

Abstract
Systematic efforts to sequence the cancer genome have identified large numbers of relevant mutations and copy number alterations in human cancers; however, elucidating their functional consequences, and their interactions to drive or maintain oncogenic states, is still a significant challenge. Here we introduce REVEALER, a computational method that identifies combinations of mutually exclusive genomic alterations correlated with functional phenotypes, such as the activation or gene-dependency of oncogenic pathways or the sensitivity to a drug treatment. We use REVEALER to uncover complementary genomic alterations associated with the transcriptional activation of β-catenin and NRF2, MEK-inhibitor sensitivity, and KRAS dependency. REVEALER successfully identified both known and new associations demonstrating the power of combining functional profiles with extensive characterization of genomic alterations in cancer genomes.
The Cancer Genome Atlas (TCGA) and other large-scale genome sequencing projects are providing ever-increasing catalogs of somatic and epigenetic alterations in cancer1–4. A major challenge moving forward is to be able to identify subsets of functionally relevant lesions for further study and eventual therapeutic targeting5–6. These “driver” lesions, in synergistic combinations, are responsible for the generation and maintenance of the oncogenic state and may determine the characteristics of each tumor or tumor type. However, the identification of such drivers is complicated by genomic instability, which increases the number of genomic alterations including low penetrance events with uncertain functional roles.
Genome-wide functional studies of cancer cell lines and tumors, have proven useful in identifying associations between gene dependencies and genomic abnormalities.7–11. Associating recurrent genomic abnormalities with their matching therapeutic agent is a common strategy under the ‘oncogene addiction’ paradigm. However, the challenge of effectively mapping molecular alterations to pathway activity and drug response can be quite difficult as those relationships are not one-to-one. Indeed, some driver mutations only partially predict drug response because of functional heterogeneity and the rise of resistance mechanisms.
One way to address these difficulties is to systematically explore the landscape of mutually exclusive genomic abnormalities along so called functional axes that represent the activation of oncogenic pathways or sensitivity to genetic or chemical perturbations. The use of appropriate functional profiles is important because the complementary nature of genomic alterations is only clearly delineated in the right context, for example, where the relevant oncogenic programs, and other synergies such as immune or stress responses, are co-activated to drive or maintain the oncogenic state.
Here we present REVEALER (Repeated Evaluation of VariablEs conditionAL Entropy and Redundancy), a method to identify groups of genomic alterations that together associate with a functional activation, gene dependency, or drug response profile. The combination of these alterations explains more of the functional target activation or sensitivity than any individual alteration considered in isolation. REVEALER can be applied to a wide variety of problems and allows prior relevant background knowledge to be incorporated into the model. We show that REVEALER can be used to identify genomic features associated with functional cancer phenotypes and demonstrate its higher sensitivity and specificity compared to other model selection methods.
Results
REVEALER Overview
The optimal execution of REVEALER requires three inputs: i) a functional “target” profile for individual samples across a given dataset, ii) a dataset containing a comprehensive collection of genomic “features,” and iii) an optional “seed” feature with which to initialize the search. The target profile is a readout from quantitative measurements, including e.g. gene expression, pathway activation, gene-dependency or drug response. Ideally, the seed is a feature that has a known effect on the target profile. REVEALER starts by measuring the degree of association between the target and seed feature using a re-scaled mutual information metric that we call the Information Coefficient (IC, Figure 1A and Supplementary Information). The IC is a non-linear correlation coefficient that takes values between 1 (perfect match) and −1 (perfect anti-match). One key distinguishing feature of REVEALER is the ability to identify features based on both: target profile and seed. Features that match the target profile but are correlated with the seed are penalized, while features that associate with the target, and are also complementary to the seed, are scored higher. In this way, only genomic features that explain activation or sensitivity in the target profile that is not already accounted for, will be included in the model. REVEALER achieves this by computing the conditional mutual information of the target profile and each feature, conditioned on the seed feature. We refer to this as the Conditional Information Coefficient (CIC) (Figure 1B and Supplementary Information). REVEALER then iterates this process (Figure 1C).
Figure 1.
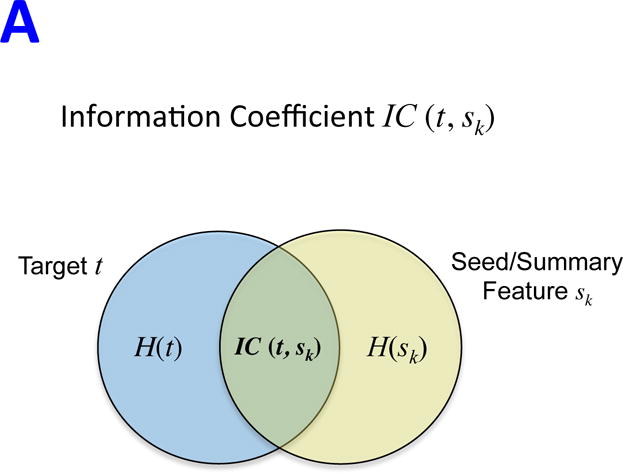
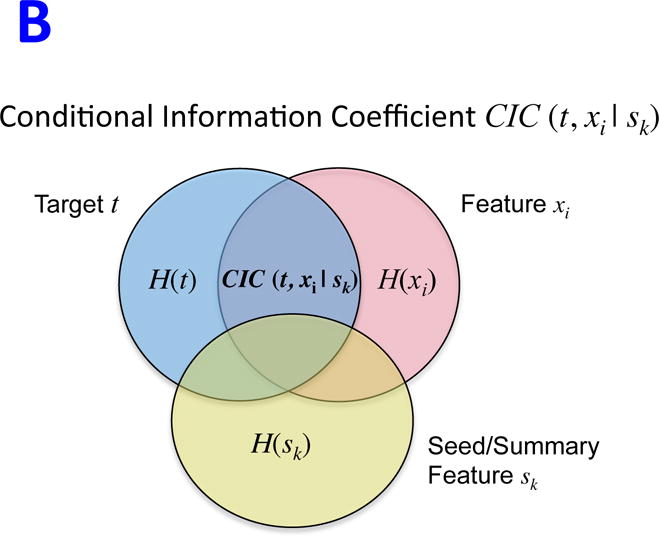
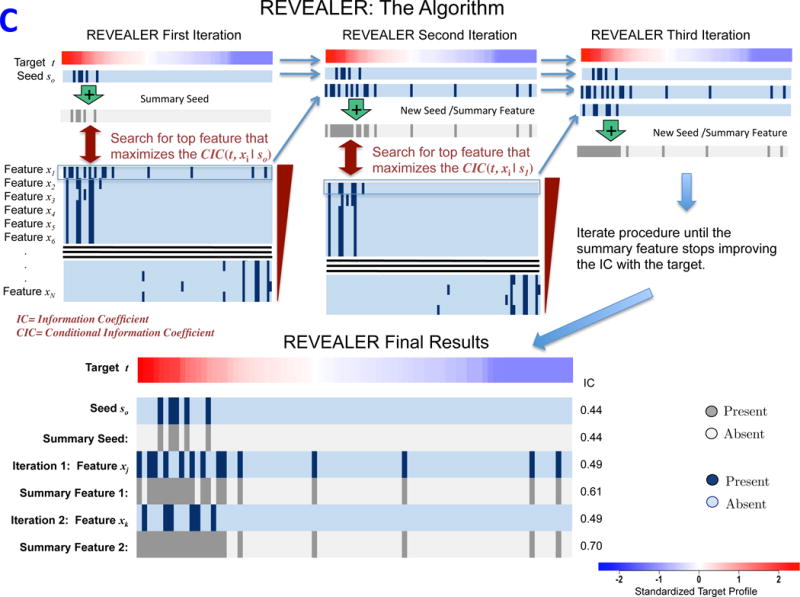
REVEALER Information-based metrics. A)The Information Coefficient IC(t, sk) represents the information shared by the target and the seed or summary feature. B) The Conditional Information Coefficient CIC(t, xi | sk) represents the information shared by the target and a feature, such as a genomic alteration, conditional to the seed feature. C) Detailed schematics of the REVEALER algorithm.
REVEALER uncovers alterations associated with β-catenin activation
We first used REVEALER to identify genomic features associated with the oncogenic activation of β-catenin.12 In Figure 2A, the target t is a β-catenin activation TCF4 reporter13 assayed across 83 cancer cell lines whose mutations and copy number profiles have previously been reported.14 The seed feature so, corresponds to activating mutations in β-catenin (S33, S34, S37, T45, T41) and the target profile in dark blue represents its presence in each sample. The seed feature associates strongly with the target (IC = 0.44) with all of the β-catenin mutations located where the reporter readout is high (left side of heatmap), consistent with the known activating role for these events. However, about half of the samples with high β-catenin activation cannot be explained by alterations in β-catenin. Therefore, we used REVEALER to find additional genomic features from a large set of candidates to explain the target profile.
Figure 2.
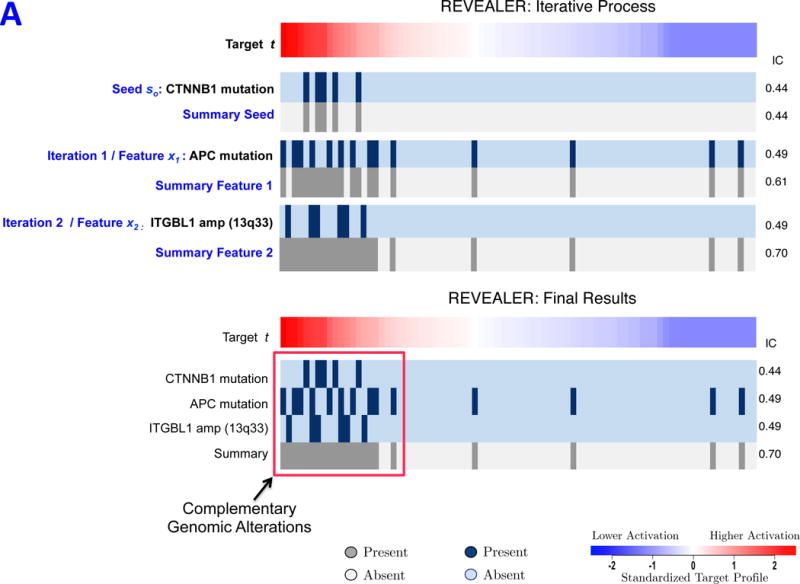
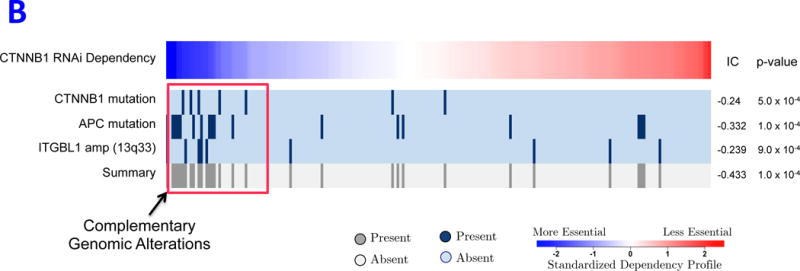
REVEALER results for transcriptional activation of β-catenin in cancer. A) This heatmap illustrates the use of the REVEALER approach to find complementary genomic alterations that match the transcriptional activation of β-catenin in cancer. The target profile is a TCF4 reporter that provides an estimate of the degree of activation of β-catenin. The “seed” is the β-catenin activating mutations, the known “cause” of high values in the target. REVEALER iterates two times and finds APC mutations and the amplification of 13q33 as complementary alterations. At the bottom the heatmap shows the complete set genomic alterations associated with activation of β-catenin found by REVEALER. As can be seen in the figure the features are highly complementary and account for 17 out of the top 20 samples with highest reporter values. B) Profiles of the features shown in Figure 2A, compared with an shRNA profile of β-catenin dependence in 209 cell lines (Supplementary Information). The 3 features are associated with a high degree of β-catenin essentiality but are also highly complementary to each other. The IC scores and nominal p-values with respect to the target are shown on the right side of the heatmap.
The top-scoring genomic feature of the first REVEALER iteration (CIC=0.49) is APC mutations (Figure 2A). REVEALER found this specific alteration from 17,721 feature candidates consisting of 671 mutations and 17,050 amplifications/deletions (Supplementary Fig. 1A). These were generated after filtering out low/high frequency features (Supplementary Information) from an initial set of 48,270 features. APC mutations are known to be associated with an uncontrolled stabilization and transcriptional activation of β-catenin15 and are mutually exclusive with β-catenin mutations. Combining β-catenin and functional APC mutations to obtain the summary feature increases the IC with the target to 0.61 (Figure 2A). REVEALER then proceeds to a second iteration and finds the amplification of chr13q33 (ITGBL1_AMP), as the top scoring feature (CIC= 0.49, Figure 2A). Several other features in the same region chr13q11–34 also attain almost the same CIC (Supplementary Fig. 2A). Recurrent amplifications in 13q are indeed common in colon cancer, and notably, one of our previous studies identified CDK8 in chr13q12.13, as a colon oncogene that regulates β-catenin activity16. Attempts to perform a third iteration fail to find any feature that will increase the IC with the target and thus REVEALER has completed the task. In this case REVEALER performed two iterations before completion but in other cases it may require a smaller or larger number of iterations.
The complete REVEALER results are summarized at the bottom of Figure 2A. The three features have high complementarity and attain a collective IC of 0.70, accounting for 17 out of the top 20 samples with the highest β-catenin. (Supplementary Figs. 1 and 2). In addition to finding the best scoring abnormalities at every iteration, REVEALER also clusters them to facilitate the identification of alternative or “second best” hits (Supplementary Figs. 1B and 2A, Supplementary Information).
We also investigated whether these features associate with shRNA β-catenin dependence in cancer cell lines (Supplementary Information). The samples harboring REVEALER’s features indeed display a much higher degree of β-catenin dependency (P values: 0.0005, 0.0001 and 0.0009, respectively) and are also highly complementary to each other (Figure 2B). This significant mutual exclusivity and association with both: transcriptional and dependency targets provides strong evidence that these alterations indeed activate β-catenin.
To investigate REVEALER’s results robustness, we randomly subsampled 80% of the samples, re-ran REVEALER, and found that APC mutations and the 13q-12–34 amplicon re-appear in 8 out of 10 runs, suggesting these results are reasonably robust (Supplementary Information).
Transcriptional NRF2/NFE2L2 activation in lung cancer
Here we show how REVEALER can also be used with a combined seed feature.
The transcription factor NRF2 (NFE2L2), induces a cytoprotective response to oxidative stresses and its mutations confer constitutive activation in cancer17. We generated a target profile using the single-sample GSEA8 scores of NRF2-driven genes18 across 182 lung cancer cell lines from the Broad-Novartis Cancer Cell Line Encyclopedia, hereafter referred as CCLE14. We selected lung cancer cell lines due to the higher frequency of NFE2L2 alterations,19,20 and used as seed both: NFE2L2 mutations and amplifications (Figure 3A). REVEALER merges multiple seeds(logical OR function) to produce a single summary seed. The input genomic features consisted of a set of 32,154 alterations (991 mutations and 31,253 amplification/deletions after filtering from an original set of 48,270).
Figure 3.
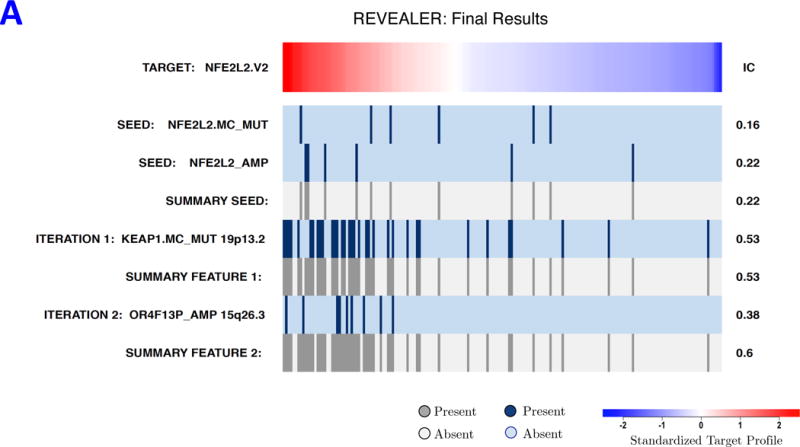
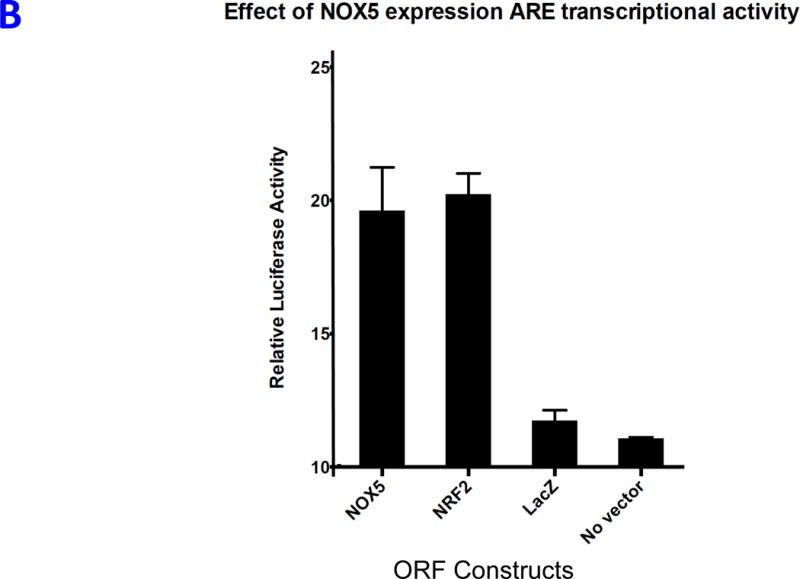
REVEALER results for transcriptional NRF2 activation in lung cancer. A) The target profile is the single-sample GSEA profile of a group of NRF2-driven genes in a group of 182 lung cancer cell lines. The seed feature was defined as the status of NRF2 mutation or amplification. The first iteration of REVEALER identifies KEAP1 mutation, a known co-activator of NRF2, as a potential cause of activation of NRF2 complementary to the seed feature. The second iteration identifies amplification of chr15q22/26 containing the locus of NOX5 (NADPH oxidase 5). B) Results of luciferase assay using antioxidant response element (ARE) reporter as readout of NRF2 pathway activation and open-reading frame (ORF) constructs for NOX5 (REVEALER result), NRF2 (positive control) and LacZ and no vector as negative controls (two tailed unpaired t-test: NOX5 vs. LacZ *p>0.01, NRF2 vs. LacZ **p>0.001).
The first REVEALER iteration yielded KEAP1 mutations, which is an established direct mediator of NRF2 and its targets21 (Figure 3A and Supplementary Fig. 3). The second iteration yielded features encompassing amplification of chr15q22/26 (OR4F13P_AMP, Figure 3A and Supplementary Fig. 4). Besides these two no other features improved the match with the target. Of special interest within this amplicon is NOX5 (NADPH oxidase 5) because its α and β isoforms have been implicated in the production of extracellular superoxide, H2O2 or other reactive oxygen species (ROS)22–24.
To experimentally assess whether NOX5 indeed regulates NRF2 transcriptional activity, we used an antioxidant response element (ARE) luciferase reporter as readout of the NRF2 pathway25. We co-transfected ARE-driven luciferase reporter construct with NOX5, NRF2 (positive control), and LacZ (negative control) open-reading frame (ORFs) constructs. We found that NRF2 and NOX5 ORF constructs led to significant increases in the ARE-driven luciferase activity relative to LacZ or no vector indicating that NOX5 expression indeed regulates ARE (Figure 3B). To test whether these results comport with biological behavior in vivo, we examined a TCGA lung cancer dataset26 and found that these genomic features are enriched in tumors with higher NRF2 activation suggesting REVEALER’s results generalize to tumors (Supplementary Fig. 5).
Drug sensitivity: the MEK-inhibitor PD-0325901 and MAPK Activation
In this example, we show REVEALER de novo discovery without a seed and with a drug sensitivity target.
MEK (MAP2K1), a member of the MAPK signaling pathway, is constitutively activated as a result of oncogenic mutations in e.g. BRAF, RAS and MEK1.27 As a target, we used the sensitivity profile to the MEK inhibitor PD-032590128 in 493 cancer cell lines from the CCLE. As MEK itself is rarely mutated, we ran REVEALER without a seed. The first iteration of REVEALER yielded BRAF mutations as the top-scoring feature (Figure 4 and Supplementary Fig. 6). The next two iterations yielded mutations in KRAS and NRAS. These 3 genes are well-known oncogenic activators of MAPK signaling and their combination explains a large fraction of PD-0325901 sensitive samples in the CCLE (Figure 4 and Supplementary Figs. 7–8).
Figure 4.
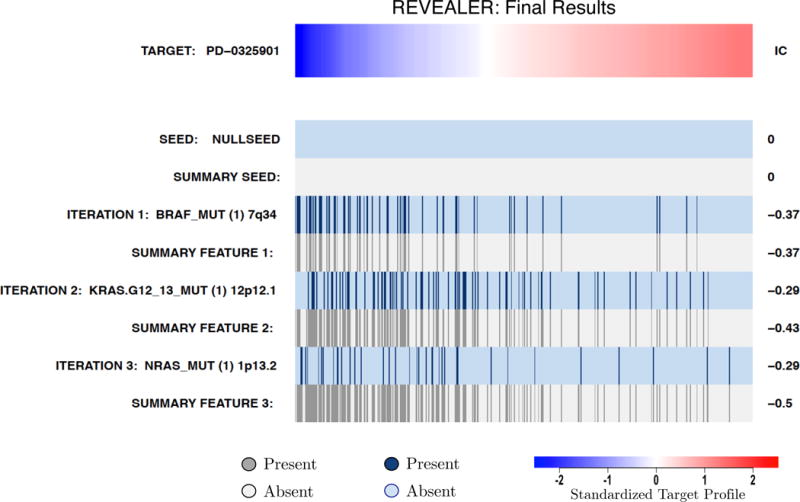
REVEALER results for the drug sensitivity to a MEK-inhibitor example. The target is the MEK-inhibitor PD0325901 sensitivity profile in cancer cell lines and no seed feature (NULLSEED). REVEALER iterates 3 times and identifies BRAF, KRAS and NRAS mutations, all well-known oncogenes upstream of MEK, as complementary “causes” of MEK-inhibitor sensitivity.
Example 4. KRAS dependency
Lastly, we show how REVEALER can be used with a gene dependency target.
The high frequency of KRAS mutations highlights its significance as a major oncogene. Besides studies linking KRAS mutations with dependency29, there is growing evidence for KRAS wild-type states that are also KRAS-dependent.8,9 Consistent with these findings, our examination of KRAS dependency profile across cancer cell lines,30 found evidence that while KRAS dependency associates with KRAS mutation status (IC = −0.41), a significant number of wild-type samples were also dependent on KRAS. We used REVEALER to assess if any other genomic alteration besides KRAS mutation, might account for these unexplained KRAS dependencies. We used as target the shRNA KRAS dependency score and KRAS mutations as seed (Figure 5A). Strikingly, REVEALER found a copy number gain (CNG) in chr8q23–4 (NSMCE2_AMP) as the top-scoring feature in the first iteration (Figure 5A and Supplementary Fig. 9A). This feature is followed by amplifications in chr9p21 and chr12p12 (KRAS locus), and deletions in chr9q12 as potentially complementary alterations with lesser incremental benefit (Figures 5A and Supplementary Figs. 10–12). These features together explain the majority of the KRAS dependent cell lines: 30 out of the top 35 samples with higher KRAS dependency (Figure 5A).
Figure 5.
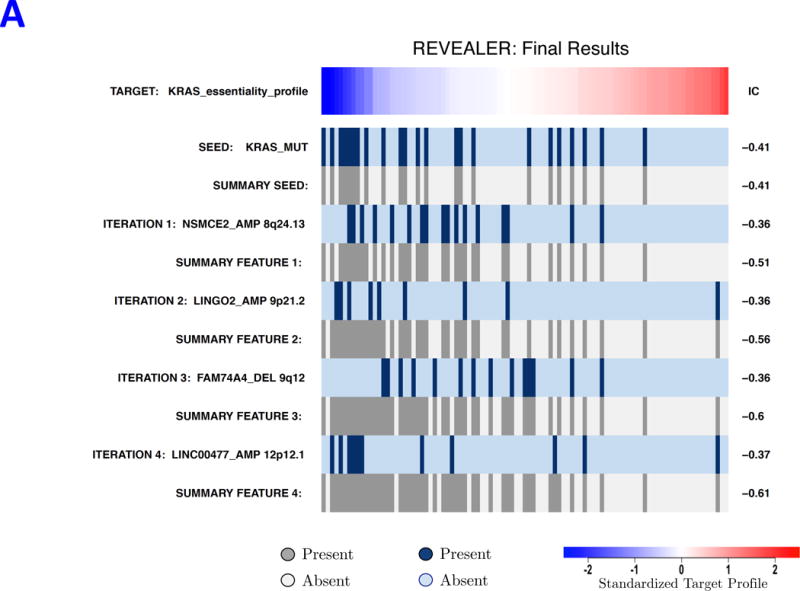
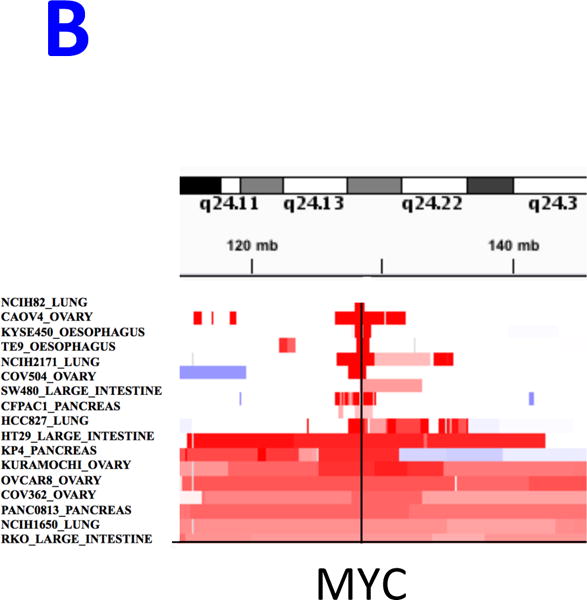
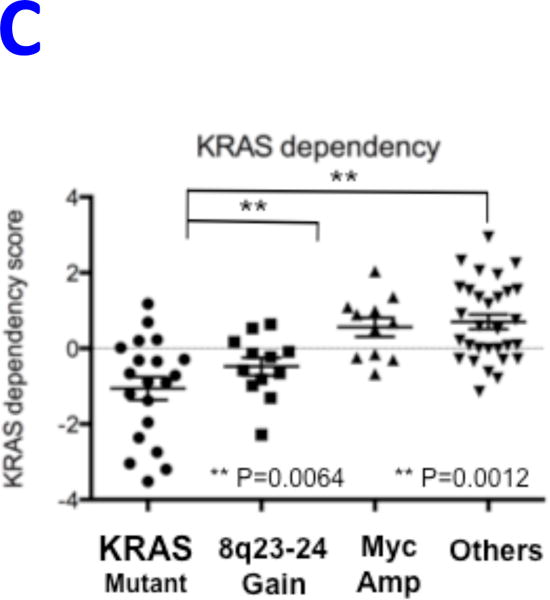
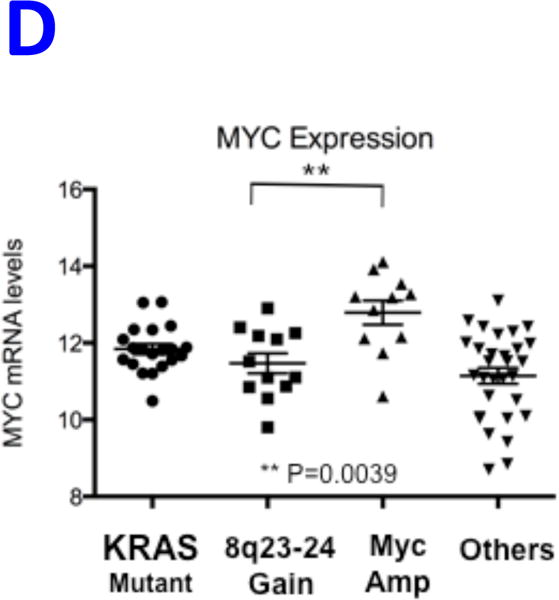
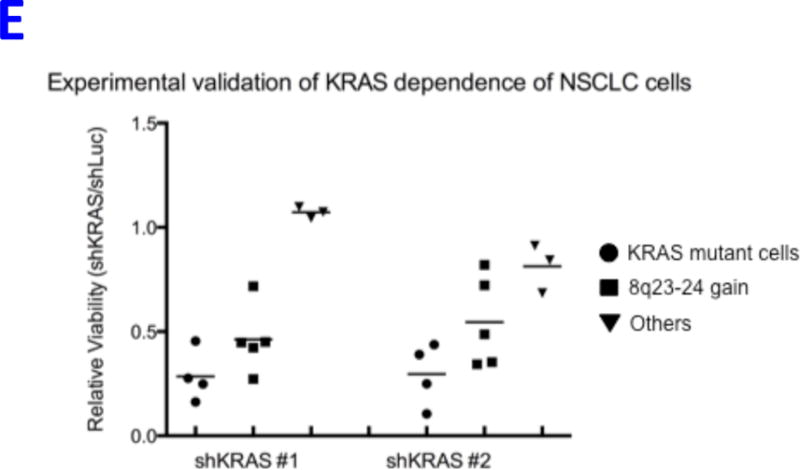
REVEALER results for KRAS-dependency. A) The target profile is the relative KRAS-dependence score in 100 cancer cell lines. The seed feature is the mutation status of KRAS, a well-known cause of activation, and the genomic features matrix represents mutations and copy number alterations in the same cell lines. REVEALER identifies a copy number gain (CNG) across a region on chromosome 8q23–24 as the most complementary genomic alteration to KRAS mutation in order to explain KRAS dependency. Other features such as amplifications in chr9p21, and chr12p12 and deletions in chr9q12 are also identified but with lesser incremental benefit. B) Pattern of copy number changes in cancer cells that have gain in 8q23–24 show that copy number changes centromeric to MYC have two distinct patterns. Red indicates regions of chromosomal gain (log2 ratio >0.6). C) Dot plot of relative KRAS dependence across cell lines with various genotypes (X-axis). Differential KRAS dependence between cells were examined between cells with copy number gain on 8q23–24 relative to cells with other genotype (student t-test with Welch’s Correction ***p<0.0001). D) Dot plot of relative MYC mRNA expression across cell lines with various genotypes (X-axis). Differential MYC mRNA levels were assessed between cells with copy number gain on 8q23–24 vs. MYC amplification (student t-test with Welch’s Correction ***p<0.0001). E) Validation of KRAS dependence in non-small cell lung cancer cells with indicated genotypic status. Cancer cells which harbor 8q23–24 gain from the CCLE were chosen and their relative KRAS dependence was assessed for cells that either have mutations in KRAS or those that harbor 8q23–24 alteration (KRAS mutant cells: NCIH2009, NCIH1944, A549, NCIH1792), 8q23–24 gain: NCIH2110, NCIH1781, NCIH1648, NCIH2126, NCIH2342, Others: NCIH28, NCIH1437, NCIH2228). Relative viability was assessed using CellTiter-Glo assay (Promega) and by normalizing the luminescence values of shKRAS infected cells with shLuciferase controls 7 days post-infection.
Alterations in chr8q23–24 are frequent events in cancer,31 and the REVEALER finding corresponds to a broad region of chr8q23–24 (“chr8q24 gain”) instead of the more specific focal MYC amplification (“MYC amplification”, Figure 5B). To assess differences in KRAS dependence we grouped cell lines based on: MYC amplification, chr8q24 gain, KRAS mutations or none of the above. We found statistically significant differences between cells that harbor chr8q24 gain and cells that either have MYC amplification or other genotypes (Figure 5C). As both events are centromeric with respect to, and potentially target MYC itself, we asked if there were differences in MYC expression between these events. MYC amplified cell lines had significantly higher expression of MYC compared to cell lines with the 8q24 gain (Figure 5D), which perhaps can be explained by high copy number value of MYC amplification region (data not shown). This is consistent with previous studies that show tumors with low MYC expression display increased dependence on KRAS32.
To further validate these findings we selected an independent panel of NSCLC cell lines with either mutations in KRAS, chr8q24 gain, or controls, and assayed them for relative viability upon suppression of KRAS (Figure 5E). Validated shRNAs against KRAS8,9 were used to assess if 8q24-gain predicts sensitivity to KRAS suppression. As expected, cells with mutant KRAS status were highly dependent on KRAS. Consistent with previous observations,8, 33 we also found that cells that do not have alterations in KRAS or chr8q24 are less dependent on KRAS; however, cells that harbor 8q24 gain were significantly more sensitive to KRAS suppression, suggesting that these samples indeed require KRAS for their survival.
REVEALER: Simulated data analysis
The objective of this benchmark was to investigate how well the CIC metric used by REVEALER could find a known complementary feature in controlled circumstances where we know the answer. We were also interested in comparing the CIC with other alternative approaches including the partial correlation coefficient and two other methods: the ElasticNet34 and mRMR35 (Supplementary Information). We generated 5,000 simulated data instances of target, seed and complementary-feature (the “signal”) using probabilistic models parameterized to fit to the empirical data using skew-t distributions and random sampling. We also generate a set of 2,000 random features (the “noise”) (Supplementary Information and Supplementary Figs. 13–15). We use each method to find the correct complementary feature in each instance and evaluated the results using ROC (Receiving Operating Characteristics) curves, which we can estimate because we know the correct complementary feature in each case. The results show that the CIC is the most sensitive at finding the correct complementary feature and attains an area under ROC equal to 0.872, compared with 0.674 for partial correlation, 0.633 for ElasticNet, and 0.672 for mRMR (Figure 6A and Supplementary Fig. 15E).
Figure 6.
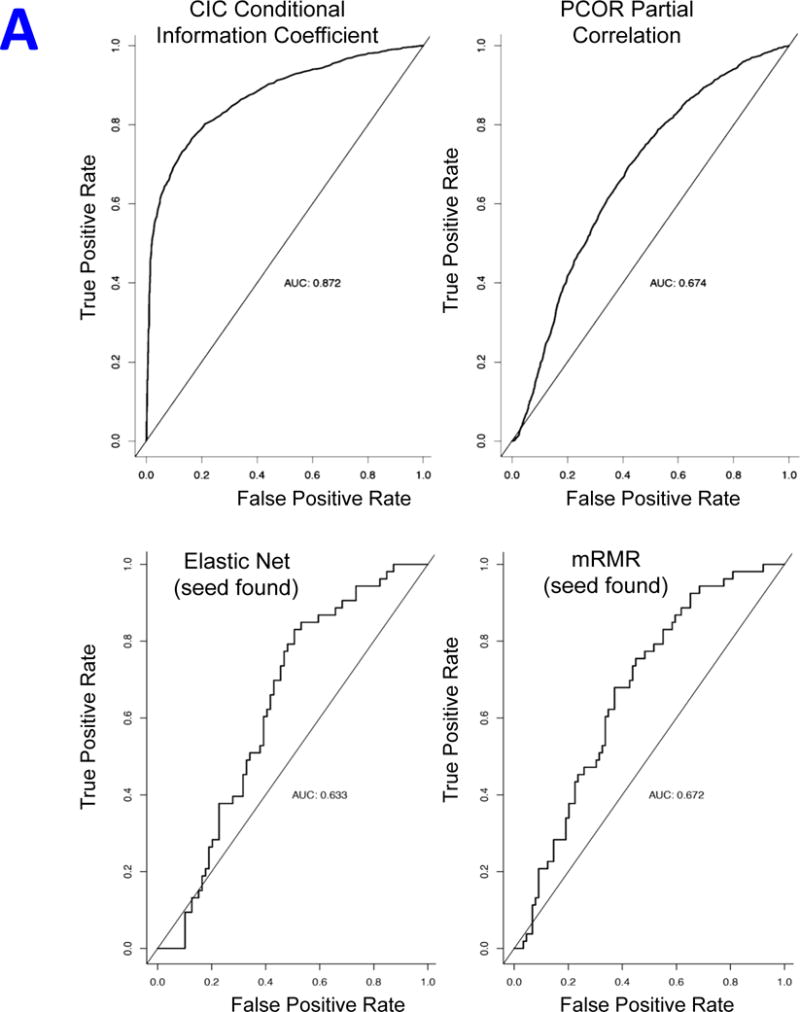
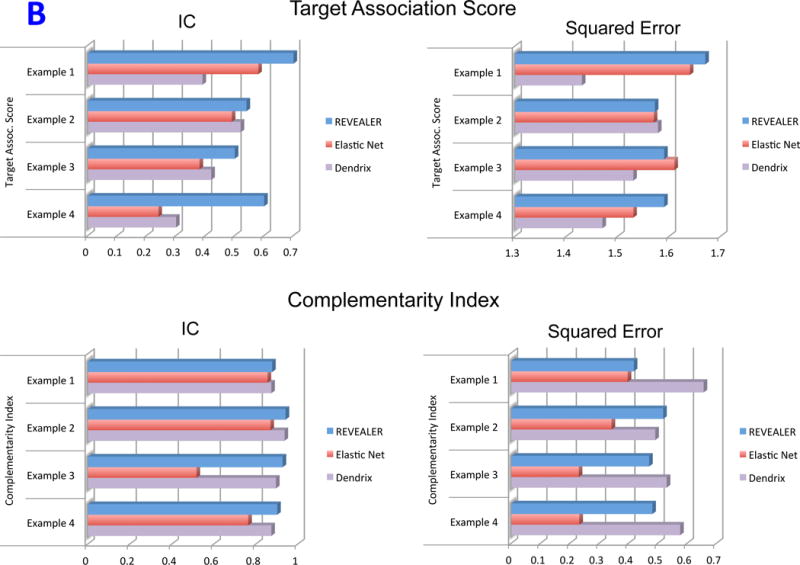
Simulated data results. A) Summary ROC curves for the simulated data benchmark using the CIC/information-based metric, the PCOR/partial correlation the Elastic Net and mRMR feature selection. B) Bar plot of the across-method comparative analysis of top features shown in Table 1 (IC metric), and the corresponding results using the square error metric instead of the IC.
REVEALER: Comparisons with other methods
Methods to search for complementary genomic alterations36–42 or general non-redundant features have been proposed in the past;34,35,43 however, REVEALER is different in several aspects: i) it incorporates 3 inputs: a target profile, a features dataset and a seed feature(s), ii) it uses a sequential search process where the features found in subsequent iterations are influenced by the choice of features in early iterations, and iii) it uses the conditional differential mutual information. These distinctions make it difficult to directly compare REVEALER with other methods; however, if one restricts the comparison to cases with no seed REVEALER can be compared with other methods such as the ElasticNet34 and Dendrix.41
We ran REVALER side to side with those methods using the data for Examples 1–4 without seeds and compared the results to provide insights into the characteristics of each method and delineate their potential suitability to different problem settings. We present below a summary of results and refer the reader to the Supplementary Information for details.
Table 1 and Figure 6B summarize the results using: the Target Association Score, the absolute value of the IC of a summary feature consisting of the combination of all the top selected features, and the Feature Complementarity Index, one minus the average IC across pairs of features. Table 1 shows that several of the features found by the ElasticNet overlap with REVEALER’s, suggesting that strong feature-target associations are retrieved by both methods. Examination of the differences appear to show that the features selected by the ElasticNet, while correlated with the target profile, were less complementary with each other than the ones selected by REVEALER (Figure 6B). This is likely a consequence of ElasticNet’s cost function34 which favors fitting the target and finds features with low correlation with each other but not necessarily mutual exclusivity. Dendrix produces rather different sets of features compared with the other methods (Table 1 and Supplementary Figs. 15A–D) and performs a more comprehensive search of feature complementarity without using the sample-per-sample target. As a consequence Dendrix appears to find features with high complementarity to each other but somewhat less association with the target (Supplementary Fig. 15A–D).
Table 1.
Comparative summary of top features results in the four examples
| Example 1 WNT/Beta-catenin Activation |
Example 2 NRF2 Activation |
Example 3 MEK-inhibitor Sensitivity |
Example 4 KRAS Essentiality |
|
|---|---|---|---|---|
|
| ||||
|
REVEALER (seed) Top Features |
CTNNB1 mut (seed) APC mut ITGBL1 amp (13q33) |
NFE2L2 mut (seed) NFE2L2 amp (seed) KEAP1 mut OR4F13P amp(15q26.3)1 |
N/A (this example had no seed) | KRAS mut (seed) NSMCE2 amp (8q24.13) LINGO2 amp (9p21.2) FAM74A4 del (9q12) LINC00477 amp (12p12.1)2 |
| Target Assoc Score (IC) Feature Comp Index |
0.7 0.877 |
0.6 0.929 |
– – |
0.54 0.847 |
|
| ||||
|
REVEALER (no seed) Top Features |
ITGBL1 amp (13q33) CTNNB1 mut APC mut |
KEAP1 mut LRP1B del (2q21.2) OR4F13P amp (15q26.3)1 |
BRAF mut KRAS.G12-13 mut NRAS mut |
KRAS.G12-13 mut LINC00340 del (6p22.3) ZNF385B amp (2q31.3) NUP153 mut |
| Target Assoc Score (IC) Feature Comp Index |
0.7 0.877 |
0.54 0.941 |
0.5 0.9268 |
0.6 0.901 |
|
| ||||
|
ElasticNet Top Features |
CTNNB1 mut APC mut FAM69A del (1p22) |
KEAP1 mut (19p13.2) PICALM mut (11q14) DOCK10 del (2q36.3) |
BRAF mut BRAF.V600E mut BRAF.MC mut |
BICD1 del (12p11.1)3 ZNF385B amp (2q31.3) FAM69 del (1p22) EMB del (5q11.1) |
| Target Assoc Score (IC) Feature Comp Index |
0.58 0.855 |
0.49 0.869 |
0.38 0.517 |
0.24 0.763 |
|
| ||||
|
Dendrix Top Features |
OR2T11 amp (1q44) PTCD1 amp (7q22.1) SLC25A37 amp (8p21.2) |
KEAP1 mut (19p13.2) LOC100505687 amp (3q26) TAB2 del (6q25.1) |
BRAF mut KRAS mut SHISA6 del (17p12) |
GSTM2 del (1p13.3) KCNJ12 amp (17p11.1) MACROD2 del (20p12.1) UGT3A2 amp (5p13.2) |
| Target Assoc Score (IC) Feature Comp Index |
0.39 0.873 |
0.52 0.936 |
0.42 0.895 |
0.30 0.873 |
Each row corresponds to one method’s results. The first method is REVEALER as described in the examples in the main text, the second is REVEALER without the seed, the third is the ElasticNet and the fourth is Dendrix. The quantities shown are the target association score, the absolute value of the IC of the summary feature consisting of the combination of all the top selected features, and the feature complementarity index, 1 minus the average IC across pairs of features. A higher complementary index means that the features are more mutually exclusive.
Confirmed experimentally (gene NOX5, this study).
KRAS locus.
Potentially representing loss of wild-type KRAS.
REVEALER is available in GenePattern (www.genepattern.org).
Discussion
In the examples presented above we demonstrated how REVEALER effectively maps genomic alterations to their relevant functional profiles. The identification of i) APC and KEAP1 mutations, as alternative causes of activation of β–catenin and NRF2 respectively; ii) the association of BRAF, KRAS and NRAS mutations with MEK-inhibition sensitivity, and iii) our successful validation of the role of NOX5 in NRF2 activation, and the chr8q23–4 amplicon in predicting KRAS dependency, all provide a direct confirmation of REVEALER’s utility and effectiveness.
The use of mutual information for estimating genomic feature association is not new,44–46 however, REVEALER makes innovative use of conditional mutual information based on continuous distributions and avoids the need for discretization and other simplifying assumptions. The simulated benchmark shows that REVEALER can identify a complementary feature reasonably well where its CIC is above 0.30 for a wide range of IC values between target and seed The results also show that the conditional mutual information is more sensitive than the partial correlation, and other selection methods, to discriminate subtler relationships between genomic features.
The comparative results across methods (Table 1 and Supplementary Fig. 15A–D) suggest that REVEALER strikes a good balance between weighting the features’ complementarity and their association with the target. REVEALER is particularly well suited in cases where there is: a) an accurate sample-per-sample functional profile representing a biological state of interest, b) prior information to guide the choice of seed(s), c) a comprehensive characterization of genomic abnormalities.
The differences between approaches are likely produced by the different emphasis of each algorithm. The ElasticNet emphasizes finding uncorrelated features that primarily “predict” the target, and are not strictly restricted to be complementary, It is well suited for cases where matching the target profile is more important than strict feature complementarity. Dendrix, on the other hand, is more appropriate to find multiple sets of features that are highly complementary in a subset of samples with less emphasis in fitting the target. These methods are all complementary approaches that emphasize different aspects of feature selection and have potential applicability depending on the problem setting.
Supplementary Material
Acknowledgments
This project was supported in part by NIH grants R01 CA154480, R01 CA121941, U01 CA176058 and R01 CA109467.
Footnotes
- Designed and conceptualized the method: JWK, OBB, JPM, JSB, WH, PT.
- Implemented the algorithm: OBB, OA, CB, PT.
- Analyzed and interpreted analysis results: JWK, DAB, JR, YS, MEA, PSH, AA, HG, FV, BAW, EMVA, DDS, TIZ, RB, LAG, JPM, PT.
- Provided expertise or work on specific issues regarding algorithmic approaches, data analysis, data preparation, data resources, benchmarking, validation datasets, and method comparisons: ARAR, MA, CR, GS, DD, GG, MG, GA, AT, PT.
- Wrote the manuscript: JWK, OBB, MEA, JR, JPM and PT.
Competing financial interests statement: I declare that the authors have no competing interest as defined by Nature Publishing Group, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
- 1.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 2.Lawrence MS, et al. 2013, Mutational heterogeneity in cancer and the search for new cancer- associated genes. Nature. 2013 Jul 11;499(7457):214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014 Jan 23;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013 Oct;45(10):1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beroukhim R, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chin L, Hahn WC, Getz G, Meyerson M. Making sense of cancer genomic data. Genes Dev. 2011;25:534–555. doi: 10.1101/gad.2017311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo B, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci U S A. 2008;105:20380–20385. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung HW, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108:12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boehm JS, Hahn WC. Towards systematic functional characterization of cancer genomes. Nat Rev Genet. 2011;12:487–498. doi: 10.1038/nrg3013. [DOI] [PubMed] [Google Scholar]
- 11.Nijhawan D, et al. Cancer vulnerabilities unveiled by genomic loss. Cell. 2012;150:842–854. doi: 10.1016/j.cell.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. EMBO J. 2012;31:2714–2736. doi: 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenbluh J, et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151:1457–1473. doi: 10.1016/j.cell.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minde DP, Anvarian Z, Rudiger SG, Maurice MM. Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer? Mol Cancer. 2011;10:101. doi: 10.1186/1476-4598-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Firestein R, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455:547–551. doi: 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeNicola GM, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malhotra D, et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38:5718–5734. doi: 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh A, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solis LM, et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 2010;16:3743–3753. doi: 10.1158/1078-0432.CCR-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brar SS, Corbin Z, Kennedy TP, Hemendinger R, Thornton L, Bommarius B, Arnold RS, Whorton AR, Sturrock AB, Huecksteadt TP, Quinn MT, Krenitsky K, Ardie KG, Lambeth JD, Hoidal JR. NOX5 NAD(P)H oxidase regulates growth and apoptosis in DU145 prostate cancer cells. Am J Physiol Cell Physiol. 2003 Aug;285(2):C353–69. doi: 10.1152/ajpcell.00525.2002. [DOI] [PubMed] [Google Scholar]
- 23.Pendyala S, Natarajan V. Redox regulation of Nox proteins. Respir Physiol Neurobiol. 2010 Dec 31;174(3):265–71. doi: 10.1016/j.resp.2010.09.016. Epub 2010 Sep 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandey D, Patel A, Patel V, Chen F, Qian J, Wang Y, Barman SA, Venema RC, Stepp DW, Rudic RD, Fulton DJ. Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am J Physiol Heart Circ Physiol. 2012 May 15;302(10):H1919–28. doi: 10.1152/ajpheart.00910.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abazeed ME, et al. Integrative radiogenomic profiling of squamous cell lung cancer. Cancer Res. 2013 Oct 15;73(20):6289–98. doi: 10.1158/0008-5472.CAN-13-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The Cancer Genome Atlas. 2012 http://cancergenome.nih.gov/
- 27.McCubrey JA, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149–2182. doi: 10.1101/gad.1437206. [DOI] [PubMed] [Google Scholar]
- 29.Young A, et al. Ras signaling and therapies. Adv Cancer Res. 2009;102:1–17. doi: 10.1016/S0065-230X(09)02001-6. [DOI] [PubMed] [Google Scholar]
- 30.Shao DD, et al. ATARiS: computational quantification of gene suppression phenotypes from multisample RNAi screens. Genome Res. 2013;23:665–678. doi: 10.1101/gr.143586.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raeder MB, et al. Integrated genomic analysis of the 8q24 amplification in endometrial cancers identifies ATAD2 as essential to MYC-dependent cancers. PLoS One. 2013;8(2):e54873. doi: 10.1371/journal.pone.0054873. Epub 2013 Feb 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hollern DP, Yuwanita I, Andrechek ER. A mouse model with T58A mutations in Myc reduces the dependence on KRas mutations and has similarities to claudin-low human breast cancer. Oncogene. 2013;32:1296–1304. doi: 10.1038/onc.2012.142. [DOI] [PubMed] [Google Scholar]
- 33.Scholl C, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 34.Zou H, Hastie T. Regularization and variable selection via the elastic net. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 2005;67:301–320. [Google Scholar]
- 35.Peng H, Long F, Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell. 2005;27:1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 36.Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22:398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller CA, Settle SH, Sulman EP, Aldape KD, Milosavljevic A. Discovering functional modules by identifying recurrent and mutually exclusive mutational patterns in tumors. BMC Med Genomics. 2011;4:34. doi: 10.1186/1755-8794-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vandin F, Upfal E, Raphael BJ. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012;22:375–385. doi: 10.1101/gr.120477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leiserson MD, Blokh D, Sharan R, Raphael BJ. Simultaneous identification of multiple driver pathways in cancer. PLoS Comput Biol. 2013;9:e1003054. doi: 10.1371/journal.pcbi.1003054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu Y, et al. Network analysis of genomic alteration profiles reveals co-altered functional modules and driver genes for glioblastoma. Mol Biosyst. 2013;9:467–477. doi: 10.1039/c2mb25528f. [DOI] [PubMed] [Google Scholar]
- 41.Vandin F, Upfal E, Raphael BJ. Algorithms for detecting significantly mutated pathways in cancer. J Comput Biol. 2011;18:507–522. doi: 10.1089/cmb.2010.0265. [DOI] [PubMed] [Google Scholar]
- 42.Vaske CJ, et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26:i237–245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waldron L, et al. Optimized application of penalized regression methods to diverse genomic data. Bioinformatics. 2011;27:3399–3406. doi: 10.1093/bioinformatics/btr591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Butte AJ, Kohane IS. Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. Pac Symp Biocomput. 2000:418–429. doi: 10.1142/9789814447331_0040. [DOI] [PubMed] [Google Scholar]
- 45.Steuer R, Kurths J, Daub CO, Weise J, Selbig J. The mutual information: detecting and evaluating dependencies between variables. Bioinformatics. 2002;18(Suppl 2):S231–240. doi: 10.1093/bioinformatics/18.suppl_2.s231. [DOI] [PubMed] [Google Scholar]
- 46.Margolin AA, et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.