

Significance
Human DEAD-box polypeptide 3 (DDX3) is an ATPase/RNA helicase involved in the replication of many viral pathogens. We reported herein the first inhibitor, to our knowledge, of the helicase binding site of DDX3 endowed with a broad spectrum antiviral activity [HIV-1 WT, HIV drug-resistant strains, Hepatitis C virus (HCV), Dengue virus (DENV), and West Nile virus (WNV)]. The good toxicity profile suggests that the DDX3 activity, although essential for viruses, could be dispensable to the cells, validating DDX3 as a pharmaceutical target. Our results clearly showed that DDX3 inhibitors could be exploited to treat HIV/HCV coinfections, emerging infectious diseases (such as DENV and WNV), and HIV-1 patients carrying drug-resistant strains. Each of these three medical conditions currently represents a major challenge for clinical treatment.
Keywords: broad spectrum antivirals, DDX3, host factors, resistance, coinfections
Abstract
Targeting a host factor essential for the replication of different viruses but not for the cells offers a higher genetic barrier to the development of resistance, may simplify therapy regimens for coinfections, and facilitates management of emerging viral diseases. DEAD-box polypeptide 3 (DDX3) is a human host factor required for the replication of several DNA and RNA viruses, including some of the most challenging human pathogens currently circulating, such as HIV-1, Hepatitis C virus, Dengue virus, and West Nile virus. Herein, we showed for the first time, to our knowledge, that the inhibition of DDX3 by a small molecule could be successfully exploited for the development of a broad spectrum antiviral agent. In addition to the multiple antiviral activities, hit compound 16d retained full activity against drug-resistant HIV-1 strains in the absence of cellular toxicity. Pharmacokinetics and toxicity studies in rats confirmed a good safety profile and bioavailability of 16d. Thus, DDX3 is here validated as a valuable therapeutic target.
Most of the current therapeutic approaches to fight viral diseases target unique components or enzymes of a given virus with direct-acting antivirals. Although therapeutically highly successful, direct-acting antivirals are still suffering shortcomings, such as drug resistance, poor adherence in selected patient groups, and associated toxicity (1, 2). In addition, virtually no broad spectrum antivirals are currently available. An alternative approach currently exploited in antiviral research is the targeting of host proteins. Examples include Maraviroc (approved for the treatment of HIV-1 infection) and Alisporivir [in phase III trials for anti-Hepatitis C virus (HCV) therapy] (3, 4). Viruses as obligatory parasites dependent on the host cell machinery for replication, protein expression, and assembly of progeny particles (5–7). RNA viruses, in particular, have high mutation rates owing to their error-prone polymerases and rapidly generating drug-resistant variants (8–10). On this basis, blocking host cell factors required by different viruses for their replication might be a low cost and short time but effective route to develop broad spectrum antivirals able to limit the occurrence of drug resistance (11, 12).
Recent studies have revealed that the cellular ATPase/RNA helicase X-linked DEAD-box polypeptide 3 (DDX3) is an essential host factor for the replication of viruses belonging to different families: CMV (13) (Herpesviridae), HIV-1 (14) (Retroviridae), HCV (15–18), Japanese Encephalitis virus (19), Dengue virus (DENV) (20), West Nile virus (WNV; Flaviviridae) (21), Vaccinia virus (22–24) (Poxviridae), and Norovirus (25) (Caliciviridae). The exact mechanisms of interaction of this protein with individual pathogens are still poorly understood (26). Nonetheless, DDX3 is being regarded as an interesting target for the development of novel antiviral compounds (27, 28).
For example, targeting DDX3 might offer the possibility of simultaneously treating patients for HIV and HCV coinfections. Several inhibitors of the ATPase activity of DDX3 have been recently identified (29–34); however, an ATP-mimetic compound may have a low degree of selectivity in vivo because of the possibility of interacting with many targets (35).
Results and Discussion
In a recent work, we designed and validated the first small molecule DDX3 inhibitors, to our knowledge, specifically targeting its RNA binding site (Fig. 1) (36). The most promising molecule was compound 2, with both antihelicase activity against DDX3 (IC50 = 1 µM) and inhibitory effects on HIV-1 replication in peripheral blood mononuclear cells [half-maximal effective concentration (EC50) = 10 µM].
Fig. 1.

2D structures of the known human DDX3 inhibitors targeting the RNA binding pocket.
Pursuing this research line, a structure-based optimization process was prosecuted herein on compound 2, resulting in the identification of a new family of more potent DDX3 inhibitors.
Because of the lack of a crystal structure of human DDX3 bound to RNA (closed conformation), we exploited an homology model previously built by us using the closed conformation of Drosophila Vasa DEAD-box helicase (Protein Data Bank ID code 2DB3) as a template (Fig. 2) (37).
Fig. 2.

Graphical representation of the DDX3 RNA binding site. The RNA strand is represented as yellow carbon sticks. The binding mode of compound 2 (green carbon sticks) was predicted by docking studies. Hydrogen bond interactions are visualized as black dashed lines. Compound 2 occupies a little part of the large pocket, and the two regions circled in magenta and cyan are unexplored by this ligand.
Similar to the RNA strand (Fig. 2, yellow stick), compound 2 (Fig. 2, green stick) establishes important polar contacts with Arg276, Arg480, and Pro274. Furthermore, it makes hydrophobic interactions with Phe357, His472, Lys451, and Val500. However, our modeling analysis revealed the presence in the binding pocket of two unexplored areas that could be exploited in search of additional interactions (Fig. 2, cyan and magenta circles). Thus, a small library of compound 2 derivatives has been designed and synthesized, introducing modifications to probe these two regions and expand available structure–activity relationship (SAR) data. Slight modifications included the replacement of the nitro group with the isosteric carboxyl group and the substitution of the methyl–phenyl ring with a cyclohexyl moiety. Furthermore, a naphthyl ring was inserted in place of the tolyl terminus to make additional interactions with Arg503 and Val500. Next, more pronounced substitutions have been made by inserting a substituted triazole ring instead of the nitro group, which allowed the exploration of additional interactions involving residues Arg326 and Gly302. The para position was predicted by docking studies as the most appropriate for such kinds of substitutions, and side chains at four positions were selected, taking into account the interactions into the pocket.
Synthesis of compounds 2–9 (Fig. S1) and 16a–16g (Fig. S2) is reported in Supporting Information.
Fig. S1.

Synthesis of derivatives 9–12. Reagents and conditions: a, CH2Cl2, 5 h, room temperature (r.t.); b, HCl/AcOH, 6 h, 100 °C.
Fig. S2.

Synthesis of derivatives 16a–16g: a, CH2Cl2, 5 h reflux; b, H2, Pd/C, MeOH, 1 h; c, t-BuONO, CH3CN, 20 min, 0 °C, then TMSiN3, CH3CN, 2 h room temperature (r.t.); d, opportune alkyne, CuSO4○5 H2O, sodium ascorbate, H2O t-BuOH (1:1), microwave (MW) 120 °C, 10 min; e, opportune alkynoic acid, CuCl, l-proline, K2CO3, DMSO, MW 65 °C, 20 min.
The anti-DDX3 activity of compounds was assayed, and results are shown in Table 1. Remarkably, the modifications aimed at optimizing the van der Waals contacts of the ligands with Arg276 and Arg326 (derivatives 16a–16g) resulted in an increase of the helicase inhibitory activity, with the best inhibitors 16d and 16g having IC50 values of 0.3 and 0.98 µM, respectively.
Table 1.
DDX3 antienzymatic activity
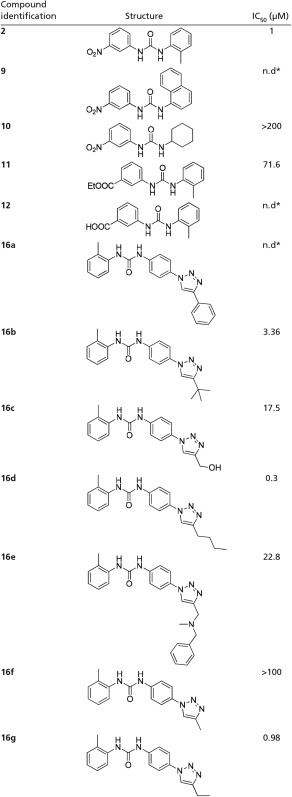 |
n.d., Not determined.
Compound precipitated during assays.
The inactivity of compound 10 suggests that the presence of an aromatic moiety is fundamental for the helicase inhibitory activity, and is probably due to a crucial cation–π interaction with Arg503. Compounds 9 and 12 unexpectedly precipitated from medium in enzymatic assays and were, thus, found inactive, whereas the ester analog 11 was found moderately active.
The predicted binding mode of 16d within the DDX3 RNA binding pocket is shown in Fig. 3. Some of the ligand contacts are coincident with the key interactions made by the parent compound 2. Indeed, the urea NH groups were involved in hydrogen bonds with the backbone carbonyl oxygen of Pro274, whereas the triazole ring interacted with the guanidine group of Arg276. Moreover, the tolyl terminus established hydrophobic interactions with residues Arg503 and Pro274, and the phenyl ring made hydrophobic contacts with the aromatic side chain of Phe357. Finally, the butyl substituent at the four position of the triazole ring made profitable interactions with residues Arg326 and Gly302, was unexploited by compound 2, and was probably responsible for the improved antihelicase activity.
Fig. 3.

Binding mode of compound 16d (purple sticks). For comparison purpose, compound 2 (green sticks) was also visualized.
According to the proposed mode of binding, 16d behaved as a competitive inhibitor with respect to the RNA substrate, which can be seen by the decrease in its inhibition potency as a function of increasing RNA substrate concentrations (Fig. 4).
Fig. 4.

Dose–response curves for DDX3 helicase activity inhibition by 16d at increasing dsRNA substrate concentrations.
The most active DDX3 inhibitor 16d was then tested against HIV, HCV, WNV, and DENV. Results are summarized in Table 2. Remarkably, 16d showed a broad spectrum of antiviral activity, being able to inhibit the replication of all of these viruses with EC50 values ranging from 0.97 to 16.5 µM. No toxicity was found in three different cell culture systems (LucUbiNeo-ET cells and Hepato cellular carcinoma cells) as a demonstration that the inhibition of a cellular cofactor essential for the viruses but not for the cells can represent a successful strategy for the development of novel antiviral agents. For comparison purpose, we also tested hit compound 2 that was previously published (36) against HCV-infected cells, but despite its activity against DDX3, it was found inactive (EC50 > 86 µM).
Table 2.
Antiviral activities
| Virus | Strain | Cell type | EC50 16d (μM) | CC50 16d (μM) | EC50 10 (μM) | CC50 10 (μM) | EC50 16f (μM) | CC50 16f (μM) |
| HIV* | NL4-3 | PBMCs | 1.1 | >200 | 110 | >200 | >50 | 50 |
| HCV† | Replicon | LucUbiNeo-ET | 0.97 | 49.77 | >80 | 80 | >36 | 36 |
| DENV‡ | 2, New Guinea C | Huh7 | 2.55 | >200 | nt | nt | nt | nt |
| WNV‡ | NY99 | Huh7 | 16.5 | >200 | nt | nt | nt | nt |
CC50, half-maximal cytotoxic concentration; EC50, half-maximal effective concentration; nt, Not tested; PBMC, peripheral blood mononuclear cell.
Evaluated in PBMCs.
Evaluated in LUNET:LucUbiNeo-ET cells.
Evaluated in Huh7:Hepato cellular carcinoma cells.
The NS3 protein of both HCV and DENV is an RNA helicase that belongs to the same family of DDX3. To exclude the involvement of such viral enzymes in the activities of 16d, we tested this compound against NS3 proteins of HCV and DENV as well as against another cellular member of the DEAD-box family, namely DDX1. We also tested the ability of 16d in the inhibition of the ATPase activity of DDX3. As shown in Table 3, 16d was found to be completely inactive against the ATPase of DDX3, DDX1 helicase, and DENV NS3 helicase. It showed moderate activity against the HCV NS3 helicase, which was, however, 56- and 17-fold lower than the activity against DDX3 helicase and HCV proliferation, respectively.
Table 3.
Enzymatic data on compound 16d
| ATPase DDX3 IC50 (µM) | DDX1 IC50 (µM) | NS3 (DENV) IC50 (µM) | NS3 (HCV) IC50 (µM) |
| >200* | >200 | >200 | 16.8 |
The value >200 indicates that less than 20% of inhibition was observed at 200 µM, the highest concentration that was tested.
The antiviral activity of compound 16d was also evaluated against HIV-1 strains carrying clinically relevant mutations conferring high-level resistance to most classes of antivirals approved to treat HIV infection (Table 4 and Table S1). Compound 16d retained full activity against all of the resistant viruses tested, confirming its novel mechanism of action and the potential to overcome HIV resistance.
Table 4.
Antiviral activity of compound 16d against HIV-1 strains carrying the most common patterns of resistance mutations selected by drugs currently used to treat HIV-1 infection
| HIV-1 strain* | Drug resistance class | IC50 (95% confidence interval; μM) | Fold change† |
| 114‡ | WT | 1.11 (0.31–3.90) | — |
| 11808 | PIs | 0.23 (0.08–0.65) | 0.2 |
| 7406 | NRTIs | 0.33 (0.13–0.87) | 0.3 |
| 7404 | NRTIs | 0.22 (0.11–0.47) | 0.2 |
| 12227 | NNRTIs | 0.94 (0.21–1.34) | 0.8 |
| 12235 | NNRTIs | 0.36 (0.15–0.87) | 0.3 |
| 11845 | INIs | 0.37 (0.26–0.52) | 0.3 |
INI, integrase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; PI, protease inhibitor. —, no fold change.
NIH AIDS Reagent Program catalog number (https://www.aidsreagent.org).
Resistant strain IC50 to WT strain IC50 ratio.
NL4-3 HIV-1 WT reference strain.
Table S1.
Resistant viruses tested
| Virus (ARP catalog no.)/drug resistance class | Mutations | Degree of resistance to most common drugs |
| 114 | ||
| WT virus | None | Full susceptibility to all drugs |
| 11808 | ||
| PIs | Major: V32I, I54V, I84V, L90M; minor: L10F, V11I, K20T, L33F, E35G, A71I, G73S, L89V | High-level resistance to atazanavir and lopinavir; intermediate resistance to darunavir |
| 7406 | ||
| NRTIs | M41L, L74V, M184V, L210W, T215Y | High-level resistance to lamivudine, emtricitabine, zidovudine, and abacavir; intermediate resistance to tenofovir |
| NNRTIs | A98G, G190C | High-level resistance to efavirenz and nevirapine; low-level resistance to etravirine and rilpivirine |
| 7404 | ||
| NRTIs | A62V, V75I, F77L, F116Y, Q151M | High-level resistance to abacavir and zidovudine; intermediate resistance to lamivudine, emtricitabine, and tenofovir |
| 12227 | ||
| NRTIs | M41L, T215Y | High-level resistance to zidovudine; intermediate resistance to abacavir and tenofovir; low-level resistance to lamivudine and emtricitabine |
| NNRTIs | K101P, K103N | High-level resistance to efavirenz, nevirapine, and rilpivirine; intermediate resistance to etravirine |
| 12235 | ||
| NRTIs | M41L, D67N, T69D, L74I, L210W, T215Y | High-level resistance to zidovudine, tenofovir, and abacavir; intermediate resistance to lamivudine and emtricitabine |
| NNRTIs | A98G, K101E, Y181C, G190A | High-level resistance to efavirenz, nevirapine, rilpivirine, and etravirine |
| 11845 | ||
| INIs | G140S, Q148H | High-level resistance to raltegravir and elvitegravir; intermediate resistance to dolutegravir |
ARP, AIDS Reagent Program; INI, integrase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; PI, protease inhibitor.
A few in vitro experiments were then conducted to quickly establish the absorption/stability of compound 16d: aqueous solubility (pH 7.4 buffer), parallel artificial membrane permeability assay, and human liver microsome stability determination (Table 5). Despite the fact that the aqueous solubility is just outside of the range of recommended values for a drug (−6 < LogS < −1), the excellent metabolic stability and the good passive membrane permeability make 16d a good lead candidate for additional development.
Table 5.
16d in vitro absorption, distribution, metabolism, and excretion (ADME) studies
HLM, human liver microsome; Papp, apparent permeability.
Papp reported in centimeters·seconds−1.
Membrane retention expressed as percentage.
Aqueous solubility.
HLM metabolic stability expressed as percentage of unmodified parent drug.
To better evaluate toxicity and biodistribution of compound 16d, we finally performed several preliminary in vivo studies; 16d was administered to Wistar rats by tail vein in a single systemic dose (0.05 mL per dose) at high-dose levels of 20 mg/kg. There were no treatment-related adverse effects on clinical chemistry analysis of serum for the measurement of biochemical parameters (including liver enzymes, such as alanine aminotransferase, aspartate aminotransferase, and glutamate dehydrogenase), urea, creatinine, triglycerides, LDLs, and HDLs. Urine analysis parameters for treated rats did not show any significant difference from the respective control groups. Histopathological analysis of the liver was free from any pathological abnormality, and H&E-stained sections appeared normal, with regular cellular architecture. The hepatic cells had intact cytoplasm, sinusoidal spaces, prominent nucleus, and nucleolus (Fig. 5). Renal tissue of all animal groups showed preserved renal parenchyma with normal appearance of glomerular tuft and urinary space (Fig. 5). Also, histological analyses of brain tissue samples showed no obvious uncharacteristic changes in all animal groups. In conclusion, 16d was found to possess excellent biocompatibility, and Wistar rats showed a good tolerance to the dose of 20 mg/kg.
Fig. 5.

Representative images of the histological examination of H&E-stained sections of (A1–E1) livers, (A2–E2) kidneys, and (A3–E3) brains from three groups of rats. (A1–A3 and B1–B3) VG. (C1–C3 and D1–D3) TG. (E1–E3) WT group. Treated rats do not exhibit abnormal histopathological changes compared with the control group. The microphotographs were taken using a digital camera (Nikon SLR-D3000) at original magnifications of 100× and 200×. gt, Glomerular tuft; U, urinary space.
The pharmacokinetic analysis of compound 16d was finally conducted. The main pharmacokinetic parameters from single-compartment model analysis are summarized in Table 6. The half-life elimination and the plasmatic clearance values denoted that 16d was rapidly eliminated after i.v. administration.
Table 6.
Pharmacokinetic parameters of 16d after i.v. administration at a dose of 10 mg·kg−1
| Pharmacokinetic parameters | 16d* |
| Dose (mg/kg) | 10.00 |
| MRT (h) | 3.98 ± 0.38 |
| AUC0–∞ (μg × h/mL) | 39.95 ± 13.50 |
| AUC0–24 (μg × h/mL) | 39.77 ± 13.51 |
| CL (mL/min) | 1.29 ± 0.54 |
| t1/2β (h) | 3.19 ± 0.24 |
AUC, area under the plasma concentration–time curve; CL, plasma clearance; MRT, mean residence time; t1/2β, plasma half-life.
All data are expressed as means ± SDs (n = 3).
The mean plasma concentration–time curves after i.v. administration are illustrated in Fig. 6A. Tissue distribution of 16d in rat after i.v. administration is presented in Fig. 6B. Higher concentration of 16d was found in adipose tissue followed by kidney, testicle, liver, and brain in that order, which can be attributed to the blood flow in this organs.
Fig. 6.

Pharmacokinetic parameters and tissue distribution in rats; 16d was administered as a single i.v. bolus injection of 10 mg/kg per group (n = 3). Data points represent the means ± SDs. (A) Plasma level curves of 16d. The semilogarithmic plot is shown in Inset. The elimination curve showed a first-order kinetic that showed an exponential decrease in the semilogarithmic plot. (B) Concentration levels of 16d in rat tissues at 24 h after single-dose administration.
In conclusion, we report herein the discovery of a novel series of human helicase DDX3 inhibitors. Among them, compound 16d represents the first compound, to our knowledge, achieving broad spectrum antiviral activity (HIV, HCV, DENV, and WNV) in infected cells, targeting a host factor. Compound 16d was active against HIV-1 drug-resistant strains, suggesting that DDX3 targeting agents may be able to treat HIV/HCV coinfections, patients harboring drug-resistant viruses, and emerging viral diseases, for which no specific drugs are available. Moreover, the good toxicity profile suggests that the DDX3 activity, although essential for viral replication, may be dispensable to the cell as shown by preclinical studies. This result represents a step forward in the fight against infectious diseases and opens a new scenario in the drug development process.
Materials and Methods
Detailed procedures for the synthesis and characterization of compounds are provided in Supporting Information.
Absorption, distribution, metabolism, and excretion (ADME) experiments, assay protocols, and pharmacokinetic and toxicity studies can be found in Supporting Information.
The rat experiments were performed under a protocol approved by the Institutional Animal Use and Care Committee at the Università Cattolica del Sacro Cuore (permit no. EE21; March 18, 2014) and authorized by the Italian Ministry of Health according to the Legislative Decree 116/92, which implemented the European Directive 86/609/EEC on laboratory animal protection in Italy. Animal welfare was routinely checked by veterinarians of the Service for Animal Welfare.
SI General and Materials
Reagents were obtained from commercial suppliers (for example, Sigma-Aldrich and Alfa Aesar). All commercially available chemicals were used as purchased without additional purification. CH2Cl2 and MeOH were dried before use by distilling from calcium hydride or magnesium methoxide. Anhydrous reactions were run under a positive pressure of dry N2 or argon. TLC was carried out using Merck TLC Plates Silica Gel 60 F254. Chromatographic purifications were performed on columns packed with Merk 60 Silica Gel (23–400 mesh) for flash technique.
All NMR spectra were recorded on a Bruker Avance DPX400 Spectrometer at 400 MHz for 1H NMR or 100 MHz for 13C NMR. Chemical shifts are reported relative to tetramethylsilane at 0.00 ppm. 1H patterns are described using the following abbreviations: singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint), sextet (sx), septet (sept), multiplet (m), broad signal (br), and broad singlet (br s).
Mass spectra data were obtained using an Agilent 1100 LC/Mass Spectra Detection (MSD) VL System (G1946C) with a 0.4-mL/min flow rate using a binary solvent system 25 of 95:5 methyl alcohol:water. UV detection was monitored at 254 nm. Mass spectra were acquired in positive and negative mode scanning over the mass range. Microwave irradiation experiments were conducted using CEM Discover Synthesis Unit (CEM Corp.).
For the quantitative analysis, was used a UV/liquid chromatography (LC) -MS system. LC analyses were performed by an Agilent 1100 LC/MSD VL System (G1946C; Agilent Technologies). Spectra were acquired over the scan range m/z 50–1,500 using a step size of 0.1 U. Chromatographic analysis was performed using a Varian Polaris 5 C18-A Column (150 × 4.6 mm; 5-µm particle size) at room temperature (r.t.). Analysis was carried out using gradient elution of a binary solution; eluent A was acetonitrile (ACN), whereas eluent B consisted of water. The analysis started at 0% A for 3 min, then rapidly increased up to 98% in 12 min, and finally, remained at 98% A until 18 min. The analysis was performed at a flow rate of 0.8 mL min−1, and injection volume was 20 µL. LC retention times, molecular ion (m/z), and LC purity (by UV) were based on the method below. Purity of compounds (as measured by peak area ratio) was >97%.
SI General Procedures and Product Characterization
General Procedure for the Synthesis of Compounds 9–11 (Fig. S1).
The opportune isocyanate 4 or 5 (1 equivalent) was added to a solution of the corresponding amine (1 equivalent) in anhydrous CH2Cl2 (10 mL) in one portion. The solution was stirred for 2 h at room temperature under a nitrogen atmosphere. The white precipitate was filtered, washed with petroleum ether, and dried under high vacuum, affording the desired product as a white solid.
1-(Naphthalen-1-yl)-(3-nitrophenyl)urea (9) 1H NMR: [400 MHz (CD3)2SO]: δ 9.28 (s, 1H), 9.03 (s, 1H), 8.61 (s, 1H), 8.13 (s, 1H), 7.85–7.80 (d, 4H), 7.73–7.71 (d, 1H), 7.58–7.54 (t, 1H), 7.51–7.49 (d, 1H), 7.46–7.42 (t, 1H), 7.37–7.34 (t, 1H) ppm. 13C NMR [100 MHz (CD3)2SO]: δ 153.00, 148.66, 141.50, 137.34, 134.13, 130.54, 129.77, 128.92, 127.92, 127.53, 126.87, 124.83, 124.65, 120.28, 116.81, 114.50, 112.66 ppm.
1-Cyclohexyl-3-(3-nitrophenyl)urea (10) 1H NMR (400 MHz MeOD): δ 8.41 (s, 1H), 7.80–7.77 (d, J = 12 Hz, 1H), 7.62–7.60 (d, J = 8.0 Hz, 1H), 7.46–7.42 (t, J = 8.0 Hz, 1H), 3.60–3.54 (m, 1H), 1.93–1.90 (m, 2H), 1.77–1.72 (m, 2H), 1.64–1.61 (m, 2H), 1.44–1.27 (m, 2H), 1.25–1.19 (m, 2H) ppm. 13C NMR (100 MHz MeOD): δ 156.69, 149.74, 140.18, 127.91, 127.01, 117.81, 113.70, 50.00, 33.51, 25.60, 24.72 ppm. MS electrospray ionization (ESI) m/z 264 [M + H]+, 286 [M + Na]+.
Ethyl 3-(3-o-tolylureido)benzoate (11): Aniline 8 (50 mg, 0.30 mmol) was added to a solution of o-tolyl-isocyanate (38 μL, 0.30 mmol) in 6 mL of anhydrous dichloromethane (DCM). The reaction mixture was stirred at r.t. for 5 h. After this time, the solution became a suspension, and the precipitate was filtered off, washed with DCM several times, and crystallized from MeOH to give 11 as a white solid. Yield = 91%. 1H NMR [400 MHz (CD3)2SO]: δ 9.25 (s, 1H), 8.12 (s, 1H), 7.91 (s, 1H), 8.13 (s, 1H), 7.81–7.79 (d, J = 8.0 Hz, 1H), 7.67–7.65 (d, J = 8.0 Hz, 1H), 7.55–7.53 (t, J = 6.0 Hz, 1H), 7.43–7.39 (d, J = 8.0 Hz, 1H), 7.17–7.12 (m, 2H), 6.96–6.93 (t, J = 6.0 Hz, 1H), 4.32–4.27 (q, J = 4.0 Hz, 2H), 2.23 (s, 3H) 1.32–1.29 (t, J = 6.0 Hz, 2H) ppm. 13C NMR [100 MHz (CD3)2SO]: δ 166.18, 153.09, 140.79, 137.65, 130.96, 130.67, 129.70, 128.28, 126.64, 123.40, 122.83, 122.77, 121.74, 118.78, 61.20, 18.29, 14.64 ppm. MS (ESI) m/z 299 [M + H]+, 321 [M + Na]+.
3-(3-o-Tolylureido)benzoic acid (12): A solution of 11 (70 mg, 0.21 mmol) in a mixture of concentrated HCl (3 mL) and concentrated HOAc (2 mL) was heated at 100 °C for 4 h. After this time, it was cooled to room temperature, the separated white solid was collected by filtration, and the solid cake was washed with water and MeOH and dried in vacuum to give 12 as a white solid. Yield = 82%. 1H NMR [400 MHz (CD3)2SO]: δ 9.21 (s, 1H), 8.12 (s, 1H), 7.91 (s, 1H,), 8.13 (s, 1H), 7.81–7.79 (d, J = 8.0 Hz, 1H), 7.63–7.61 (d, J = 8.0 Hz, 1H), 7.54–7.52 (t, J = 4.0 Hz, 1H), 7.40–7.36 (d, J = 8.0 Hz, 1H), 7.17–7.11 (m, 2H), 6.96–6.93 (t, J = 6.0 Hz, 1H), 2.23 (s, 3H) ppm. 13C NMR [100 MHz (CD3)2SO]: δ 167.77, 153.11, 140.64, 137.70, 131.87, 130.68, 129.51, 128.27, 126.64, 123.38, 122.00, 122.55, 121.72, 119.14, 18.32 ppm. MS (ESI) m/z 269 [M − H]−, 305 [M + Cl]−.
1-(4-Nitrophenyl)-3-o-tolylurea: Aniline 13 (500 mg, 3.62 mmol) was added to a solution of o-tolyl-isocyanate (673 µL, 5.43 mmol) in anhydrous CH2Cl2 (10 mL) in one portion. The solution was stirred for 4 h at 60 °C under a nitrogen atmosphere. The yellow precipitate was filtered, washed with DCM and petroleum ether, and dried under high vacuum to afford the desired product as a white solid. Yield = 63%. 1H NMR [400 MHz (CD3)2SO]: δ 9.7 (s, 1H), 8.19–8.16 (d, J = 9.2 Hz, 2H), 8.13 (s, 1H), 7.78–7.76 (d, J = 8.0 Hz, 1H), 7.69–7.66 (d, J = 12.0 Hz, 2H), 7.19–7.13 (m, 2H), 7.00–6.97 (t, 1H, J = 12.0 Hz), 2.24 (s, 3H) ppm. MS (ESI) m/z 270 [M − H]−, 306 [M + Cl]−.
1-(4-Aminophenyl)-3-o-tolylurea (14): 1-(4-nitrophenyl)-3-o-tolylurea (300 mg, 1.10 mmol) was solubilized in 30 mL anhydrous MeOH, and palladium on charcoal (50 mg) was added. The reaction mixture was stirred under hydrogen atmosphere for 1 h; then, the mixture was filtered off on a celite pad, the solvent was evaporated at reduced pressure, and the residue was crystallized from acetonitrile. Yield = 70%; white solid. 1H NMR [400 MHz (CD3)2SO]: δ 8.48 (s, 1H), 7.83–7.81 (d, J = 8.0 Hz, 2H), 7.67 (s, 1H), 7.15–7.05 (m, 4H), 6.89–6.87 (d, J = 8.0 Hz, 1H), 6.50–6.48 (d, J = 8.0 Hz, 2H), 4.72 (s, 2H), 2.20, (s, 3H) ppm. MS (ESI) m/z 242.0 [M + H]+, 264 [M + Na]+, 505 [2M + Na]+.
1-(4-Azidophenyl)-3-o-tolylurea (15): Aniline 14 (100 mg, 0.41 mmol) was dissolved in CH3CN and cooled to 0 °C in an ice-salt bath. To this stirred solution was added t-BuONO (73 µL, 0.61 mmol), and the mixture was stirred for 10 min; after this time, TMSN3 (65 µL, 0.49 mmol) was added dropwise for 10 min, and the resulting brown solution was stirred at r.t. One hour later, the solvent was removed at reduced pressure, and the residue was purified by flash chromatography on silica gel (DCM:MeOH at 9:1). Yield = 67%. 1H NMR (400 MHz CDCl3-d): δ 9.10 (s, 1H), 7.91 (s, 1H), 7.80–7.78 (d, J = 8.0 Hz, 1H), 7.50–7.48 (d, J = 8.0 Hz, 2H), 7.16–7.19 (m, 2H), 7.04–7.02 (d, J = 8.0 Hz, 2H), 7.95–7.91 (t, J = 8.0 Hz, 1H), 2.22 (s, 3H) ppm. MS (ESI) m/z 267 [M + Na]+, 557 [2M + Na]+.
General Procedure for the Preparation of Compounds 16a–16e (Fig. S2).
The appropriate alkyne (0.10 mmol) and azide 15 (25 mg, 0.10 mmol) were suspended in a 1:1 mixture of water and t-BuOH (1.5 mL each) in a 10-mL glass vial equipped with a small magnetic stirring bar. To this was added sodium ascorbate (0.10 mmol) and copper(II) sulfate pentahydrate (0.10 mmol). The mixture was then heated for 10 min at 125 °C under microwave irradiation using an irradiation power of 300 W. After this time, the precipitate was filtered off and purified on silica to give final products of 16a–16e.
1-(4-(4-Phenyl-1H-1,2,3-triazol-1-yl)phenyl)-3-o-tolylurea (16a): The residue was purified by flash chromatography on silica gel (DCM:MeOH at 98:2). Yield = 97%, white solid. 1H NMR [400 MHz (CD3)2SO]: δ 9.42 (s, 1H), 9.18 (s, 1H), 8.08 (s, 1H), 7.93–7.91 (d, J = 8.0 Hz, 2H), 7–85 to 7–80 (m, 3H), 7.70–7.68 (d, J = 8.0 Hz, 2H), 7.50–7.46 (m, 3H), 7.38–7.34 (t, J = 8.0 Hz, 1H), 7.18–7.14 (m, 2H), 6.97–6.94 (t, J = 12.0 Hz, 1H), 2.25 (s, 3H) ppm. 13C NMR (100 MHz (CD3)2SO): δ 153.09, 148.24, 140.57, 137.65, 131.33, 130.87, 130.71, 129.45, 128.63, 126.66, 125.79, 123.48, 121.83, 121.35, 119.88, 119.12 ppm. MS (ESI) m/z 368 [M − H]−, 404 [M + Cl]−.
1-(4-(4-Tert-butyl-1H-1,2,3-triazol-1-yl)phenyl)-3-o-tolylurea (16b): The residue was purified by flash chromatography on silica gel (DCM:MeOH at 98:2). Yield = 91%, white solid. 1H NMR [400 MHz (CD3)2SO]: δ 9.23 (s, 1H), 8.45 (s, 1H), 7.96 (s, 1H), 7.80–7.75 (m, 3H), 7.63–7.61 (d, J = 8.0 Hz, 2H), 7.17–7.11 (m, 2H), 6.96–6.93 (t, J = 12.0 Hz, 1H), 2.23 (s, 3H), 1.32 (s, 9H) ppm. 13C NMR [100 MHz (CD3)2SO]: δ 158.38, 155.30, 139.59, 135.73, 131.87, 131.75,130.66, 126,64, 125.48, 124.84, 121.22, 120.18, 117, 50, 30.33, 17.83 ppm. MS (ESI) m/z 348 [M − H]−, 384 [M + Cl]−.
1-(4-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)phenyl)-3-o-tolylurea (16c): The residue was purified by flash chromatography on silica gel DCM:methanol at 95:5. Yield = 79%, white solid. 1H NMR [400 MHz (CD3)2SO]: δ 9.23 (s, 1H), 8.54 (s, 1H), 7.97 (s, 1H), 7.80–7.76 (d, J = 8 Hz, 2H), 7.80–7.76 (m, 3H), 7.64–7.62 (d, J = 8.0 Hz, 2H), 7.17–7.12 (m, 2H), 6.96–6.93 (t, J = 6.0 Hz, 1H), 4.58 (s, 2H), 2.24 (s, 3H) ppm. 13C NMR [100 MHz (CD3)2SO]: δ 153.09, 140.57, 137.78, 131.50, 131.09, 128.46, 127.04, 123.60, 122.38, 121.27, 120.49, 118.84, 55.44, 18.36 ppm. MS (ESI) m/z 322.1 [M − H]−, 358 [M + Cl]−.
1-(4-(4-Methyl-1H-1,2,3-triazol-1-yl)phenyl)-3-o-tolylurea (16d): The residue was purified by flash chromatography on silica gel (DCM:MeOH at 98:2). Yield = 72%, white solid. 1H NMR (400 MHz MeOD-d4): δ 8.18 (s, 1H), 7.72–7.70 (d, J = 8.0 Hz, 2H), 7.63–7.61 (d, J = 8.0 Hz, 2H), 7.20–7.16 (m, 2H), 7.05–7.01 (t, J = 8.0 Hz, 1H), 2.78–2.74 (t, J = 8.0 Hz, 2H), 2.3 (d, J = 8.0 Hz, 2H). 13C NMR (100 MHz MeOD-d4): δ 153.09, 140.57, 137.65, 131.33, 130.69, 128.42, 126.62, 123.44, 121.86, 121.09, 120.49, 119.08, 31.41, 25.18, 22.17, 18.34, 14.15 ppm. MS (ESI) m/z 348 [M − H]−, 384 [M + Cl]−.
1-(4-(4-((Benzyl(methyl)amino)methyl)-1H-1,2,3-triazol-1-yl)phenyl)-3-(o-tolyl)urea (16e): The residue was purified by flash chromatography on silica gel (DCM:MeOH to 98:2). Yield = 90%, white solid. 1H NMR (400 MHz CDCl3-d): δ 8.43 (s, 1H), 7.80 (s, 1H), 7.57 (s, 1H), 7.51–7.43 (m, 6H), 7.31–7.23 (m, 4H), 7.12–7.07 (m, 2H), 7.00–6.99 (t, J = 12.0 Hz, 1H), 3.75 (s, 2H), 3.58 (s, 2H), 2.25 (s, 3H), 2.14 (s, 1H) ppm. 13C NMR (100 MHz CDCl3-d): δ 154.31, 145.73, 139.98, 137.90, 135.90, 131.48, 130.60, 129.15, 128.39, 127.36, 126.58, 125.26, 124.61, 121.13, 120.08, 61.58, 51.88, 42.11, 17.89 ppm. MS (ESI) m/z 425.0 [M − H]−, 461.1 [M + Cl]−.
General Procedure for the Preparation of Compounds 16f and 16g (Fig. S2).
l-Proline (1.9 mg, 0.01 mmol), CuCl (8.2 mg, 0.08 mmol), K2CO3 (13.7 mg, 0.10 mmol) azide 15 (20 mg, 0.08 mmol), and the appropriate alkynoic acid (0.08 mmol) were sequentially added to a 10-mL glass vial equipped with a magnetic stirrer. The vial was closed with a septum and irradiated at 65 °C. After 15 min, the mixture was partitioned between water (20 mL) and AcOEt (40 mL), the organic layer was separated and dried (Na2SO4), and the solvent was removed in vacuo to furnish a brown residue that was purified by flash chromatography on silica gel (DCM:MeOH at 98:2) to give the desired triazole compounds 16f or 16g.
1-(4-(4-Methyl-1H-1,2,3-triazol-1-yl)phenyl)-3-o-tolylurea (16f): The residue was purified by flash chromatography on silica gel (DCM:MeOH at 98:2). Yield = 77%, white solid. 1H NMR (400 MHz MeOD-d4): δ 8.15 (s, 1H), 7.72–7.69 (d, J = 8.0 Hz, 2H), 7.64–7.62 (m, 3H), 7.21–7.15 (m, 2H), 7.05–7.02 (t, J = 8.0 Hz, 1H), 2.38 (s, 1H), 2.30 (s, 3H) ppm. 13C NMR (100 MHz MeOD-d4): δ 153.20, 151.00, 141.20, 138.2, 133.01, 131.8, 126.08, 124.23, 123.13, 120.77, 120.28, 119.23, 16.60, 9.06 ppm. MS (ESI) m/z 306 [M − H]−, 342 [M + Cl]−.
1-(4-(4-Ethyl-1H-1,2,3-triazol-1-yl)phenyl)-3-o-tolylurea (16g): The residue was purified by flash chromatography on silica gel (DCM:MeOH at 98:2).Yield = 82%, white solid. 1H NMR (400 MHz MeOD-d4): δ 8.21 (s, 1H), 7.73–7.71 (d, J = 8.0 Hz, 2H), 7.65–7.63 (m, 3H), 7.21–7.15 (m, 2H), 7.05–7.02 (t, J = 8.0 Hz, 1H), 2.82–2.76 (q, J = 6.0 Hz, 2H), 2.30 (s, 3H), 1.35–1.31 (t, J = 8.0 Hz, 3H) ppm. 13C NMR (100 MHz MeOD-d4): δ 152.60, 149.76, 140.30, 137.38, 131.86, 130.21, 128.32, 126.25, 123.33, 121.92, 120.59, 118.95, 118.77, 18.75, 17.20, 13.17 ppm. MS (ESI) m/z 320 [M − H]−, 356 [M + Cl]−.
SI Docking Studies
All compounds studied herein were docked within the RNA binding site of the modeled hDDX3 closed conformation previously published (37) using the software package GOLD 4.1 (38, 39). The pocket under investigation was inserted into a grid box centered on residue Phe357 and enclosing residues lying within 10 Å from such amino acid. The genetic algorithm parameter settings were used with the search efficiency set at 100%, and 100 runs were carried out for each ligand. Chemscore was chosen as the fitness function. Finally, results differing less than 1.5 Å in ligand–all atom rmsd were clustered together. For each inhibitor, the first ranked solution was selected for additional analysis. Pictures of the modeled ligand–enzyme complexes together with graphic manipulations were rendered using the PyMOL molecular graphic system (40).
SI Enzymatic Assays
Helicase Assay Based on FRET.
Helicase assay using dsRNA substrate was performed in 20 mM Tris⋅HCl (pH 8), 70 mM KCl, 2 mM MgCl2, 2 mM DTT, 12 U RNasin, 2 mM ATP, 50 nM dsRNA, and 100 nM capture strand in 20 μL reaction volume. The unwinding reaction was started by adding 60 pmol DDX3 or 200 pmol DDX1 recombinant protein and carried out at 37 °C for 40 min using a LightCycler 480 (Roche). The fluorescence intensity was recorded every 30 s. Data of fluorescence signal were analyzed with the Excel program, and the slope value was used to determine enzyme activity.
Gel-Based Helicase Assay for NS3 HCV.
The helicase activity was monitored by measuring the conversion of a dsRNA [labeled at the 5′ end of one strand with a 6-FAM (6-carboxyfluorescein) fluorescent group] into single-stranded nucleic acid. For the RNA helicase assay, an 18/36mer dsRNA oligonucleotide with both 3′ and 5′ protruding single-stranded ends was used. Reactions were performed in 50 mM Tris⋅HCl (pH 7.5), 1 mM DTT, 0.2 mg/mL BSA, 5% (vol/vol) glycerol and 1 mM ATP, 3 mM MgCl2, and 20 nM dsRNA. Reaction was started by adding 30 pmol NS3 HCV protein at 37 °C for 30 min and stopped by adding 50 mM EDTA (pH 8). Products were separated on a native 7% PAGE containing 0.1% (wt/vol) SDS at 5 W in Tris/borate/EDTA (TBE) buffer with 0.1% SDS at 4 °C for 2 h. Substrates and products were quantified by laser scanning densitometry (Thyphoon-TRIO; GE Healthcare).
Gel-Based Helicase Assay for NS3 DENV.
The helicase activity was monitored by measuring the conversion of a dsDNA-RNA (DNA labeled at the 5′ end of a 6-FAM fluorescent group) into single-stranded nucleic acid. For the helicase assay, an 18/38mer dsRNA oligonucleotide with 3′-protruding single-stranded ends was used. Reactions were performed in 50 mM Tris⋅HCl (pH 7.4), 1 mM DTT, 0.25 mg/mL BSA, 0.5% Tween 20, 2 mM MgCl2, 20 U RNasin, 5 mM ATP, and 2.5 nM dsRNA-DNA. Reaction was performed with a preincubation of 40 pmol NS3-DENV protein with compounds for 5 min on ice. After this preincubation, mix was added into tubes, and reactions were performed at 30 °C for 40 min and stopped by adding 50 mM EDTA (pH 8). Products were separated on a native 7% PAGE containing 0.1% (wt/vol) SDS at 5 W in TBE buffer with 0.1% SDS at 4 °C for 2 h. Substrates and products were quantified by laser scanning densitometry (Thyphoon-TRIO; GE Healthcare).
ATPase Assay.
The ATPase assay was carried out by using the commercial kit Promega ADP-Glo Kinase Assay. Reaction was performed in 30 mM Tris⋅HCl, 9 mM MgCl2, 0.05 mg/mL BSA, 50 μM ATP, and 4 μM DDX3. Reaction was performed following the ADP-Glo Kinase Assay Protocol, and luminescence was measured with MicroBeta TriLux Perkin-Elmer.
SI Cellular Assays
In Vitro Antiviral Activity (Half-Maximal Effective Concentration) and Cytotoxicity (Half-Maximal Cytotoxic Concentration) of DENV and WNV.
Around 104 Huh7 cells (provided by Apath, LLC) cultured in DMEM (Invitrogen) supplemented with 10% FBS, 1% nonessential amino acids, 100 U/mL penicillin, and 100 U/mL streptomycin (DMEM complete) per well were infected for 1 h with DENV-2 [New Guinea-C Strain; National Collection of Pathogenic Viruses (NCPV) reference no. 0006041v] or WNV-NY99 (NCPV reference no. 0209291v) at a multiplicity of infection of 0.5 in 96-well plates. After washing viral input, 10-fold serial dilutions of DMSO control or DDX3 inhibitors were immediately added in duplicates to the corresponding wells together with fresh DMEM complete medium (final concentrations of 100, 10, 1, 0.1, and 0.01 μM); 48 h after initial infection, cells were fixed with 4% paraformaldehyde and immunostained against the viral E protein (envelope) using specific anti-E protein primary antibodies (Anti-DENV; catalog no. ab41349; Abcam; Anti-WNV; catalog no. ab156843; Abcam) and an AlexaFluor488 secondary antibody (catalog no. A11029; Invitrogen). DAPI was used for nuclei staining. Plates were analyzed with the ImageXpress Automated Microscope using the 20× Plan Fluor objective. The microscope was set for two channels (FITC and DAPI). Between four and nine images were taken per well per condition at arbitrary objective fields. Between 500 and 1,000 images were analyzed using the CellProfiler Software (Broad Institute of the Massachusetts Institute of Technology IT; cellprofiler.org). Images were thresholded for background subtraction, and target fluorescent dot sizes and intensities were analyzed. Positive green counts and nuclei blue counts were estimated per image per condition.
Relative percentages of infectivity and toxicity for each compound were estimated by normalizing the number of green signals (positive counts) and blue signals (nuclei counts) to DMSO-treated controls, respectively. Half-maximal effective concentration (EC50) and half-maximal cytotoxic concentration (CC50) values were calculated in GraphPad (www.graphpad.com) by applying a nonlinear regression with log dose vs. normalized response to the CellProfiler data.
Evaluation of the in Vitro Antiviral Activity (EC50) and Cytotoxicity (CC50) in the HCV Replicon System.
Antiviral activity was carried out in white clear-bottom 96-well plates, and cytotoxicity assays were performed in clear 96-well plates. LucUbiNeo-ET cells were seeded at a density of 104 per well in 100 μL DMEM without selection of antibiotics. After 24 h, ninefold serial dilutions of compounds, with three replicates at each dilution, were prepared in DMEM and added to the appropriate wells, yielding concentrations of 125–0.00032 µg/mL in a final volume of 100 µL DMEM. After 2 d of incubation, the inventors determined the antiviral activity (EC50) by quantifying the luciferase with the BriteLite Plus Luciferase (Perkin-Elmer). Briefly, LucUbiNeo-ET cells were harvested with the addition of 50 µL luciferase per well, and after 2 min of shaking, the light was read in a Luminometer Fluoroskan Ascent FL.
Toxicity (CC50) was analyzed in the Lunet/HuH7 cells by the addition of 10 μL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide per well. After 4 h of incubation at 37 °C in a humidified atmosphere with 5% CO2, the amount of formazan produced was quantified spectrophotometrically at 550/620 nm (41). The EC50 and CC50 values were calculated for each compound in GraphPad by applying a nonlinear regression with log dose vs. normalized response to the CellProfiler data. These assays were done in duplicate.
Evaluation of Antiviral Activity on HIV-1 Strains Carrying Mutations Conferring Resistance to Drugs Currently Used to Treat HIV Infection.
MT-2 and TZM-bl cell lines and the infectious clones used to evaluate the antiviral activity of compound 16d were all obtained through the AIDS Reagent Program [Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), NIH]. Drug susceptibility of NIH clones has been previously characterized through the “Phenosense” phenotypic assay (available at Quest Diagnostics, Monogram Biosciences Laboratory). The features of the reference and resistant viruses are showed in Table S1.
The IC50 values of the HIV-1 WT reference strain NL4-3 and viruses carrying resistant mutations were determined in a phenotypic assay consisting of a first cycle of replication in the MT-2 cell line followed by an additional round of replication in TZM-bl cells. MT-2 cells were seeded at a concentration of 50,000 cells per well in a 96-well plate and infected with the reference strain NL4-3 in the presence of fivefold dilutions of the compounds. After 48–72 h, 50 µL supernatant from each well containing the virus produced in the first round of infection was used to infect TZM-bl cells seeded in a 96-plate well at a concentration of 30,000 cells per well. Two days later, cells were lysed by adding 40 µL Glo Lysis Buffer (Promega) to each well for 5 min; then, 40 µL Bright-Glo Luciferase Reagent (Promega) was added to each well for relative luminescence units counting using the Glo-Max Multi Detection System (Promega). Relative luminescence unit values from each well were elaborated using the GraphPad v5.0 software to calculate the IC50 of each compound.
SI in Vitro ADME Studies
Chemicals.
All solvents and reagents were from Sigma-Aldrich. Dodecane was purchased from Fluka. Pooled male donors human liver microsomes (20 mg mL−1) were from BD Gentest-Biosciences. Milli-Q Quality Water (Millipore) was used. Hydrophobic filter plates (MultiScreen-IP; Clear Plates; 0.45-μm-diameter pore size), 96-well microplates, and 96-well UV-transparent microplates were obtained from Millipore.
UV/LC-MS Method.
For the quantitative analysis, a UV/LC-MS system was used. LC analyses were performed by a Agilent 1100 LC/MSD VL System (G1946C; Agilent Technologies) of a vacuum solvent degassing unit, a binary high-pressure gradient pump, an 1100 series UV detector, and a 1100 MSD model VL benchtop mass spectrometer. The Agilent 1100 Series MSD Single-Quadrupole Instrument was equipped with the orthogonal spray atmospheric pressure ionization electrospray (API-ES) (Agilent Technologies). Nitrogen was used as nebulizing and drying gas. The pressure of the nebulizing gas, the flow of the drying gas, the capillary voltage, the fragmentor voltage, and the vaporization temperature were set at 40 psi, 9 L min−1, 3,000 V, 70 V, and 350 °C, respectively. UV detection was monitored at 280 nm. The LC-ESI-MS determination was performed by operating the MSD in the positive ion mode. Spectra were acquired over the scan range m/z 50–1,500 using a step size of 0.1 U. Chromatographic analysis was performed using a Varian Polaris 5 C18-A Column (150 × 4.6 mm; 5-µm particle size) at room temperature. Analysis was carried out using gradient elution of a binary solution; eluent A was ACN, whereas eluent B consisted of water. The analysis started at 0% A for 3 min, then rapidly increased up to 98% in 12 min, and finally, remained at 98% A until 18 min. The analysis was performed at a flow rate of 0.8 mL min−1, and injection volume was 20 µL.
Parallel Artificial Membrane Permeability Assay.
Donor solution (0.5 mM) was prepared by diluting 1 mM DMSO compound stock solution using phosphate buffer (pH 7.4, 0.025 M). Filters were coated with 5 μL 1% (wt/vol) dodecane solution of phosphatidylcholine prepared from CHCl3 solution (10% wt/vol) for intestinal permeability. Donor solution (150 μL) was added to each well of the filter plate. To each well of the acceptor plate was added 300 μL solution (50% DMSO in phosphate buffer). All compounds were tested in three different plates on different days. The sandwich was incubated for 5 h at room temperature under gentle shaking. After the incubation time, the plates were separated, and samples were taken from both receiver and donor sides and analyzed using LC with UV detection at 280 nm.
LC analyses were performed with a Varian Prostar HPLC System (Varian Analytical Instruments) equipped with a binary pump with a manual injection valve and a model Prostar 325 UV-VIS Detector. Chromatographic separation was conducted using a Polaris C18-A Column (150–4.6 mm; 5-μm particle size) at a flow rate of 0.8 mL min−1 with a mobile phase composed of 60% ACN and 40% H2O−.
Permeability (Papp) for parallel artificial membrane permeability assay was calculated according to the following equation obtained from equations by Wohnsland and Faller (42) and Sugano et al. (43) with some modification to obtain permeability values in centimeters second−1:
where VA is the volume in the acceptor well, VD is the volume in the donor well (centimeter3), A is the “effective area” of the membrane (centimeter2), t is the incubation time (seconds), and r is the ratio between drug concentration in the acceptor and equilibrium concentration of the drug in the total volume (VD + VA). Drug concentration is estimated by using the peak area integration.
Membrane retentions (MRs; percentages) were calculated according to the following equation:
where r is the ratio between drug concentrations in the acceptor and equilibrium concentrations, and D, A, and Eq represent drug concentrations in the donor, acceptor, and equilibrium solutions, respectively.
Water Solubility Assay.
Each solid compound (1 mg) was added to 1 mL water. The samples were shaken in a shaker bath at room temperature for 24–36 h. The suspensions were filtered through a 0.45-μm nylon filter (Acrodisc), and the solubilized compound was determined by LC-MS-MS assay. For each compound, the determination was performed in triplicate.
For the quantification, an LC-MS system consisting of a Varian apparatus (Varian Inc.), including a vacuum solvent degassing unit, two pumps (212-LC), a Triple Quadrupole MSD (Model 320-LC) Mass Spectrometer with ES Interface, and Varian MS Workstation System Control, Version 6.9 software was used. Chromatographic separation was obtained using a Pursuit C18 Column (50 × 2.0 mm; Varian) with 3-μm particle size and gradient elution: eluent A was ACN, and eluent B consisted of water. The analysis started with 0% of eluent A, which was linearly increased up to 70% in 10 min and then slowly increased up to 98% up to 15 min. The flow rate was 0.2 mL min−1, and injection volume was 5 μL. The instrument operated in positive mode, and parameters were detector, 1,850 V; drying gas pressure, 25.0 psi; desolvation temperature, 300.0 °C; nebulizing gas, 40.0 psi; needle, 5,000 V; and shield, 600 V. Nitrogen was used as the nebulizer gas and the drying gas. Collision-induced dissociation was performed using argon as the collision gas at a pressure of 1.8 mTorr in the collision cell.
Microsomal Stability Assay.
Each compound in DMSO solution was incubated at 37 °C for 60 min in 125 mM phosphate buffer (pH 7.4) and 5 μL human liver microsomal protein (0.2 mg mL−1) in the presence of an NADPH-generating system at a final volume of 0.5 mL (compound final concentration of 50 μM); DMSO did not exceed 2% (final solution). The reaction was stopped by cooling on ice and adding 1.0 mL acetonitrile. The reaction mixtures were then centrifuged, and the parent drug and metabolites were subsequently determined by LC-UV-MS.
Chromatographic analysis was performed with an Agilent 1100 LC/MSD VL System (G1946C; Agilent Technologies) consisting of a vacuum solvent degassing unit, a binary high-pressure gradient pump, an 1100 series UV detector, and an 1100 MSD model VL benchtop mass spectrometer.
Chromatographic separation was obtained using a Varian Polaris C18-A Column (150–4.6 mm; 5-μm particle size) and gradient elution: eluent A was ACN, and eluent B consisted of water. The analysis started with 2% of eluent A, which was rapidly increased up to 70% in 12 min and then slowly increased up to 98% in 20 min. The flow rate was 0.8 mL min−1, and injection volume was 20 μL.
The Agilent 1100 Series MSD Single-Quadrupole Instrument was equipped with the orthogonal spray API-ES (Agilent Technologies). Nitrogen was used as the nebulizing gas and the drying gas. The pressure of the nebulizing gas, the flow of the drying gas, the capillary voltage, the fragmentor voltage, and the vaporization temperature were set at 40 psi, 9 L/min, 3,000 V, 70 V, and 350 °C, respectively. UV detection was monitored at 280 nm. The LC-ESI-MS determination was performed by operating the MSD in the positive ion mode. Spectra were acquired over the scan range m/z 100–1,500 using a step size of 0.1 U. The percentage of not metabolized compound was calculated by comparison with reference solutions.
SI Animal Studies
Healthy Wistar rats (Harlan Italy S.r.l.) were housed with free access to water and standard rat food at animal facilities of the Sacred Heart University. All animals used for experiments were males at 8 wk of age weighing ∼180 ± 15 g. The animals were housed individually in metabolic cages at room temperature (22 °C ± 2 °C) with 50–60% humidity under a 12-h light/dark cycle. For the experiments, animals were killed by ketamine i.p. injection. Animal welfare was routinely checked by veterinarians of the Service for Animal Welfare.
In Vivo Toxicity of 16d.
Healthy Wistar rats were used to assess the in vivo toxicity of 16d by behavioral and histopathological analysis. Rats were divided into three groups designated as treated group (TG), vehicle group (VG), and WT group. Five rats of TG group were injected with compound through the tail vein in a single systemic dose (0.05 mL per dose) at high-dose levels of 20 mg/kg, whereas the VG group of five rats was inoculated with only vehicle (DMSO) as control. All of the rats were monitored for survival and the presence of any drug-related adverse effect (local signs of inflammation, weight loss, diarrhea, and behavioral alterations) throughout the experimental period. The rats were killed after 5 d of treatment, and blood samples were used for the evaluation of clinical chemistry biochemical parameters. Samples were immediately frozen at −80 °C until the time of analysis. Liver, kidney, and brain tissue specimens were aseptically removed from all animals, fixed in 4% formaldehyde solution, and processed for paraffin embedding. Histopathological changes in tissue specimens were assessed in at least 10 randomly selected tissue sections from each organ for all rats of the groups under study. Sections cut at 2 μm were stained with H&E staining for light microscopic observation.
Statistics.
In vivo experiments were performed in duplicate, and the results were subjected to statistical analysis by using one-way ANOVA with a Bonferroni correction posttest using GraphPad Prism, version 5.04 for Windows (GraphPad Software).
Pharmacokinetic Studies.
Animals were divided into two groups designated as TG and VG. Three rats from the TG group were injected with compound by tail vein in a single systemic dose (0.05 mL per dose) at high-dose levels of 10 mg/kg, whereas the VG group was inoculated with only vehicle (DMSO) as control.
Blood samples (0.5 mL) were collected from the catheter of jugular vein implant into heparinized tubes at 5 and 30 min and 1, 2, 4, 6, and 24 h. Plasma samples were obtained from whole-blood samples by centrifugation at 6,000 rpm (Eppendorf centrifuge 5804, rotor A-4-44) for 10 min. A 1:1 mixture of acetonitrile:methanol (1 mL; with standard compound at the concentration of 5 µM) was added to each plasma sample to denature proteins. Samples were centrifuged at 4,000 rpm for 20 min, and the supernatants were recovered, dried under vacuum, and analyzed using the HPLC method reported above.
Tissue Distribution Study.
Tissues (liver, brain, kidney, testicle, and fat) were collected at 24 h after administration of 10 mg/kg 16d. Tissue samples were accurately weighted and then homogenized in water. Subsequently, the samples were processed in a mixture of ACN:MeOH (1:1) in the presence of an internal standard to extract the distributed compound. The solvent was removed by rotary evaporator and analyzed.
The results are reported in Fig. 6.
UV/LC Method.
LC analyses were performed by a Varian ProStar 210 System consisting of a binary high-pressure gradient pump and a ProStar 340 UV-Vis detector. UV detection was monitored at 280 nm. Chromatographic analysis was performed using a Kinetex EVO C18 100A Column (150 × 4.6 mm; 5-µm particle size) at room temperature. Analysis was carried out using a gradient elution of methanol (MeOH) and water (H2O): t = 0 min, MeOH 0%; t = 3 min, MeOH 0%; t = 12 min, MeOH 98%; t =25 min, MeOH 98%. The analysis was performed at a flow rate of 0.6 mL/min, and injection volume was 20 µL. The quantitative analysis was carried out with appropriate calibration curve in the presence of 16c as an internal standard at a concentration of 5 µM.
Statistical Analysis.
Pharmacokinetic parameters of 16d, including half-life (t1/2β), area under curve (AUC0–t and AUC0–∞), plasma clearance, and mean residence time, were analyzed by PKCALC software (44). Data were expressed as means ± SDs.
Acknowledgments
The authors gratefully acknowledge use of the facilities provided by Lead Discovery Siena S.r.l. in collaboration with First Health Pharmaceuticals B.V. of Amsterdam. This research was supported by First Health Pharmaceuticals B.V., Tuscany Region Grant DD3242/2009 Bando Salute 2009, and Prin 2010 Research Project Grant 2010W2KM5L. This work was also supported by Spanish Ministry of Economy and Competitiveness (MINECO) Fondo Europeo de Desarrollo Regional (FEDER) Grants SAF2013-46077-R and BFU2015-63800R.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1522987113/-/DCSupplemental.
References
- 1.Lou Z, Sun Y, Rao Z. Current progress in antiviral strategies. Trends Pharmacol Sci. 2014;35(2):86–102. doi: 10.1016/j.tips.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bekerman E, Einav S. Infectious disease. Combating emerging viral threats. Science. 2015;348(6232):282–283. doi: 10.1126/science.aaa3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorr P, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49(11):4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallay PA, Lin K. Profile of alisporivir and its potential in the treatment of hepatitis C. Drug Des Devel Ther. 2013;7:105–115. doi: 10.2147/DDDT.S30946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahlquist P, Noueiry AO, Lee WM, Kushner DB, Dye BT. Host factors in positive-strand RNA virus genome replication. J Virol. 2003;77(15):8181–8186. doi: 10.1128/JVI.77.15.8181-8186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prussia A, Thepchatri P, Snyder JP, Plemper RK. Systematic approaches towards the development of host-directed antiviral therapeutics. Int J Mol Sci. 2011;12(6):4027–4052. doi: 10.3390/ijms12064027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheller N, et al. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc Natl Acad Sci USA. 2009;106(32):13517–13522. doi: 10.1073/pnas.0906413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drake JW, Holland JJ. Mutation rates among RNA viruses. Proc Natl Acad Sci USA. 1999;96(24):13910–13913. doi: 10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gianella S, Richman DD. Minority variants of drug-resistant HIV. J Infect Dis. 2010;202(5):657–666. doi: 10.1086/655397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duffy S, Shackelton LA, Holmes EC. Rates of evolutionary change in viruses: Patterns and determinants. Nat Rev Genet. 2008;9(4):267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 11.Ruiz A, Russell SJ. A new paradigm in viral resistance. Cell Res. 2012;22(11):1515–1517. doi: 10.1038/cr.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez JP, Sasse F, Brönstrup M, Diez J, Meyerhans A. Antiviral drug discovery: Broad-spectrum drugs from nature. Nat Prod Rep. 2015;32(1):29–48. doi: 10.1039/c4np00085d. [DOI] [PubMed] [Google Scholar]
- 13.Lenarcic EM, Ziehr BJ, Moorman NJ. An unbiased proteomics approach to identify human cytomegalovirus RNA-associated proteins. Virology. 2015;481:13–23. doi: 10.1016/j.virol.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yedavalli VS, Neuveut C, Chi YH, Kleiman L, Jeang KT. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell. 2004;119(3):381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 15.Owsianka AM, Patel AH. Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology. 1999;257(2):330–340. doi: 10.1006/viro.1999.9659. [DOI] [PubMed] [Google Scholar]
- 16.Mamiya N, Worman HJ. Hepatitis C virus core protein binds to a DEAD box RNA helicase. J Biol Chem. 1999;274(22):15751–15756. doi: 10.1074/jbc.274.22.15751. [DOI] [PubMed] [Google Scholar]
- 17.You LR, et al. Hepatitis C virus core protein interacts with cellular putative RNA helicase. J Virol. 1999;73(4):2841–2853. doi: 10.1128/jvi.73.4.2841-2853.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ariumi Y, et al. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J Virol. 2007;81(24):13922–13926. doi: 10.1128/JVI.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li C, et al. Cellular DDX3 regulates Japanese encephalitis virus replication by interacting with viral un-translated regions. Virology. 2014;449:70–81. doi: 10.1016/j.virol.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noble CG, et al. Strategies for development of Dengue virus inhibitors. Antiviral Res. 2010;85(3):450–462. doi: 10.1016/j.antiviral.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 21.Chahar HS, Chen S, Manjunath N. P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology. 2013;436(1):1–7. doi: 10.1016/j.virol.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schröder M, Baran M, Bowie AG. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008;27(15):2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalverda AP, et al. Poxvirus K7 protein adopts a Bcl-2 fold: Biochemical mapping of its interactions with human DEAD box RNA helicase DDX3. J Mol Biol. 2009;385(3):843–853. doi: 10.1016/j.jmb.2008.09.048. [DOI] [PubMed] [Google Scholar]
- 24.Benfield CT, Ren H, Lucas SJ, Bahsoun B, Smith GL. Vaccinia virus protein K7 is a virulence factor that alters the acute immune response to infection. J Gen Virol. 2013;94(Pt 7):1647–1657. doi: 10.1099/vir.0.052670-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vashist S, Urena L, Chaudhry Y, Goodfellow I. Identification of RNA-protein interaction networks involved in the norovirus life cycle. J Virol. 2012;86(22):11977–11990. doi: 10.1128/JVI.00432-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schröder M. Viruses and the human DEAD-box helicase DDX3: Inhibition or exploitation? Biochem Soc Trans. 2011;39(2):679–683. doi: 10.1042/BST0390679. [DOI] [PubMed] [Google Scholar]
- 27.Tintori C, et al. Protein-protein interactions and human cellular cofactors as new targets for HIV therapy. Curr Opin Pharmacol. 2014;18:1–8. doi: 10.1016/j.coph.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Dürr R, et al. Targeting cellular cofactors in HIV therapy. In: Diederich WE, Steuber H, editors. Topics in Medicinal Chemistry. Springer; Heidelberg: 2014. pp. 183–222. [Google Scholar]
- 29.Yedavalli VS, et al. Ring expanded nucleoside analogues inhibit RNA helicase and intracellular human immunodeficiency virus type 1 replication. J Med Chem. 2008;51(16):5043–5051. doi: 10.1021/jm800332m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang N, Zhang P, Baier A, Cova L, Hosmane RS. Dual inhibition of HCV and HIV by ring-expanded nucleosides containing the 5:7-fused imidazo[4,5-e][1,3]diazepine ring system. In vitro results and implications. Bioorg Med Chem Lett. 2014;24(4):1154–1157. doi: 10.1016/j.bmcl.2013.12.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garbelli A, et al. Targeting the human DEAD-box polypeptide 3 (DDX3) RNA helicase as a novel strategy to inhibit viral replication. Curr Med Chem. 2011;18(20):3015–3027. doi: 10.2174/092986711796391688. [DOI] [PubMed] [Google Scholar]
- 32.Maga G, et al. Pharmacophore modeling and molecular docking led to the discovery of inhibitors of human immunodeficiency virus-1 replication targeting the human cellular aspartic acid-glutamic acid-alanine-aspartic acid box polypeptide 3. J Med Chem. 2008;51(21):6635–6638. doi: 10.1021/jm8008844. [DOI] [PubMed] [Google Scholar]
- 33.Samal SK, Routray S, Veeramachaneni GK, Dash R, Botlagunta M. Ketorolac salt is a newly discovered DDX3 inhibitor to treat oral cancer. Sci Rep. 2015;5:9982. doi: 10.1038/srep09982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bol GM, et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol Med. 2015;7(5):648–669. doi: 10.15252/emmm.201404368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bantscheff M, Scholten A, Heck AJ. Revealing promiscuous drug-target interactions by chemical proteomics. Drug Discov Today. 2009;14(21-22):1021–1029. doi: 10.1016/j.drudis.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Radi M, et al. Discovery of the first small molecule inhibitor of human DDX3 specifically designed to target the RNA binding site: Towards the next generation HIV-1 inhibitors. Bioorg Med Chem Lett. 2012;22(5):2094–2098. doi: 10.1016/j.bmcl.2011.12.135. [DOI] [PubMed] [Google Scholar]
- 37.Fazi R, et al. Homology model-based virtual screening for the identification of human helicase DDX3 inhibitors. J Chem Inf Model. 2015;55(11):2443–2454. doi: 10.1021/acs.jcim.5b00419. [DOI] [PubMed] [Google Scholar]
- 38.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52(4):609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 39.Cole JC, Nissink JWM, Taylor R. Protein-ligand docking and virtual screening with GOLD. In: Shoichet B, Alvarez J, editors. Virtual Screening in Drug Discovery. Taylor & Francis CRC Press; Boca Raton, FL: 2005. [Google Scholar]
- 40.PyMOL . PyMOL Molecular Graphics System, Version 0.99. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 41.Pannecouque C, Daelemans D, De Clercq E. Tetrazolium-based colorimetric assay for the detection of HIV replication inhibitors: Revisited 20 years later. Nat Protoc. 2008;3(3):427–434. doi: 10.1038/nprot.2007.517. [DOI] [PubMed] [Google Scholar]
- 42.Wohnsland F, Faller B. High-throughput permeability pH profile and high-throughput alkane/water log P with artificial membranes. J Med Chem. 2001;44(6):923–930. doi: 10.1021/jm001020e. [DOI] [PubMed] [Google Scholar]
- 43.Sugano K, et al. High throughput prediction of oral absorption: Improvement of the composition of the lipid solution used in parallel artificial membrane permeation assay. J Biomol Screen. 2001;6(3):189–196. doi: 10.1177/108705710100600309. [DOI] [PubMed] [Google Scholar]
- 44.Shumaker RC. PKCALC: A BASIC interactive computer program for statistical and pharmacokinetic analysis of data. Drug Metab Rev. 1986;17(3-4):331–348. doi: 10.3109/03602538608998295. [DOI] [PubMed] [Google Scholar]