

Abstract
Melatonin, has been shown repeatedly to inhibit the growth of human breast tumor cells in vitro and in vivo. Its anti-proliferative effects have been well-studied in MCF-7 human breast cancer cells and several other estrogen receptor α (ERα)-positive human breast cancer cell lines. However, the MDA-MB-231 breast cancer cell line, an ERα negative cell line widely used in breast cancer research, has been shown to be unresponsive to melatonin’s growth-suppressive effect in vitro. Here we examined the effect of melatonin on the cell proliferation of several ERα-negative breast cancer cell lines including MDA-MB-231, BT-20 and SK-BR-3 cells. Although the MT1 G-protein-coupled receptor is expressed in all three cell lines, melatonin significantly suppressed the proliferation of SK-BR-3 cells without having any significant effect on the growth of MDA-MB-231 and BT-20 cells. We confirmed that the MT1-associated Gα proteins are expressed in MDA-MB-231 cells. Further studies demonstrated that the melatonin-unresponsiveness in MDA-MB-231 cells may be caused by aberrant signaling downstream of the Gαi proteins, resulting in differential regulation of ERK1/2 activity.
Keywords: Melatonin, melatonin receptor (MT1)
Introduction
Melatonin, the main secretory product of the pineal gland, is involved in the regulation of a wide variety of biological processes in target tissues, which include the regulation of reproduction in seasonally-breeding animals, synchronization of the circadian rhythms, and sleep-promotion [1–4]. Like the other hormones, melatonin exerts these physiological effects in target tissues via specific binding proteins or receptors. Two membrane-associated melatonin receptors, which belong to the G-protein-coupled receptor (GPCR) superfamily, have been cloned and sequenced in mammals [5]. The MT1 receptor is distributed in the central nervous system, numerous peripheral tissues, including ovary and uterus, and mammary tumor cells including the MCF-7 human breast cancer cell line [5–10]. In contrast, expression of the MT2 receptor is limited primarily to the central nervous system [11, 12].
In addition to its physiologic effects, studies in the last decade have revealed that melatonin exerts an inhibitory influence on the development and growth of breast tumors [9, 13]. It has been proposed that melatonin acts on breast tumor growth by directly suppressing the proliferation of breast tumor cells. Numerous studies have shown that melatonin administration inhibits the in vitro proliferation of several breast cancer cell types including the estrogen receptor α (ERα)-positive MCF-7 human breast cancer cells [14, 15].
As demonstrated in our previous studies, melatonin administration at physiologic concentrations (10−9 M), which correspond to the human peak plasma nighttime concentration of melatonin, significantly inhibit the in vitro proliferation of MCF-7 cells by 30–50% [16]. Further studies have shown that the anti-proliferative actions of melatonin are mediated primarily through activation of the MT1 G-protein-coupled receptor. Overexpression of MT1 receptor in MCF-7 cells potentiates melatonin-induced growth-suppression in vitro, enhances melatonin’s growth-inhibitory effect in vivo in athymic nude mice implanted with MT1-overexpressing MCF-7 cells [17, 18], and suppresses mammary gland development in mice [19]. Additionally, melatonin-induced growth-inhibition is blocked by pre-treatment with antagonists to the MT1/MT2 receptors [17, 18]. Although the precise intracellular mechanism(s) underlying melatonin’s anti-proliferative effects are not yet fully elucidated, it has been suggested that the MT1-mediated growth-suppressive actions of melatonin involve “cross talk” with specific growth-regulatory signaling pathways, such as the steroid/thyroid hormone nuclear receptor family, and some growth factor signaling pathways, via selective activation of multiple G-proteins.
Melatonin’s growth-suppressive effects are not limited to ERα-positive breast cancer cells as melatonin also impacts the growth of mammary gland epithelial cells and inhibits the proliferative activities in ERα-negative MCF-7 tumor xenografts [19–21]. However, in our studies, administration of physiologic concentrations of melatonin failed to inhibit the proliferation of ERα-negative MDA-MB-231 breast cancer cells [22], despite the expression of the MT1 receptor at both mRNA and protein levels in these cells [23].
As a GPCR, the MT1 receptor has been shown to associate with a wide variety of G-proteins in various tissues [24–27]. The pattern of responses of a particular cell type to GPCR activation depends on the G-proteins associated with the receptor, specific effector molecules expressed, and the relative concentration of the various components in the signaling cascade. The ERα-negative MDA-MB-231 breast cancer cells exhibit a distinctive malignant phenotype including loss of nuclear receptors and loss of responsiveness to estradiol, resistance to anti-estrogenic treatment, high invasive/metastatic potential, and constitutive-activation of the MAPK signaling pathway. Since the MT1 receptor, which mediates much of melatonin’s growth-suppressive action in MCF-7 breast cancer cells [18], is expressed in MDA-MB-231 cells, we hypothesize that, a molecular deficiency in the MT1 signaling pathway leads to the uncoupling of the melatonin signal transduction and the unresponsive phenotype in MDA-MB-231 human breast cancer cells.
MATERIALS AND METHODS
Chemicals and reagents
All chemicals and tissue culture reagents were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Cell culture medium, RPMI 1640, and fetal bovine serum (FBS) were purchased from Gibco BRL (Grand Island, NY, USA). The FuGENE 6 transfection reagent was purchased from Roche (Indianapolis, IN, USA).
Cell lines and cell culture
MCF-7, BT-20, SK-BR-3 and MDA-MB-231 cells were purchased from American Tissue Culture Collection (ATCC, MD, USA). These cells were routinely maintained in RPMI-1640 medium supplemented with 10% FBS (Gibco BRL), 2 mM glutamine, 50 mM MEM non-essential amino acids, 1 mM sodium pyruvate and 10 mM BME. Cells were cultured as monolayer in 150 cm2 flasks, at 37° C in a humidified atmosphere of 5% CO2 and 95% air.
Transient transfection
For cell proliferation assays, MDA-MB-231 cells were plated onto 35 mm2 6-well plates at a density of 2.0 × 104 cells/well, in RPMI-1640 medium supplemented with 10% FBS. Twenty-four hours after plating, cells were transiently transfected with 50 ng/well of the control vector (pcDNA3.1), dominant-positive (DP) or –negative (DN) G-protein constructs, using Fugene 6 transfection reagent (Roche). Eight hours following transfection, cells were re-fed with fresh medium supplemented with 10% FBS. Cell numbers were counted on a hemacytometer 6 days following transfection. For Western blot analyses, MCF-7 and MDA-MB-231 cells were plated onto 10-cm petri dishes at a density of 1.2 × 106 cells/dish and were transiently transfected with 600 ng/dish of control vector (pcDNA3.1) or dominant-positive Gαi2 protein plasmid in RPMI medium (6 ml/dish) supplemented with 10% FBS, using Fugene 6 transfection reagent. Six hours following transfection, another 4 ml of fresh medium containing 10% FBS was added to each dish. Cells were harvested at 3, 18, 24 and 36 hours following transfection.
Cell proliferation assay
For cell proliferation studies, MCF-7, BT-20, SK-BR-3 and MDA-MB-231 cells were plated onto 35 mm2 6-well plates at a density of 2.0 × 104 cells per well, in RPMI-1640 medium supplemented with 10% FBS. Cells were serum-starved for 24 hours prior to treatment. For melatonin response studies, BT-20, SK-BR-3 and MDA-MB-231 cells were treated with either melatonin (10−9 M), or diluent (0.0001% ethanol). For 8-Br-cAMP dose response assays, MCF-7 and MDA-MB-231 cells were treated with 8-Br-cAMP (10−10, 10−8, 10−6, and 10−4 M), or diluent (ddH2O). After 6 days, cells were counted on a hemacytometer, using the trypan blue exclusion test.
Western blot analysis
Cells were harvested and then lysed in a protein extraction buffer containing Tris (50 mM, pH 7.4), EDTA (20 mM), NP-40 (0.5%), NaCl (150 mM), phenylmethylsulfonyl fluoride (0.3 mM), NaF (1 mM), NaVO4 (1 mM), dithiothreitol (1mM), aprotinin (1 μg/ml), leupeptine (1 μg/ml), and pepstatin (1 μg/ml). The cell lysates were centrifuged for 10 min at 10,000 × g, at 4° C. Protein concentrations of the supernatants were determined by Bio-Rad assay (Bio-Rad, Hercules, CA, USA). Total protein (50 ×g per sample) was electrophorectically separated on a 10% SDS polyacrylamide gel, and electroblotted onto a Hybond membrane. After incubation with 5% nonfat milk in a Tris-buffered saline containing 0.1% Tween-20, the blots were probed with anti-G protein polyclonal rabbit antibodies (Gαi2, Gαi3, Gαq and Gα11) (Santa Cruz, CA, USA), phospho-p38 (Thr180/Tyr182) or phospho-p44/42 MAPK (Thr183/Tyr185) antibodies (Cell Signaling, Beverly, MA, USA). The same blots were stripped and re-probed with anti-actin antibody (Sigma, St. Louis, MO, USA).
Statistical analysis
Data are represented as the mean ± the standard error of the mean. The statistical significance at 95% confidence level was determined by one-way ANOVA followed by Newman-Kuels post-hoc test analyses using the Statview software.
RESULTS
To determine if melatonin’s growth-suppressive effects are limited to ERα-positive breast cancer cells, cell proliferation assays were conducted to determine melatonin’s effect on the in vitro growth of ERα-negative MDA-MB-231, BT-20 and SK-BR-3 breast cancer cells. As shown in Fig. 1, melatonin treatment (10−9 M) significantly inhibited the proliferation of ERα-negative SK-BR-3 cells by 50%, but did not significantly inhibit the proliferation of MDA-MB-231 cells. In BT-20 cells, melatonin appears to induce a trend of growth-inhibition but its effect on cell growth was not statistically significant.
Figure 1. Effect of melatonin on the proliferation of BT-20, SK-BR-3, MDA-MBA-231 human breast cancer cells.

BT-20, SK-BR-3 and MDA-MB-231 breast cancer cells were plated into 35 mm2 6-well Costar plates, at a density of 2.0 × 104 cells/well, in RPMI-1640 supplemented with 10% FBS. Cells were serum-starved for 24 hours and then treated with melatonin (Mlt, 10−9 M) or diluent (Control, 0.0001% ethanol). Six days following treatment cells were harvested by brief trypsinization and counted on a hemacytometer using the trypan blue exclusion test. Data is presented as percent of mean cell count in ethanol-treated MDA-MB-231 cells (100%). One-way ANOVA followed by Newman-Kuels post-hoc test analyses was used to determine statistically significant differences in the percentage among different groups. n=3, * P < 0.05.
As demonstrated in previous studies, the membrane-associated MT1 melatonin receptor, but not MT2, is expressed in breast cancer cells [9, 10], and mediates melatonin’s growth-suppressive effects in breast cancer cells [17, 18]. To determine if loss of melatonin-responsiveness in MDA-MB-231 cells is due to the lack of the MT1 melatonin receptor, qPCR analyses were conducted to examine the steady state expression of the MT1 receptor mRNA in MDA-MB-231 cells. As shown in Fig. 2, expression of MT1 mRNA was highest in MCF-7 cells and at comparable levels in BT-20, SK-BR-3 and MDA-MB-231 cells.
Figure 2. mRNA expression of MT1 melatonin receptor in MCF-7, BT-20, SK-BR-3, MDA-MBA-231 human breast cancer cells.
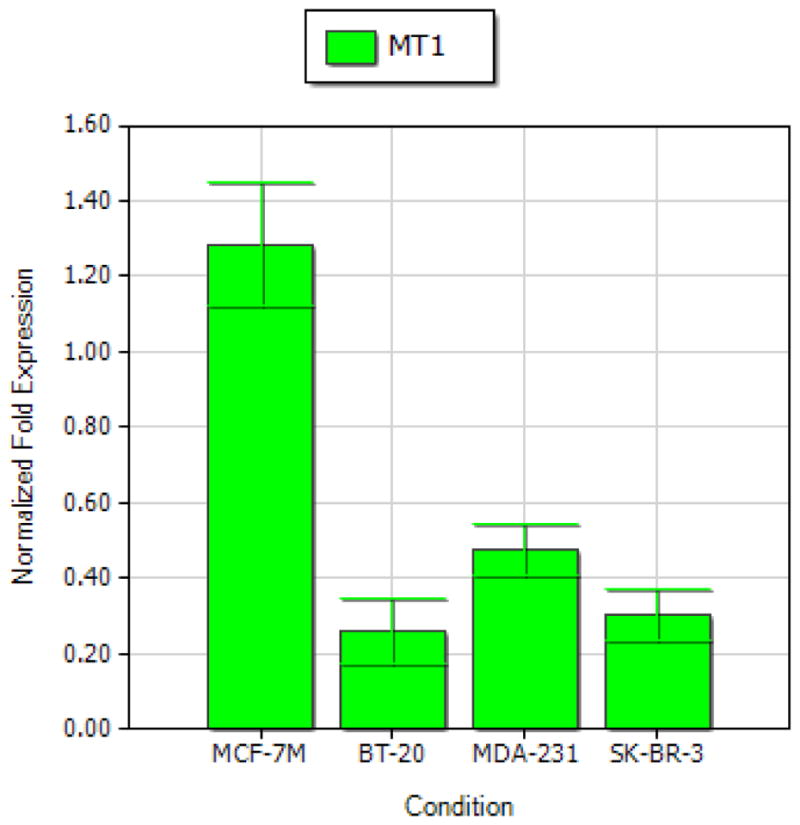
Expression levels of MT1 were determined by qPCR analysis.
To address if uncoupling of the MT1 signal transduction cascade in MDA-MB-231 cells is caused by the absence of certain G-proteins which associate with MT1 receptor in MCF-7 cells, protein expression of Gαi2, Gαi3, Gαq and Gα11 proteins in MDA-MB-231 cells was determined by Western blot analyses. As shown in Fig. 3, all of these G-proteins are expressed in MDA-MB-231 breast cancer cells.
Figure 3. Expression of G-proteins in MDA-MB-231 human breast cancer cells.

MDA-MB-231 breast cancer cells were plated into 150 mm2 flasks in RMPI-1640 supplemented with 10% FBS. Four days after seeding cells were harvested by scraping and whole cells lysates were extracted. Fifty micrograms of total cellular protein from MDA-MB-231 cells was electrophoretically separated on 10% SDS polyacrylamide gel, and transferred onto nitrocellulose membrane. Expression of specific G-proteins was detected by Western blot analyses using anti-G protein polyclonal rabbit antibodies (Gαi2, Gαi3, Gαq and Gα11) at a 1:1000 dilution. These figures are representative of multiple (n=3) Western blot analyses of Gα-protein expression in MDA-MB-231 breast cancer cells.
As shown in Fig. 4, transfection with the vector construct had no significant effect on MDA-MB-231 cell growth, while transfection with the dominant-positive G-protein plasmids (DP-Gαi3, -Gαq and -Gα11) significantly stimulated cell proliferation by approximately 30% (non-transfected control set as 100%). In contrast, transfection with dominant-negative G-protein plasmids (DN-Gαi2, -Gαi3, -Gαq and -Gα11) suppressed cell growth, causing a significant decrease of approximately 20% in cell growth (non-transfected control set as 100%). The above data suggests that the G-proteins (Gαi2, Gαi3, Gαq and Gα11) that couple to MT1 receptor in MCF-7 cells and inhibit their proliferation are also expressed in MDA-MB-231 cells, but act as mitogenic signaling pathways in MDA-MB-231 cells.
Figure 4. Effects of dominant-positive and dominant-negative G-proteins on the proliferation of MDA-MB-231 human breast cancer cells.

MDA-MB-231 breast cancer cells were plated into 35 mm2 6-well Costar plates, at a density of 2.0 × 104 cells/well, in RPMI-1640 supplemented with 10% FBS. Twenty-four hours after seeding, cells were sham transfected (Parental), or transfected with 50 ng/well of pcDNA3.1 (Vector), dominant-positive G-protein plasmids (Gαi2, Gαi3, Gαq and Gα11), or dominant-negative G-protein plasmids (Gαi2, Gαi3, Gαq and Gα11). Six days following transfection cells were harvested by brief trypsinization and counted on a hemacytometer using the trypan blue exclusion test. Data is presented as percent of mean cell count in parental MDA-MB-231 cells (100%). One-way ANOVA followed by Newman-Kuels post-hoc test analyses was used to determine statistically significant differences in the percentage among different groups. * P < 0.05 vs. parental cells, n=3.
In the subsequent studies, we examined the effects of DP-Gαi2 on ERK1/2 phospho-activation in MCF-7 and MDA-MB-231 cells by Western blot analyses. As shown in Fig. 5, transfection with the vector construct for up to 36 hours did not affect ERK1/2 phosphorylation in either cell line. Transfection with DP-Gαi2 plasmid at 3 hours almost completely blocked the phosphorylation of ERK1/2 in MCF-7 cells (by ~90%), but had no effect on ERK1/2 phosphorylation in MDA-MB-231 cells.
Figure 5. Effects of the dominant-positive Gαi2 protein on the activation of ERK1/2 in MCF-7 and MDA-MB-231 human breast cancer cells.

(A) MCF-7 and (B) MDA-MB-231 breast cancer cells were plated onto 10-cm Petri dishes at a density of 1.2 × 106 cells/dish in RPMI-1640 supplemented with 10% FBS. Twenty-four hours after seeding, cells were sham transfected (Parental), or transfected with 600 ng/dish of pcDNA3.1 (Vector) or dominant-positive Gαi2 protein (DP-Gαi2) plasmid for 6 hours and then harvested at 3, 18, 24, and 36 hours following transfection. Whole cell lysates were extracted from MCF-7 and MDA-MB-231 cells. Fifty micrograms of total cellular protein was electrophoretically separated on 10% SDS polyacrylamide gel, and transferred onto nitrocellulose membrane. The phosphorylation of ERK1/2 was examined by Western blot analyses using anti-phospho-p44/42 MAPK (Thr183/Tyr185) antibody, at a 1:1000 dilution. The same blots were stripped and reprobed with anti-actin antibody. Western blot signal intensities were measured by phosphoimage densitometry and then normalized to the actin levels (bottom graph). Results were expressed as relative arbitrary units with a value of 100% for the parental control groups. This figure is a Western blot analysis of ERK1/2 phosphorylation in MCF-7 and MDA-MB-231 breast cancer cells, and it is representative of 3 independent experiments.
A well-established second messenger that transduces signals from the Gαi-protein is cAMP. Therefore, we further compared the effects of 8-Br-cAMP, a cAMP analog, on the proliferation of MCF-7 and MDA-MB-231 cells by cell proliferation assays. As shown in Fig. 6, 8-Br-cAMP at low concentrations (10−10 to 10−6 M) caused a general trend of growth-suppression in MCF-7 cells but growth-stimulation in MDA-MB-231 cells. The most obvious effects were observed at the lowest concentration used (10−10 M), at which, 8-Br-cAMP significantly inhibited cell proliferation in MCF-7 cells by approximately 40%, while significantly increasing MDA-MB-231 cell growth by approximately 40% (controls set as 100%). The highest concentration of 8-Br-cAMP (10−4 M), which is considered as a cytotoxic concentration in previous reports, however, induced a dramatic reduction (>80%) in cell growth in both cell lines.
Figure 6. Effects of 8-Br-cAMP on the proliferation of MCF-7 and MDA-MB-231 human breast cancer cells.

MDA-MB-231 cells were plated into 35 mm2 6-well Costar plates, at a density of 2.0 × 104 cells/well, in RPMI-1640 supplemented with 10% FBS. Cells were treated with either ddH2O (Control) or 8-Br-cAMP (10−10, 10−8, 10−6, 10−4 M). Six days following treatment cells were harvested by brief trypsinization and counted on a hemacytometer using the trypan blue exclusion test. Data is presented as percent of mean cell count in vehicle-treated control MCF-7 and MDA-MB-231 cells (100%). One-way ANOVA followed by Newman-Kuels post-hoc test analyses was used to determine statistically significant differences in the percentage among different groups. * P < 0.05 vs. vehicle treated cells, n=3.
To determine if the difference in Gαi2 regulation of ERK1/2 activity in MCF-7 and MDA-MB-231 cells is caused by alterations in B-Raf expression, which has been shown to be critical for cAMP to regulate ERK1/2 [28, 29], we examined the protein expression of B-Raf by Western blot analyses. Figure 7 clearly shows that both MCF-7 and MDA-MB-231 cells express B-Raf.
Figure 7. Expression of B-Raf in MCF-7 and MDA-MB-231 human breast cancer cells.
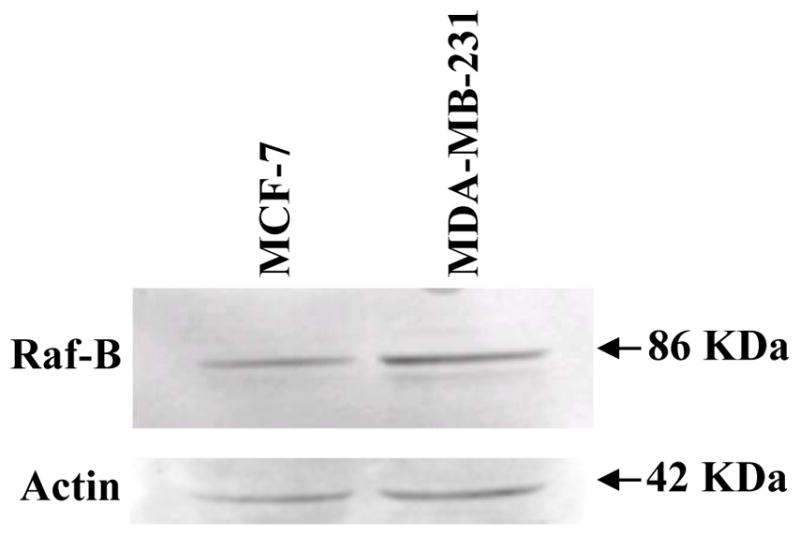
Whole cell lysates were extracted from MCF-7 and MDA-MB-231 breast cancer cells incubated in RPMI-1640 medium supplemented with 10% FBS. Fifty micrograms of total cellular protein from MDA-MB-231 cells was electrophoretically separated on 10% SDS polyacrylamide gel, and transferred onto nitrocellulose membrane. Expression of B-Raf was detected by Western blot analyses using anti-B-Raf antibody at a 1:1000 dilution. The same blots were stripped and reprobed with anti-actin antibody. This figure is a Western blot analysis of B-Raf expression in MCF-7 and MDA-MB-231 breast cancer cells, and it is representative of 3 independent experiments.
Discussion
Melatonin has been repeatedly shown to directly inhibit the in vitro growth of a wide variety of cancer cell types including the MCF-7 ERα-positive human breast cancer cells [14, 15]. Previous studies have shown that, at physiologic concentrations (10−9 M to 10−11 M), melatonin significantly inhibits the proliferation of MCF-7 cells by 30–50% [16]. It has also been shown that melatonin down-regulates the estrogen-response pathway by decreasing ERα expression, suppressing estrogen-induced ERα transactivation, thus modulating the expression of the estrogen-responsive growth-regulatory genes in MCF-7 cells [10, 30, 31]. However, down-regulation of the ERα signaling may not be the only mechanism underlying the growth-suppressive actions of melatonin since melatonin has also been shown to differentially regulate the transcriptional activity of other members of the steroid/thyroid hormone receptor superfamily, including the retinoic acid receptor alpha (RARα), glucocorticoid receptor (GR), retinoic acid related orphan receptor alpha (RORα), and vitamin D receptor (VDR) [24, 32], which, like the ERα, play an important role in the development and growth of breast tumors. It has been suggested that melatonin inhibits breast cancer cell proliferation partially through both suppression of the mitogenic pathways (ERα, GR, and RORα) and potentiation of the growth-inhibitory pathway (RARα). As shown in Figure 1, melatonin also exhibits an inhibitory effect on cell proliferation in the SK-BR-3 ERα-negative breast cancer cells. This result indicates that melatonin may act through other mechanism(s) to regulate cell growth in ERα-negative cell lines. Further studies are needed to delineate the signaling pathways involved in melatonin’s actions on cell growth in SK-BR-3 cells.
In contrast to SK-BR-3 cells, the MDA-MB-231 cell line was unresponsive to the anti-proliferative effect of melatonin (Fig. 1), even though it expresses the MT1 melatonin receptor at levels comparable to MCF-7 cells (Fig. 2). These results are consistent with previous reports [22] and demonstrate that the loss of responsiveness to melatonin is not due to the absence of the MT1 melatonin receptor. A recent study reported that melatonin showed weak cytotoxicity in MDA-MB-231 cells as measured by MTT assay [33]. The concentrations of melatonin examined in their report were at pharmacological levels (8 and 16 mM). The discrepancy between their results and our data may be largely due to different effects of melatonin concentrations (physiological versus pharmacologic) and a result of receptor-mediated effects alone at physiologic concentrations versus combined effects of receptor-mediated and non-receptor mediated events at pharmacologic concentrations.
As a GPCR, the MT1 melatonin receptor has been previously reported to associate with a wide variety of G-proteins in a number of tissues [24–27]. By association with specific G-proteins, and thus activating specific G-protein signaling pathways, the activated MT1 can elicit particular intracellular events that may lead to different patterns of cellular responses in a given cell type. In MCF-7 cells, MT1 specifically couples to a selective subset of G-proteins upon melatonin binding, including the Gαi2, Gαi3, Gαq and Gα11 proteins. The Gαi2 and Gαq proteins appear to be involved in melatonin’s growth-suppressive effect [25]. All of these G proteins (Gαi2, Gαi3, Gαq and Gα11) are expressed in MDA-MB-231 cells as determined by Western blot analysis (Fig. 3), suggesting that uncoupling of the MT1 signal transduction cascade in MDA-MB-231 cells does not result from the absence of certain G-proteins which associate with MT1 receptor in MCF-7 cells.
To further determine if the signaling pathways downstream of the Gαi2, Gαi3, Gαq and Gα11 proteins are functional at regulating cell growth in MDA-MB-231 cells, cell proliferation assays were performed to examine the effects of Gαi2, Gαi3, Gαq and Gα11 proteins on cell growth in MDA-MB-231 breast cancer cells. We have previously shown that transfection with the DP-Gαi2 and -Gαq constructs inhibited the proliferation of MCF-7 cells [25], suggesting that these G-protein signaling pathways (Gαi2 and Gαq) act as anti-mitogenic pathways in MCF-7 cells. Interestingly, in MDA-MB-231 cells these same G-proteins act as mitogenic pathways instead as transfection with the same DP-Gα constructs (Gαi2 and Gαq) significantly stimulated cell proliferation, while transfection with DN-Gα (Gαi2 and Gαq) constructs inhibited cell growth (Fig. 4). Together, these results suggest that the same set of G-proteins differentially regulates cellular proliferation in ERα-negative MDA-MB-231 cells vs. ERα-positive MCF-7 breast tumor cells.
Melatonin repression of ERα transcriptional activity appears to be mediated specifically via Gαi2, since expression of a DN-Gαi2 protein, but not Gαi3, Gαq or Gα11 proteins, abrogated melatonin’s ability to repress estrogen-induced ERα transactivation [25]. A well-established second messenger transducing signals from the Gαi-protein is cAMP. Transcriptional activity of steroid receptors including ERα can be modulated by phosphorylation at specific residues by a wide variety of effector protein kinases in the G-protein signaling pathways, such as PKC, calmodulin (CaM), and PKA. These kinases, in turn, are under the direct control of second messengers including cAMP, cGMP, Ca2+, and diacylglycerol (DAG) [34]. Melatonin has been previously reported to block the accumulation of intracellular cAMP in various tissues, including the breast [34–36], and suppress PKA activity in tissues other than the breast [7]. In MCF-7 human breast cancer cells it has been shown that melatonin inhibits estrogen and forskolin- or pituitary activating polypeptide (PACAP)-induced elevation of intracellular cAMP [35]. This inhibitory action appears to be mediated through Gαi2. Therefore, melatonin-mediated suppression of ERα transactivation may involve Gαi2-mediated regulation of the cAMP/PKA signaling cascade.
As a key player in the signaling network, the cAMP/PKA pathway may play an important role in mediating melatonin’s growth-suppressive effects by linking MT1 signaling pathway to other central signaling pathways as well. Previous studies have shown that the cAMP/PKA pathway cross talks with the MAPK cascade at the level of Raf. Therefore, it is possible that melatonin exerts its anti-proliferative effects partially by PKA-mediated suppression of the mitogenic ERK1/2 signaling pathway via Gαi2 protein. As shown in Figure 5, transfection with DP-Gαi2 construct blocked the phosphorylation/activation of ERK1/2 in MCF-7 cells, which is consistent with the anti-mitogenic role of Gαi2 protein revealed by cell proliferation assay and our recent studies in MCF-7 triple-negative breast tumor xenografts [21]. In contrast, Gαi2 protein had no effect on ERK1/2 phosphorylation in MDA-MB-231 cells (Fig. 5). Together, these results demonstrate that Gαi2 protein inhibits MCF-7 cell growth partially through repressing ERK1/2 activity and loss of Gαi2–regulation of ERK1/2 may lead to abnormal function of Gαi2 on cell growth in MDA-MB-231 cells.
To further determine if the signal transduction components in Gαi2 pathway downstream of cAMP are functional in terms of regulating cell growth, we compared the effects of 8-Br-cAMP on the proliferation of MCF-7 and MDA-MB-231 cells. As shown in Figure 6, 8-Br-cAMP (at a concentration of 10−10 to 10−6 M) exerts opposite effects on the proliferation of MCF-7 and MDA-MB-231 cells, significantly inhibiting cell proliferation in MCF-7 cells while increasing MDA-MB-231 cell growth. This data suggests that the signaling cascade downstream of cAMP/PKA is not functioning in the same way in these two cell lines, leading to different patterns of cellular responses to cAMP, and thus, to Gαi2 protein, and ultimately, melatonin, in MCF-7 vs. MDA-MB-231 cells.
As mentioned above the cAMP/PKA pathway cross talks with the MAPK pathway. Elevation of the intracellular cAMP level represses the activity of ERK1/2 in some cells, while stimulating ERK1/2 activity in others [34]. One of the well-established mechanisms underlying cell type-specific effects of cAMP on ERK1/2 activation is the differential expression of B-Raf. According to previous results, cAMP generally inhibits ERK1/2 activity unless B-Raf is expressed [28, 29]. As shown in Figure 7, B-Raf protein is expressed at comparable levels in both MCF-7 and MDA-MB-231 cells. This data suggests that, the different pattern of cellular responses to cAMP and Gαi2 protein in MCF-7 and MDA-MB-231 cells is not caused by presence or absence of B-Raf. Further studies are needed to pinpoint the missing link in the MT1/Gαi2 signal transduction cascade downstream of cAMP, which causes abnormal regulation of ERK1/2 activity, cellular responses to cAMP and Gαi2, and ultimately responsiveness to melatonin in MDA-MB-231 cells. Finally, these studies demonstrate that melatonin at physiologic concentrations, and through receptor-mediated mechanisms, can inhibit the proliferation of both ERα-positive and ERα-negative breast tumor cell lines, but that some ERα-negative breast tumors, such as the MDA-MB-231 cell line, are not growth-inhibited by melatonin even though they express the MT1 melatonin receptor. Interestingly, uncoupling of melatonin signal transduction pathway and loss of melatonin-responsiveness has also been observed in the ERα-positive melatonin-responsive MCF-7 cells in response to exposure to magnetic field [37], which causes uncoupling of the melatonin receptor from downstream signaling (adenyl cyclase/cAMP) [37]. This study further demonstrates that insensitivity to melatonin may be caused by uncoupling of the MT1/Gαi2 signal transduction cascade.
Acknowledgments
We thank Mr. Tripp Frasch for his assistance with editing the manuscript.
Grant Support: This work was generously supported by National Institutes of Health grant R01 CA-054152-14 and Army DOD grant to SMH.
Footnotes
DISCLOSURE STATEMENT: The authors have nothing to disclose.
References
- 1.Vanecek J. Cellular mechanisms of melatonin action. Physiol Rev. 1998;78:687–721. doi: 10.1152/physrev.1998.78.3.687. [DOI] [PubMed] [Google Scholar]
- 2.Hardeland R, Madrid JA, Tan DX, et al. Melatonin, the circadian multioscillator system and health: the need for detailed analyses of peripheral melatonin signaling. J Pineal Res. 2012;52:139–66. doi: 10.1111/j.1600-079X.2011.00934.x. [DOI] [PubMed] [Google Scholar]
- 3.Cardinali DP, Srinivasan V, Brzezinski A, et al. Melatonin and its analogs in insomnia and depression. J Pineal Res. 2012;52:365–75. doi: 10.1111/j.1600-079X.2011.00962.x. [DOI] [PubMed] [Google Scholar]
- 4.Barrett P, Bolborea M. Molecular pathways involved in seasonal body weight and reproductive responses governed by melatonin. J Pineal Res. 2012;52:376–88. doi: 10.1111/j.1600-079X.2011.00963.x. [DOI] [PubMed] [Google Scholar]
- 5.Reppert SM, Weaver DR, Ebisawa T. Cloning and characterization of a mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron. 1994;13:1177–1185. doi: 10.1016/0896-6273(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 6.Zhao H, Poon AM, Pang SF. Pharmacological characterization, molecular subtyping, and autoradiographic localization of putative melatonin receptors in uterine endometrium of estrous rat. Life Sci. 2000;66:1581–1591. doi: 10.1016/s0024-3205(00)00478-1. [DOI] [PubMed] [Google Scholar]
- 7.Witt-Enderby PA, Masana MI, Dubocovich ML. Physiological exposure to melatonin supersensitizes the cyclic adenosine 3’,5’-monophosphate-dependent signal transduction cascade in Chinese hamster ovary cells expressing the human mt1 melatonin receptor. Endocrinol. 1998;139:3064–3071. doi: 10.1210/endo.139.7.6102. [DOI] [PubMed] [Google Scholar]
- 8.Burns DM, Cos CS, Blask DE. Demonstration and partial characterization of 2-iodomelatonin binding sites in experimental mammary carcinoma. Program of the 72nd annual meeting of the endocrine society; 1990; San Antonio, TX. p. 62. abstract. [Google Scholar]
- 9.Blask DE, Pelletier DB, Hill SM, et al. Pineal melatonin inhibition of tumor promotion in the N-nitroso-N-methylurea of mammary carcinogenesis: Potential involvement of antiestrogenic mechanisms in vivo. J Cancer Res Clin Oncol. 1990;117:526–532. doi: 10.1007/BF01613283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ram PT, Kiefer TL, Silverman M, et al. Estrogen receptor transactivation in MCF-7 breast cancer cells by melatonin and growth factors. Mol Cell Endocrinol. 1998;141:53–64. doi: 10.1016/s0303-7207(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 11.Reppert SM, Godson C, Mahel CD, Weaver DR, Slaugenhaupt SA, Gusella JF. Molecular characterization of a second melatonin receptor expressed in human retina and brain: the Mel(1b) melatonin receptor. Proc Natl Acad Sci USA. 1995;92:8734–8738. doi: 10.1073/pnas.92.19.8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reppert SM, Weaver DR, Godson C. Melatonin receptor step into light: cloning and classification of subtypes. Trends Pharmocol Sci. 1996;17:100–102. doi: 10.1016/0165-6147(96)10005-5. [DOI] [PubMed] [Google Scholar]
- 13.Nowfar S, Teplitzky SR, Melancon K, et al. Tumor prevention by 9-cis-retinoic acid in the N-nitroso-N-methylurea model of mammary carcinogenesis is potentiated by the pineal hormone melatonin. Breast Cancer Res Treat. 2002;72:33–43. doi: 10.1023/a:1014912919470. [DOI] [PubMed] [Google Scholar]
- 14.Hill SM, Spriggs LL, Simon MA, et al. The growth inhibitory action of melatonin on human breast cancer cells is linked to the estrogen response system. Cancer Lett. 1992;64:249–256. doi: 10.1016/0304-3835(92)90050-6. [DOI] [PubMed] [Google Scholar]
- 15.Blask DE, Hill SM. Effects of melatonin on cancer: studies on MCF-7 human breast cancer cells in culture. J Neural Transm Suppl. 1986;21:433–449. [PubMed] [Google Scholar]
- 16.Hill SM, Blask DE. Effects of the pineal gland hormone, melatonin, on the proliferation and morphological characteristics of human breast cancer cells (MCF-7) in culture. Cancer Res. 1988;48:6121–6128. [PubMed] [Google Scholar]
- 17.Yuan L, Collins AR, Dai J, et al. MT(1) melatonin receptor overexpression enhances the growth suppressive effect of melatonin in human breast cancer cells. Mol Cell Endocrionl. 2002;192:147–156. doi: 10.1016/s0303-7207(02)00029-1. [DOI] [PubMed] [Google Scholar]
- 18.Collins A, Yuan L, Kiefer TL, et al. Overexpression of the MT1 melatonin receptor in MCF-7 human breast cancer cells inhibits mammary tumor formation in nude mice. Cancer Lett. 2003;189:49–57. doi: 10.1016/s0304-3835(02)00502-5. [DOI] [PubMed] [Google Scholar]
- 19.Xiang S, Mao L, Yuan L, et al. Impaired mouse mammary gland growth and development is mediated by melatonin and its MT1 G protein-coupled receptor via repression of ERα, Akt1, and Stat5. J Pineal Res. 2012;53:307–18. doi: 10.1111/j.1600-079X.2012.01000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blask DE, Brainard GC, Dauchy RT, et al. Melatonin-depleted blood from premenopausal women exposed to light at night stimulates growth of human breast cancer xenografts in nude rats. Cancer Res. 2005;65:11174–84. doi: 10.1158/0008-5472.CAN-05-1945. [DOI] [PubMed] [Google Scholar]
- 21.Mao L, Dauchy RT, Blask DE, et al. Circadian gating of epithelial-to-mesenchymal transition in breast cancer cells via melatonin-regulation of GSK3β. Mol Endocrinol. 2012;26:1808–20. doi: 10.1210/me.2012-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill SM, Spriggs LL, Simon MA, et al. The growth inhibitory action of melatonin on human breast cancer cells is linked to the estrogen response system. Cancer Lett. 1992;64:249–256. doi: 10.1016/0304-3835(92)90050-6. [DOI] [PubMed] [Google Scholar]
- 23.Ram PT, Dai J, Yuan L, et al. Involvement of the mt1 melatonin receptor in human breast cancer. Cancer Lett. 2002;179:141–150. doi: 10.1016/s0304-3835(01)00873-4. [DOI] [PubMed] [Google Scholar]
- 24.Kiefer TL, Lai L, Yuan L, et al. Differential regulation of estrogen receptor alpha, glucocorticoid receptor and retinoic acid receptor alpha transcriptional activity by melatonin is mediated via different G proteins. J Pineal Res. 2005;38:231–239. doi: 10.1111/j.1600-079X.2004.00198.x. [DOI] [PubMed] [Google Scholar]
- 25.Lai L, Yuan L, Chen Q, et al. The Galphai and Galphaq proteins mediate the effects of melatonin on steroid/thyroid receptor transcriptional activity and breast cancer cell proliferation. J Pineal Res. 2008;45:476–88. doi: 10.1111/j.1600-079X.2008.00620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brydon L, Roka F, Petit L, et al. Dual signaling of human Mel1a melatonin receptors via Gi2, Gi3, Gq/11 proteins. Mol Endocrinol. 1999;13:2025–2038. doi: 10.1210/mend.13.12.0390. [DOI] [PubMed] [Google Scholar]
- 27.Chan AS, Lai FP, Lo RK, et al. Melatonin mt1 and MT2 receptors stimulate c-Jun N-terminal kinase via pertussis toxin-sensitive and –insensitive G proteins. Cell Signal. 2002;14:249–257. doi: 10.1016/s0898-6568(01)00240-6. [DOI] [PubMed] [Google Scholar]
- 28.Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. doi: 10.1016/s0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 29.Dumaz N, Marais R. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft fur Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J. 2005;272:3491–3504. doi: 10.1111/j.1742-4658.2005.04763.x. [DOI] [PubMed] [Google Scholar]
- 30.Molis TM, Spriggs LL, Jupiter Y, et al. Melatonin modulation of estrogen-regulated proteins, growth factors, and proto-oncogenes in human breast cancer. J Pineal Res. 1995;18:93–103. doi: 10.1111/j.1600-079x.1995.tb00146.x. [DOI] [PubMed] [Google Scholar]
- 31.Molis TM, Spriggs LL, Hill SM. Modulation of estrogen receptor mRNA expression by melatonin in MCF-7 human breast cancer cells. Mol Endocrinol. 1994;8:1683–1690. doi: 10.1210/mend.8.12.7708056. [DOI] [PubMed] [Google Scholar]
- 32.Hill SM, Blask DE, Xiang S, et al. Melatonin and associated signaling pathways that control normal breast epithelium and breast cancer. J Mammary Gland Biol Neoplasia. 2011;16:235–45. doi: 10.1007/s10911-011-9222-4. [DOI] [PubMed] [Google Scholar]
- 33.Jung JH, Sohn EJ, Shin EA, et al. Melatonin Suppresses the Expression of 45S Preribosomal RNA and Upstream Binding Factor and Enhances the Antitumor Activity of Puromycin in MDA-MB-231 Breast Cancer Cells. Evid Based Complement Alternat Med. 2013;2013:879746. doi: 10.1155/2013/879746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cabrera-Vera TM, Vanhauwe J, Thomas TO, et al. Insights into G protein structure, function, and regulation. Endocrine Rev. 2003;24:765–781. doi: 10.1210/er.2000-0026. [DOI] [PubMed] [Google Scholar]
- 35.Kiefer TL, Yuan L, Ram PT, et al. Melatonin inhibits estrogen receptor transactivation and cAMP levels in MCF-7 human breast cancer cells. Breast Cancer Res Treat. 2002;71:37–45. doi: 10.1023/a:1013301408464. [DOI] [PubMed] [Google Scholar]
- 36.Morgan PJ, Lawson W, Davidson G, et al. Guanine nucleotides regulate the affinity of melatonin receptors in the ovine pars tuberalis. Neuroendocrinol. 1989;50:359–362. doi: 10.1159/000125245. [DOI] [PubMed] [Google Scholar]
- 37.Ishido M, Nitta H, Kabuto M. Magnetic fields (MF) of 50 Hz at 1.2 microT as well as 100 microT cause uncoupling of inhibitory pathways of adenylyl cyclase mediated by melatonin 1a receptor in MF-sensitive MCF-7 cells. Carcinogenesis. 2001;22:1043–8. doi: 10.1093/carcin/22.7.1043. [DOI] [PubMed] [Google Scholar]