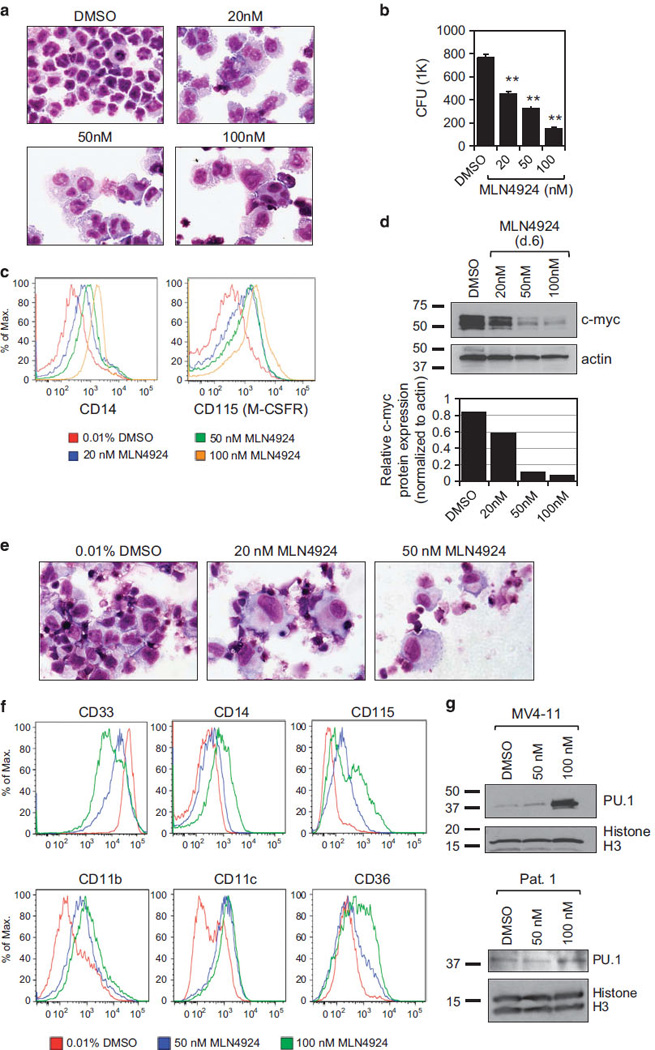
Induction of monocytic differentiation by MLN4924. (a) MV4-11 cells were cultured for 6 days in the medium supplemented with MLN4924 at the concentrations shown or vehicle control (DMSO). Cytospins were stained using the Wright–Giemsa method. (b) MV4-11 cells were treated as in a and 1000 cells were plated in methycellulose in triplicate. Colonies (CFU) were counted 7 days later. (c) Flow cytometric analyses of MV4-11 cells treated for 7 days as in a and stained with anti-CD14 (left panel), or anti-CD115 (right panel). (d) Cell aliquots treated with MLN4924 for 6 days were analyzed for c-myc protein expression by western blot of whole-cell extracts. Staining with anti-actin antibody served to normalize for loading control. Band intensities were measured using ImageJ software and the amount of c-myc normalized to actin levels was plotted (below). (e) Blasts from AML patient (Pat.1) were cultured in the presence of MLN4924 at the indicated concentrations of DMSO. Six days later, cell morphology was analyzed by Wright–Giemsa-stained cytospins. Although 100 nm MLN4924 was included in this experiment, the cell viability was very low and there was not enough material for cytospin. (f) Blast cells from patient 1 (Pat.1) were treated with 50 nm, 100 nm or 0.01% DMSO, aliquots were collected on day 6 and the expression of cell surface markers (indicated) was determined by fluorescence-activated cell sorting. (g) Western blot of nuclear extracts from MV4-11 cells (top) and AML blasts from patient 1 (Pat.1; bottom) treated with designated concentrations of MLN4924 for 24 h. Blot was stained successively with anti-PU.1 and Histone H3 antibodies.
We wondered if a short-term treatment with MLN4924 would be sufficient to set the differentiation program in motion. MV4-11 cells were treated with 100 nm MLN4924 for 24 h, washed three times and cultured in medium without MLN4924 for up to six additional days (diagrammed on top of Figure 6a). Monocytic morphology was observed as early as day 6 of the experiment (days 1+5; Figure 6a). Downregulation of c-myc protein (Figure 6b) and upregulation of CD115 and CD11c were also noted (Figure 6c). Interestingly, the activity of MLN4924 appears to require at least low levels of PU.1, as this compound had no effect on growth or morphological differentiation of a PU.1-null cell line (Supplementary Figures S9A and B). However, MLN4924 treatment of a PU.1 inducible stable transfectant, which expressed low levels of PU.1 protein in the nucleus even in the absence of the inducer (Supplementary Figure S9C), caused growth inhibition and morphological changes consistent with differentiation (Supplementary Figures S9A and B).
Figure 6.
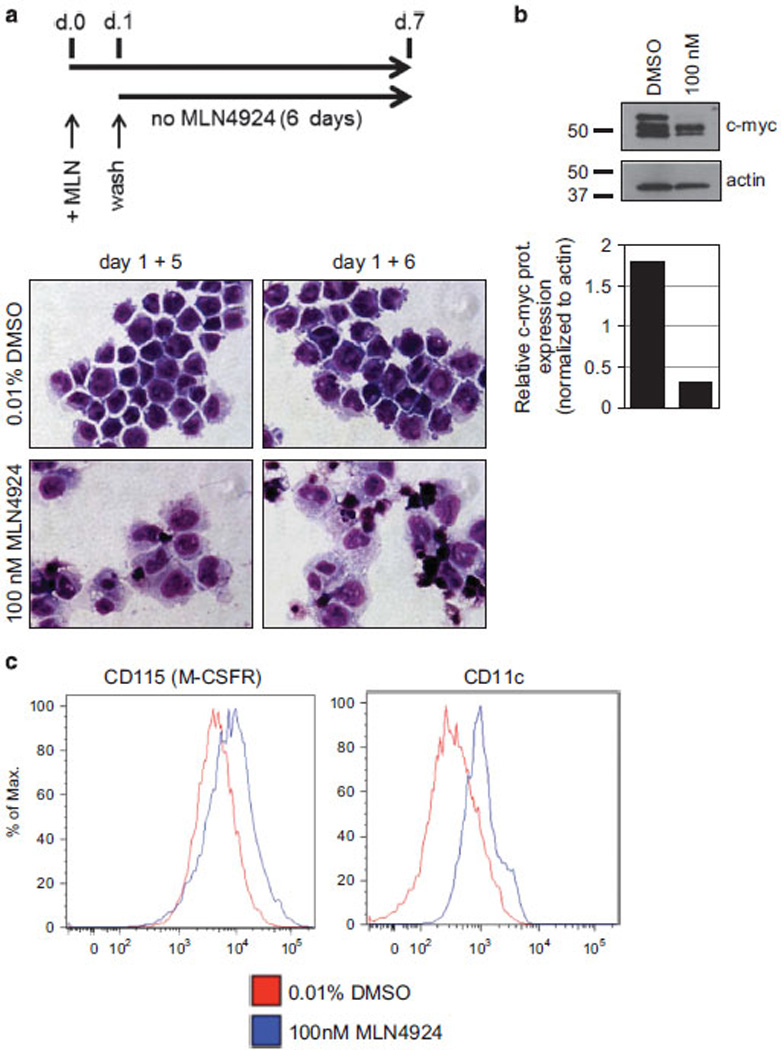
Transient treatment of MV4-11 cells with MLN4924 is sufficient for the inception of monocytic differentiation. (a) Top: diagram of a timeline of the transient treatment with MLN4924. MV4-11 cells were treated with 100 nm MLN4924 for 24 h, washed in MLN4924-free medium three times and grown for up to an additional 6 days in the absence of MLN4924. Bottom: Wright–Giemsa-stained cytospin preparations of cells collected on day 6 (1+5) or day 7 (1+6) of the culture. (b) Whole-cell extracts were tested by western blot for c-myc protein levels and the total protein levels were normalized to actin. Densitometric quantification of band intensities, using ImageJ software is shown below. (c) Cells harvested on day 7 were analyzed by fluorescence-activated cell sorting using anti-CD-115 and anti-CD11c antibodies.
In summary, our data show that low concentrations of MLN4924 can induce differentiation of AML cells. Importantly, a transient exposure of MV4-11 cells to MLN4924 is sufficient to induce monocytic differentiation, suggesting that MLN4924 can activate a molecular switch promoting differentiation of leukemic blasts.
Ectopic expression of miR-155 interferes with MLN4924-mediated antileukemic effects
Our results suggested that the downregulation of miR-155 by MLN4924 might be at least partially responsible for the compound’s activity in AML. Thus, we hypothesized that MLN4924’s activity would be abrogated by miR-155 overexpression in AML cells. We transfected AML blasts from patient 3 (Pat. 3) with miR-155 precursor and 24 h later treated them with 500 nm MLN4924, or vehicle for an additional 48 h. As compared with the scramble miRNA control, mature miR-155 expression in primary blasts transfected with miR-155 precursor was approximately 50-fold higher (Figure 7a) and ectopic expression of miR-155 significantly reduced the impact of MLN4924 on cell viability (almost twofold; P = 0.02; Figure 7b). As expected, the 24-h treatment of scramble miR-transfected cells with 500 nm MLN4924 resulted in the upregulation of known direct targets of miR-155, SHIP1 and PU.1 proteins (Figure 7c), as well as induction of apoptosis, determined by annexin V staining (Figure 7d). In contrast, the overexpression of miR-155 partially prevented the induction of SHIP1 and PU.1 proteins, as well as the impact of MLN4924 on apoptosis (Figures 7c and d).
Figure 7.
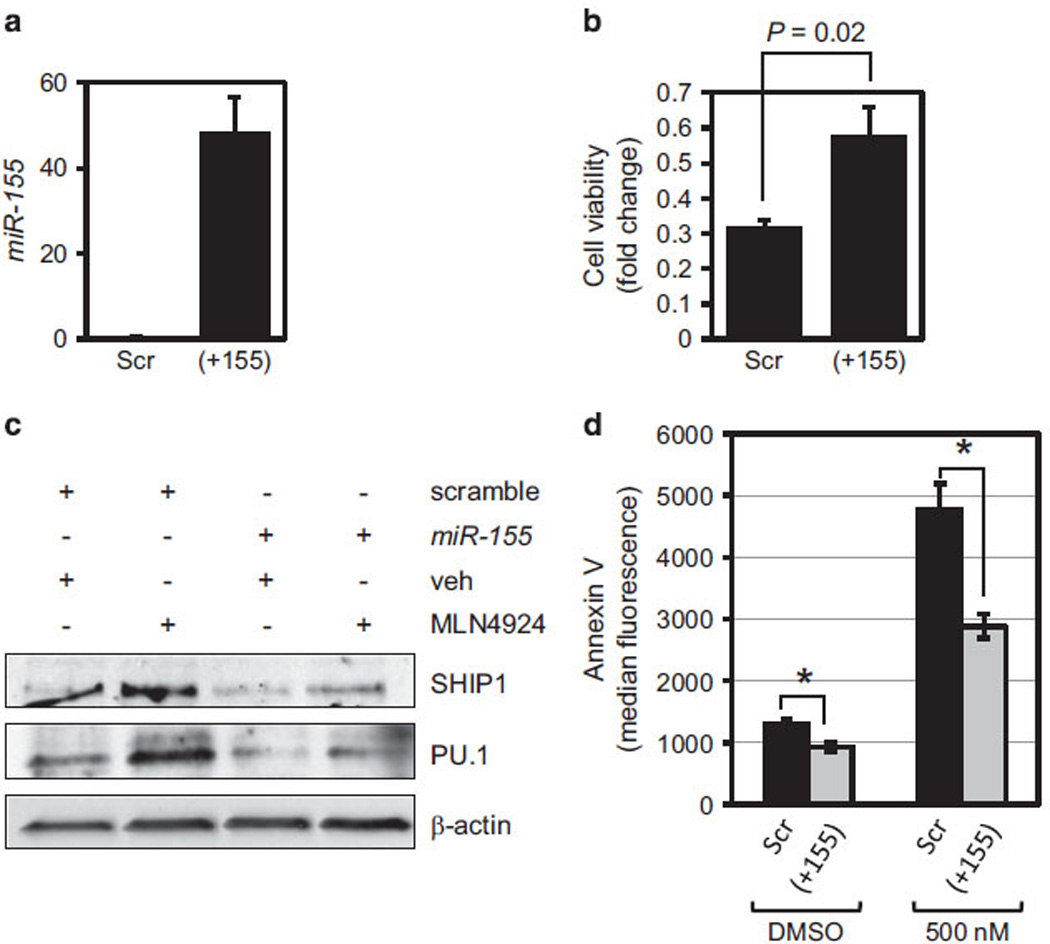
Ectopic expression of miR-155 in AML cells counteracts the antileukemic effects of MLN4924. (a) AML blasts from Patient 3 were transfected with the precursor of miR-155 and mature miR-155 expression was analyzed 24 h later. Results are shown as relative copy numbers after normalization for U44. Average of three readings is shown and error bars indicate s.d. (P <0.01). (b) Twenty-four hours after transfection with miR-155 precursor or scramble control, leukemic blasts were treated with 500 nm MLN4924 for an additional 48 h and viable cell counts were determined by Trypan blue exclusion. Results are shown as relative fold change in viable cell numbers of an average of two experiments done in triplicate. Error bars indicate s.d. (c) SHIP1, PU.1, caspase-3 and cleaved caspase-3 (indicator of apoptosis) protein levels were determined by western blot in whole-cell lysates of cells transfected with miR-155 precursor or scramble oligonucleotide and 24 h later treated for an additional 48 h with 500 nm MLN4924 or vehicle control. β-actin shows equal loading of the protein extracts. (d) Annexin V-FITC apoptosis assay of patient blasts transfected and treated as in c. Median fluorescence intensities of Annexin-V-positive cells from three independent experiments were averaged and represented in as a bar graph. Error bars indicate s.e.m.
These findings support the notion that MLN4924-induced downregulation of miR-155 is a component of its antileukemic activity.
In vivo activity of MLN4924
To test the activity of MLN4924 in AML in vivo, a human AML xenograft model was used. NOD/SCIDγ mice were inoculated via tail vein with MV4-11 AML cells. Upon engraftment, leukemic cells were harvested from the spleens and injected intravenously into secondary recipients. One week later, mice were injected intraperitoneally with 180 mg/kg of MLN4924 (n = 10), or vehicle control (n = 10) three times a week. As early as 24 and 48 h after the first dose of MLN4924, primiR and mature miR-155 expression levels in the peripheral blood of transplanted mice were decreased (by 50% at 24 h and by 80% at 48 h; P <0.01; Figure 8a). Twenty-one days following the first dose of treatment, three mice representing each group were killed and analyzed (Figures 8b–d). The average white blood cell count was lower in the MLN4924-treated group (5333 cells/µl ± 1040) compared with the vehicle-treated group (36 166 cells/µl ± 10 598; P <0.01; Figure 8b). In addition, the average spleen weight was reduced in the MLN4924-treated group compared with the control group (58.06 mg ± 12.74 vs 305.66 mg ± 51.1; P <0.01, Figure 8c). Hematoxylin and eosin staining was performed on sections from spleen, sternum and liver of xenografted mice (Figure 8d). MLN4924-treated mice showed decreased infiltration of blasts in the sternum, spleen, liver and kidney, as compared with the control animals (Figure 8d). Also, MLN4924 significantly prolonged the survival of leukemic mice (Figure 8e). The median survival was 46 vs 31 days for the MLN4924-treated group vs vehicle-treated group (P <0.0001, Log-Rank test, n = 10 per group; Figure 8e), respectively. These data demonstrate that MLN4924 treatment in vivo results in decreased miR-155 expression and significant antileukemic activity.
Figure 8.
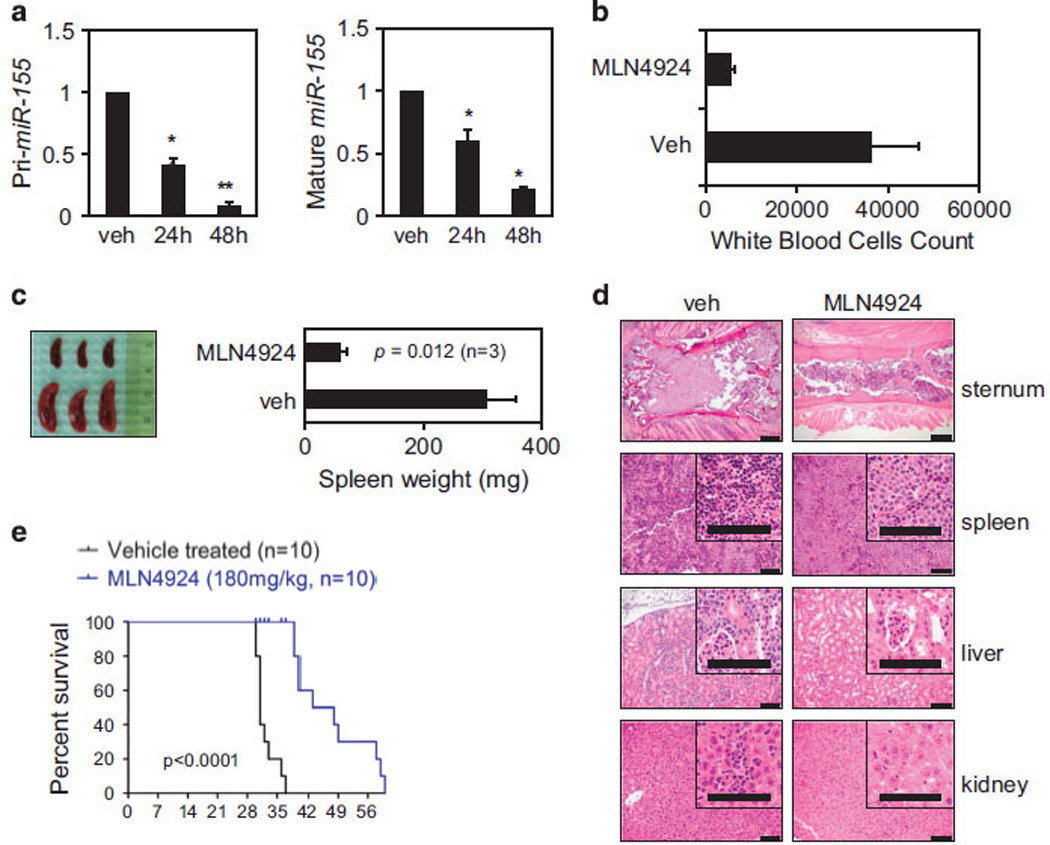
Antileukemic activity of MLN4924 in a human AML xenograft model. (a) NOD/SCID γ (NSG) mice were transplanted intravenously with MV4-11 cells. After the engraftment, splenic cells from the leukemic mice were injected into secondary recipient NSG mice. One week later, 10 mice were treated with 180 mg/kg of MLN4924 or vehicle control. Pri-miR (left) and mature miR-155 (right) expression levels in the peripheral blood of xenografted mice were measured by real-time PCR, as early as 24 and 48 h after the first dose of MLN4924 and compared with vehicle control (veh). Results of mRNA expression are shown as relative fold change after normalization to 18 S and U44, respectively, and 2 Ct calculations. Error bars represent s.d. (P <0.01 for 48 h) based on triplicate readings of average of two separate experiments. (b) Average white blood cell count (µl/ml) taken from three mice representing each group and killed 21 days after the first injection of MLN4924 or vehicle control (veh). (c) Spleen pictures of three representative cases (MLN4924 n = 3; vehicle n = 3) at 21 days. Spleens were weighed and results are shown as bar graph representing the average of three in each group. P-value obtained using t-test is shown. (d) Hematoxylin and eosin staining of sections from sternum, spleen and liver of xenografted mice treated with vehicle or MLN4924 at 21 days. (e) Survival plot of xenografted mice after the treatments with MLN4924 at 180 mg/kg (n = 10) or vehicle control (veh; n = 10). Survival comparison was made using the log-rank test.
DISCUSSION
The expression of miR-155 is low in normal hematopoietic cells and often upregulated in AML, where it likely mediates aberrant cell differentiation and proliferation.11,40 Recently, we found that high miR-155 expression is an independent predictor of adverse prognosis in cytogenetically normal AML patients.4 Thus, therapeutic silencing of this microRNA might represent a novel strategy to treat aggressive AML. One approach may eventually involve the use of the antagomiR technology;41 however, antagomiRs are not yet available for clinical use. Thus, we sought for an immediate strategy that would allow us to inhibit aberrant miR-155 expression in malignant blasts by pharmacological intervention.
We reasoned that to identify a clinically available miR-155 inhibitor, the first logical step was to understand the mechanisms leading to its dysregulation. As NF-κB transcriptionally regulates miR-155 in B cells,13,42 we postulated that it may do the same in AML and that pharmacological inhibition of NF-κB might be a potentially successful approach to downregulate miR-155 expression in AML. Among the compounds that are already in clinical trials and inhibit NF-κB,26,27,43 we selected MLN4924 because of its promising activity in AML and minimal toxicity to normal cells.27 We demonstrate that indeed MLN4924 decreases NF-κB binding to the miR-155 promoter, resulting in the downregulation of miR-155 expression, upregulation of several of its key target genes and ultimately in antileukemic activity, which we demonstrated to be mediated by miR-155 downregulation. As a neddylation inhibitor, MLN4924 is likely to act on multiple pathways; however, our data support a preferential antileukemia activity in high miR-155-expressing cells.
As expected, pharmacological downregulation of miR-155 by MLN4924 led to the upregulation of SHIP1 and PU.1, key targets of miR-155 whose aberrant expression contributes to blasts’ proliferation and differentiation block.7,9–11 SHIP1 is a tumor suppressor37,44 involved in the regulation of the PI3K/AKT pathway, which is often abnormally activated in cancer and AML.45 Previous reports showed that aberrant expression of miR-155 in natural killer cells induces dysregulation of the SHIP1/AKT signaling pathway by decreasing SHIP1 protein expression.36 Furthermore, miR-155 overexpression and knockout of SHIP1 exhibit similar myeloproliferative phenotypes in mice.6,9 This suggests that SHIP1 suppression is a key contributor to miR-155-associated leukemia. Consistent with our hypothesis, we found that SHIP1 was strongly upregulated by MLN4924 in a miR-155-dependent manner, concomitant with the downregulation of the PI3K/AKT pathway. In addition, we demonstrate that MLN4924 also mediates monocyte/macrophage differentiation of AML cells. This effect is likely facilitated by the upregulation of PU.1, a transcription factor important for maturation of myeloid cells and a known target of miR-155.10–12 Indeed, our results showed that MLN4924-dependent inhibition of miR-155 expression led to the upregulation of PU.1 protein, whereas ectopic expression of miR-155 in AML blasts correlated with lower levels of PU.1 protein. Upon MLN4924 treatment, we also observed an increase of the surface expression of macrophage colony-stimulating factor receptor, a marker of monocytic differentiation46 that is transcriptionally regulated by PU.147 and has been recently demonstrated to be one of the miR-155 targets.10 Interestingly, even a transient (that is, 24 h) exposure to a relatively low MLN4924 concentration, led to re-expression of PU.1 and was sufficient for the onset of monocytic differentiation. This result is analogous to findings that short-term induction of expression of another master regulator of myelopoiesis, C/EBPα, in myeloid progenitor cells was also sufficient for the inception of granulocytic maturation.48
MLN4924 treatment appears to have a preferential activity on cells harboring FLT3-ITD and in turn expressing higher miR-155 levels (Supplementary Figure S4). Likely, this may be related to a biologic dependence of these leukemic cells on high miR-155 expression. Altogether, our data support the idea that pharmacologic targeting of miR-155 may represent a novel therapeutic approach in high-risk AML. Changing levels of this oncomiR seemingly affects a cascade of concurrently active leukemia-promoting mechanisms that dysregulate hematopoietic cell growth, differentiation and survival. Thus, in addition to a possible use of this drug as a single agent in AML patients not candidates for intensive chemotherapy induction, our findings also provide support for the use of MLN4924 in combination with chemotherapy, as knocking down high miR-155 levels may ultimately restore chemosensitivity in otherwise resistant AML blasts.4
Over the past decades, efforts have been made to develop antileukemic small-molecule inhibitors with high biologic activity and low toxicity, specifically targeting leukemogenic proteins, including FLT3-ITD, which was shown to contribute to pharmacological downregulation of miR-155.11 However, AML patients often demonstrate primary49 or secondary50,51 resistance to these inhibitors. More recently, miR-155 has been shown itself to activate the NF-κB signaling pathway;52 thus, downregulation of miR-155 by MLN4924 could provide an additional mechanism that potentiates a primary effect of MLN4924 on NF-κB in a NEDD8-independent manner. Given promising anticancer activity of MLN4924 in preclinical models of AML and solid tumors,27,33,53,54 a recent study was designed to determine whether and how resistance might occur. It was demonstrated that heterozygous mutations in NAEβ can occur in cell lines and xenograft models exposed to high concentrations of MLN4924, thus reducing this compound’s potency.54 However, MLN4924-resistant mutations in NAEβ have not been described in human subjects. Our data showing monocytic differentiation of AML cells using transient, lower dosing of MLN4924 may decrease the chance of developing resistance to this compound.
In summary, we demonstrate for the first time the promise of targeting miR-155 in AML by an investigational drug, MLN4924. We anticipate that our discovery will better inform how to use this agent and select the most responsive patients, thereby leading to novel therapeutic strategies and eventually better outcomes for AML patients.
Supplementary Material
Acknowledgments
We thank Millennium Pharmaceuticals Inc., Cambridge, MA, USA, for supplying MLN4924 compound and Donna Bucci for providing primary AML samples from the OSU Leukemia Tissue bank. We are thankful to Dr Massimo Mallardo (University of Naples, Naples, Italy) for making sequences of EMSA probes available. Drs Daniel Tenen and Annalisa DiRuscio kindly provided 503 and 503/PUER cell lines. This work was supported by National Science Foundation (EEC-0914790) for XH, LJL and RJL and the Wilmot cancer research fellowship for JHM. Grant Numbers: U10CA180861, R01 CA135332, P50 CA140158 for GM and MAC and the Gehr Family Center for Leukemia.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–494. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- 2.Faraoni I, Antonetti FR, Cardone J, Bonmassar E. miR-155 gene: a typical multi-functional microRNA. Biochim Biophys Acta. 2009;1792:497–505. doi: 10.1016/j.bbadis.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Kong W, He L, Coppola M, Guo J, Esposito NN, Coppola D, et al. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J Biol Chem. 2010;285:17869–17879. doi: 10.1074/jbc.M110.101055. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Marcucci G, Maharry KS, Metzeler KH, Volinia S, Wu YZ, Mrózek K, et al. Clinical role of microRNAs in cytogenetically normal acute myeloid leukemia: miR-155 upregulation independently identifies high-risk patients. J Clin Oncol. 2013;31:2086–2093. doi: 10.1200/JCO.2012.45.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitman SP, Maharry K, Radmacher MD, Becker H, Mrózek K, Margeson D, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010;116:3622–3626. doi: 10.1182/blood-2010-05-283648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205:585–594. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costinean S, Sandhu SK, Pedersen IM, Tili E, Trotta R, Perrotti D, et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood. 2009;114:1374–1382. doi: 10.1182/blood-2009-05-220814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Georgantas RW, Hildreth R, Morisot S, Alder J, Liu CG, Heimfeld S, et al. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc Natl Acad Sci USA. 2007;104:2750–2755. doi: 10.1073/pnas.0610983104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci USA. 2009;106:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27:847–859. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerloff D, Grundler R, Wurm AA, Bräuer-Hartmann D, Katzerke C, Hartmann JU, et al. NF-κB/STAT5/miR-155 network targets PU.1 in FLT3-ITD-driven acute myeloid leukemia. Leukemia. 2014;29:535–547. doi: 10.1038/leu.2014.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 13.Gatto G, Rossi A, Rossi D, Kroening S, Bonatti S, Mallardo M. Epstein-Barr virus latent membrane protein 1 trans-activates miR-155 transcription through the NF-kappaB pathway. Nucleic Acids Res. 2008;36:6608–6619. doi: 10.1093/nar/gkn666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–2307. doi: 10.1182/blood.v98.8.2301. [DOI] [PubMed] [Google Scholar]
- 15.Finco TS, Beg AA, Baldwin AS. Inducible phosphorylation of I kappa B alpha is not sufficient for its dissociation from NF-kappa B and is inhibited by protease inhibitors. Proc Natl Acad Sci USA. 1994;91:11884–11888. doi: 10.1073/pnas.91.25.11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995;14:2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 18.Read MA, Brownell JE, Gladysheva TB, Hottelet M, Parent LA, Coggins MB, et al. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol. 2000;20:2326–2333. doi: 10.1128/mcb.20.7.2326-2333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 20.Podust VN, Brownell JE, Gladysheva TB, Luo RS, Wang C, Coggins MB, et al. A Nedd8 conjugation pathway is essential for proteolytic targeting of p27Kip1 by ubiquitination. Proc Natl Acad Sci USA. 2000;97:4579–4584. doi: 10.1073/pnas.090465597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem. 2008;283:29045–29052. doi: 10.1074/jbc.M806045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishitani H, Sugimoto N, Roukos V, Nakanishi Y, Saijo M, Obuse C, et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006;25:1126–1136. doi: 10.1038/sj.emboj.7601002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huh J, Piwnica-Worms H. CRL4(CDT2) targets CHK1 for PCNA-independent destruction. Mol Cell Biol. 2013;33:213–226. doi: 10.1128/MCB.00847-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37:102–111. doi: 10.1016/j.molcel.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 26.Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116:1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- 27.Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115:3796–3800. doi: 10.1182/blood-2009-11-254862. [DOI] [PubMed] [Google Scholar]
- 28.Swords RT, Savona MR, Maris MB, Erba HP, Hua Z, Faessel H, et al. Pevonedistat (MLN4924), an Investigational, first-in-class NAE inhibitor in combination with azacitidine elderly patients with acute myeloid leukemia (AML) considered unfit for conventional chemotherapy: updated results from the phase I C15009 trial. Blood. 2014;124 abstract #2313. [Google Scholar]
- 29.Munker R, Nordberg ML, Veillon D, Williams BJ, Roggero A, Kern W, et al. Characterization of a new myeloid leukemia cell line with normal cytogenetics (CG-SH) Leuk Res. 2009;33:1405–1408. doi: 10.1016/j.leukres.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Mueller BU, Pabst T, Hauser P, Gilliland G, Neuberg D, Tenen DG. Mutations of the transcription factor PU.1 are not associated with acute lymphoblastic leukaemia. Br J Cancer. 2006;94:1918–1920. doi: 10.1038/sj.bjc.6603198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garzon R, Garofalo M, Martelli MP, Briesewitz R, Wang L, Fernandez-Cymering C, et al. Distinctive microRNA signature of acute myeloid leukemia bearing cyto-plasmic mutated nucleophosmin. Proc Natl Acad Sci USA. 2008;105:3945–3950. doi: 10.1073/pnas.0800135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang X, Schwind S, Yu B, Santhanam R, Wang H, Hoellerbauer P, et al. Targeted delivery of microRNA-29b by transferrin-conjugated anionic lipopolyplex nano-particles: a novel therapeutic strategy in acute myeloid leukemia. Clin Cancer Res. 2013;19:2355–2367. doi: 10.1158/1078-0432.CCR-12-3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 34.DeAngelo DJ, Erba HP, Maris M, Swords RT, Anwer F, Altman JK, et al. MLN4924, a novel investigational inhibitor of NEDD8-activating enzyme (NAE), in adult patients with acute myeloid leukemia (AML) and myelodys-plastic syndrome (MDS): results from multiple dosing schedules in a phase 1 study. Blood. 2013;122 abstract #1443. [Google Scholar]
- 35.Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci USA. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamanaka Y, Tagawa H, Takahashi N, Watanabe A, Guo YM, Iwamoto K, et al. Aberrant overexpression of microRNAs activate AKT signaling via down-regulation of tumor suppressors in natural killer-cell lymphoma/leukemia. Blood. 2009;114:3265–3275. doi: 10.1182/blood-2009-06-222794. [DOI] [PubMed] [Google Scholar]
- 37.Luo JM, Yoshida H, Komura S, Ohishi N, Pan L, Shigeno K, et al. Possible dominant-negative mutation of the SHIP gene in acute myeloid leukemia. Leukemia. 2003;17:1–8. doi: 10.1038/sj.leu.2402725. [DOI] [PubMed] [Google Scholar]
- 38.Lu C, Huang X, Zhang X, Roensch K, Cao Q, Nakayama KI, et al. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood. 2011;117:4293–4303. doi: 10.1182/blood-2010-12-322503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johansen LM, Iwama A, Lodie TA, Sasaki K, Felsher DW, Golub TR, et al. c-Myc is a critical target for c/EBPalpha in granulopoiesis. Mol Cell Biol. 2001;21:3789–3806. doi: 10.1128/MCB.21.11.3789-3806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Babar IA, Cheng CJ, Booth CJ, Liang X, Weidhaas JB, Saltzman WM, et al. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci USA. 2012;109:E1695–E1704. doi: 10.1073/pnas.1201516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF-kappaB signaling. J Mol Cell Biol. 2011;3:159–166. doi: 10.1093/jmcb/mjr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, et al. Pevonedistat (MLN4924), a First-in-Class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukemia and myelodysplastic syndromes: a phase I study. Br J Haematol. 2015;169:534–543. doi: 10.1111/bjh.13323. [DOI] [PubMed] [Google Scholar]
- 44.Metzner A, Precht C, Fehse B, Fiedler W, Stocking C, Günther A, et al. Reduced proliferation of CD34(+) cells from patients with acute myeloid leukemia after gene transfer of INPP5D. Gene Ther. 2009;16:570–573. doi: 10.1038/gt.2008.184. [DOI] [PubMed] [Google Scholar]
- 45.Brauer H, Strauss J, Wegner W, Müller-Tidow C, Horstmann M, Jücker M. Leukemia-associated mutations in SHIP1 inhibit its enzymatic activity, interaction with the GM-CSF receptor and Grb2, and its ability to inactivate PI3K/AKT signaling. Cell Signal. 2012;24:2095–2101. doi: 10.1016/j.cellsig.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 46.Olweus J, Thompson PA, Lund-Johansen F. Granulocytic and monocytic differentiation of CD34hi cells is associated with distinct changes in the expression of the PU.1-regulated molecules, CD64 and macrophage colony-stimulating factor receptor. Blood. 1996;88:3741–3754. [PubMed] [Google Scholar]
- 47.Zhang DE, Fujioka K, Hetherington CJ, Shapiro LH, Chen HM, Look AT, et al. Identification of a region which directs the monocytic activity of the colony-stimulating factor 1 (macrophage colony-stimulating factor) receptor promoter and binds PEBP2/CBF (AML1) Mol Cell Biol. 1994;14:8085–8095. doi: 10.1128/mcb.14.12.8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT/ enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. 1998;18:4301–4314. doi: 10.1128/mcb.18.7.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Piloto O, Wright M, Brown P, Kim KT, Levis M, Small D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood. 2007;109:1643–1652. doi: 10.1182/blood-2006-05-023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107:293–300. doi: 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- 51.Moore AS, Faisal A, Gonzalez de Castro D, Bavetsias V, Sun C, Atrash B, et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: a model for emerging clinical resistance patterns. Leukemia. 2012;26:1462–1470. doi: 10.1038/leu.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang L, Zhang H, Rodriguez S, Cao L, Parish J, Mumaw C, et al. Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-κB-dependent manner. Cell Stem Cell. 2014;15:51–65. doi: 10.1016/j.stem.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71:3042–3051. doi: 10.1158/0008-5472.CAN-10-2122. [DOI] [PubMed] [Google Scholar]
- 54.Milhollen MA, Thomas MP, Narayanan U, Traore T, Riceberg J, Amidon BS, et al. Treatment-emergent mutations in NAEβ confer resistance to the NEDD8-activating enzyme inhibitor MLN4924. Cancer Cell. 2012;21:388–401. doi: 10.1016/j.ccr.2012.02.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.