

Abstract
The year 2015 has seen great progress in the renal fibrosis field, as key studies began to build a consensus on the importance of epithelial-to-mesenchymal transition, cell cycle arrest, and defective metabolism in the pathogenesis of kidney fibrosis. New findings also point to a role of developmental signalling in renal fibrogenesis.
Renal fibrosis is an important topic that attracts broad interest in nephrology owing to its status as a hallmark and common outcome across all kinds of progressive chronic kidney disease (CKD). The year 2015 saw much progress in the renal fibrosis field, with major breakthroughs and new findings markedly advancing our understanding of the fibrogenic process. These studies have laid strong foundations for the future development of novel treatments for fibrotic CKD. For the first time in more than a decade, scientists in the field have begun to build a consensus on several key issues such as the importance of partial epithelial-to-mesenchymal transition (EMT), cell cycle arrest, and defective cellular metabolism in the development and progression of kidney fibrosis.
The process of renal fibrosis is characterized by an excessive deposition of extracellular matrix in the interstitial compartment, leading to scar formation. An activated form of interstitial fibroblast — the α-smooth muscle actin-positive myofibroblast — is widely recognized as the major type of matrix- producing cell in the fibrotic kidney. However, tubular epithelial cells, which are the main constituent of renal parenchyma, often localize at the epicentre of damage and are especially vulnerable to damage after kidney injury. In this context, a key question is how tubular injury drives fibroblast activation and matrix overproduction. One hypothesis is that kidney tubular cells undergo EMT after injury, a phenotypic conversion programme that is characterized by the loss of epithelial markers and gain of mesenchymal features. Such a notion, however, has been intensely contested as studies using genetic cell lineage tracing could not find evidence of a direct contribution of epithelial cells to the myofibroblast population in the fibrotic kidney1, instigating a controversy over the relative contribution of EMT to fibroblast activation that has lasted several years.
In 2015, two back-to-back studies addressed this dispute and offered new insights into the potential role of tubular EMT in the development and progression of renal fibrosis2,3. These studies tackled the issue by generating genetically modified mice, in which Snail or Twist, two key transcription factors that regulate the EMT programme, were ablated specifically in tubules. As a result, the EMT programme is specifically inhibited in the renal tubular epithelium in vivo. Both studies demonstrated that inhibition of the EMT programme by conditional deletion of Snai1 or Twist1 in tubular epithelial cells reduced interstitial fibrosis in numerous CKD models, including unilateral ureteral obstruction, nephrotoxic serum-induced nephritis, and folic acid-induced nephropathy. Not surprisingly, inhibition of an EMT programme in the kidney also led to preservation of tubular cell integrity and function, restoration of tubular repair and regeneration, and a reduction in myofibroblast accumulation, suggesting that the EMT programme is crucial and required for initiating tubular dysfunction and driving fibrosis development after various insults.
The mechanism of EMT involvement in renal fibrosis revealed by these studies is particularly intriguing. Both studies found that tubular epithelial cells only undergo a partial EMT during renal fibrosis — the cells express markers of both epithelial and mesenchymal cells and remain associated with their basement membrane. In this respect, these observations are in harmony with earlier genetic cell linage tracing studies1, and demonstrate that a complete phenotypic conversion of tubular epithelial cells to a myofibroblast phenotype is extremely rare, if occurring at all. Nevertheless, this partial EMT is sufficient to induce tubular function impairment, triggering cell cycle arrest and promoting the release of critical fibrogenic cytokines. Lovisa et al. further demonstrated that one of the functional consequences of partial EMT is the induction of arrest in the G2 phase of the cell cycle, which compromises the potential of tubular epithelial cells to repair and regenerate3. As cell cycle arrest has been postulated as a mechanistic pathway that leads to kidney fibrosis, the linkage of EMT to cell cycle arrest is especially appealing, as it helps to form a consensus on our understanding of the mechanism of renal fibrosis.
Damage to the tubular epithelium might induce renal fibrosis via other mechanisms as well. In 2015, a landmark study used a genome-wide transcriptome approach to demonstrate that defects in fatty acid metabolism in tubular epithelial cells have a crucial role in the pathogenesis of kidney fibrosis4. This metabolic reprogramming is characterized by the decreased expression of key enzymes and regulators of fatty acid oxidation (FAO) and increased intracellular lipid deposition4. As FAO is the preferred energy source for kidney proximal tubular epithelial cells, a reduction in FAO would affect lipid metabolism by disrupting the balance between fatty acid synthesis, uptake, and consumption, leading to dysregulated intracellular lipid accumulation. Inhibition of FAO in tubular epithelial cells in vitro indeed causes ATP depletion, cell death, dedifferentiation, and intracellular lipid deposition4. Conversely, restoring fatty acid metabolism by genetic or pharmacologic approaches protects against renal fibrosis, suggesting that stimulation of metabolic pathways could be a novel strategy for preventing and treating fibrotic CKD.
Kang et al. also investigated the mechanisms behind the depressed metabolic pathways in fibrotic kidney disease4. Transforming growth factor β1 (TGF-β1), the most potent profibrotic cytokine, inhibits the expression of carnitine palmitoyltransferase 1 (CPT1), the rate-limiting enzyme in FAO, and thereby decreases fatty acid metabolism. Furthermore, TGF-β1 also represses mRNA expression of upstream regulators of CPT1 that encode the peroxisome proliferator- activated receptor-α (PPARα) and PPARγ coactivator-1α (PGC-1α). A separate study in 2015 also showed that inhibition of microRNA-21 (miR-21) enhances mitochondrial function, reduces production of reactive oxygen species, preserves tubular integrity, and attenuates renal fibrosis5. Therefore, miR-21, a downstream target of TGF-β1 signalling, contributes to kidney fibrosis by silencing metabolic pathways.
One common outcome of partial EMT, cell cycle arrest, and depressed metabolism following kidney injury is the conversion of tubular cells to a pathologic secretory phenotype. Our understanding of the secretome by injured tubular epithelial cells continued to advance in 2015. Emerging evidence suggests that injured tubular cells produce and secrete the ligands of key developmental signalling pathways, such as Wnts and sonic hedgehog (Shh)6. Tubule-derived Shh mediates epithelial–mesenchymal communication by selectively targeting interstitial fibroblasts in a paracrine manner, and induces fibroblast proliferation and myofibroblastic activation, leading to kidney fibrogenesis6. An interesting study in 2015 showed that hedgehog-responding Gli1+ cells possess features of mesenchymal stem cells in vitro, and proliferate and expand following kidney injury7. Genetic ablation of these cells substantially ameliorates kidney fibrosis. Consistent with these findings, pharmacologic inhibition of Shh/Gli signalling reduces the size of the myofibroblast population and inhibits fibrosis after injury6,8.
Wnt ligands are induced in many cell types of the injured kidney and can target both interstitial fibroblasts and tubular epithelial cells via autocrine or paracrine mechanisms. Similar to Shh, Wnts induce fibroblast proliferation and myofibroblastic activation, leading to matrix overproduction and the development of fibrosis9. In 2015, we showed that multiple genes of the renin–angiotensin system are direct targets of Wnt/β-catenin signalling in tubular epithelial cells10. These studies provide a novel mechanistic link between Wnt upregulation and activation of the intrarenal renin–angiotensin system, hypertension, and kidney fibrosis.
In summary, important studies published in 2015 highlight a central role for varying tubular responses such as partial EMT, cell cycle arrest, and defective metabolism in driving renal fibrosis (FIG. 1). Intriguingly, these tubular responses eventually converge on the acquisition of a secretory phenotype in tubular epithelial cells, leading to the release of pathological mediators that sustain fibroblast activation and inflammation. In our opinion, advances in understanding the pathogenesis of renal fibrosis in 2015 will be memorable, and should inspire more intensive studies for many years to come.
Figure 1. Studies have highlighted important roles for varying tubular responses such as partial epithelial-to-mesenchymal transition (EMT), cell cycle arrest and defective metabolism in driving renal fibrosis.
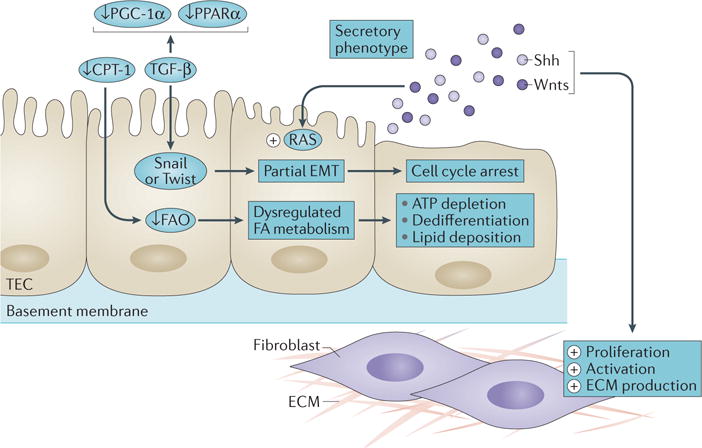
ECM, extracellular matrix; FA, fatty acid; FAO, fatty acid oxidation; TEC, tubular epithelial cell. Adapted from Nature Publishing Group © Ovadya, Y. & Krizhanovsky, V. Nat. Med. 21, 975–977 (2015).
Key advances.
Renal tubular epithelial cells undergo a partial epithelial-to-mesenchymal transition after injury, which impairs tubular repair and regeneration, induces cell cycle arrest, and drives interstitial fibroblast activation2,3
Kidney fibrosis is associated with defective cellular metabolism and mitochondrial dysfunction in renal tubular epithelial cells; approaches to restore fatty acid metabolism or stimulate metabolic pathways might represent new therapeutic strategies to combat fibrotic kidney disease4,5
Activation of key developmental pathways such as Wnt and hedgehog signalling after injury has a critical role in triggering fibroblast proliferation, as well as in activating the intrarenal renin–angiotensin system8–10
Acknowledgments
The author’s work is supported by NIH grants DK064005, DK091239 and DK106049, and National Science of Foundation of China grants 81130011 and 81521003.
Footnotes
Competing interests statement
The authors declare no competing interests.
Contributor Information
Dong Zhou, Department of Pathology, University of Pittsburgh School of Medicine, 200 Lothrop Street, Pittsburgh, Pennsylvania 15261, USA.
Youhua Liu, Department of Pathology, University of Pittsburgh School of Medicine, 200 Lothrop Street, Pittsburgh, Pennsylvania 15261, USA; State Key Laboratory of Organ Failure Research, Nanfang Hospital, Southern Medical University, 1838 North Guangzhou Avenue, Guangzhou 510515, China.
References
- 1.Humphreys BD, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grande MT, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015;21:989–997. doi: 10.1038/nm.3901. [DOI] [PubMed] [Google Scholar]
- 3.Lovisa S, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21:998–1009. doi: 10.1038/nm.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang HM, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez IG, et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest. 2015;125:141–156. doi: 10.1172/JCI75852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou D, et al. Sonic hedgehog is a novel tubule-derived growth factor for interstitial fibroblasts after kidney injury. J Am Soc Nephrol. 2014;25:2187–2200. doi: 10.1681/ASN.2013080893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kramann R, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16:51–66. doi: 10.1016/j.stem.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kramann R, et al. Pharmacological GLI2 inhibition prevents myofibroblast cell-cycle progression and reduces kidney fibrosis. J Clin Invest. 2015;125:2935–2951. doi: 10.1172/JCI74929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao L, et al. Sustained activation of Wnt/β-catenin signaling drives AKI to CKD progression. J Am Soc Nephrol. 2015;27 doi: 10.1681/ASN.2015040449. http://dx.doi.org/10.1681/ASN.2015040449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou L, et al. Multiple genes of the renin–angiotensin system are novel targets of Wnt/β-catenin signaling. J Am Soc Nephrol. 2015;26:107–120. doi: 10.1681/ASN.2014010085. [DOI] [PMC free article] [PubMed] [Google Scholar]