

Summary
HIV-1 protease is a major drug target for AIDS therapy. With the appearance of drug-resistant HIV-1 protease variants, understanding the mechanism of drug resistance becomes critical. Computational methods can provide more details about inhibitor-protease binding other than crystallography and isothermal titration calorimetry. Darunavir is the latest FDA approved HIV-1 protease inhibitor. In this context, the free energy component analysis is performed on the DRV binding to WT protease and ACT, a drug resistant variant, to evaluate contribution of each atoms of DRV to the binding affinity. This information can contribute to the rationale design of new HIV-1 protease inhibitors.
Keywords: HIV-1 Protease, Darunavir, Drug Resistance, Rationale Drug Design, Free Energy Calculation, Free energy components analysis
Introduction
The human immunodeficiency virus type 1 (HIV-1) protease is a homodimeric aspartic acid protease. It cleaves the viral Gag-Pol polyprotein to release the enzymes and structural proteins indispensable for the maturation of infectious viral particles.1 The nine FDA approved protease effectively decrease the mortality rate of HIV/AIDS patients.2,3 The exposure to protease inhibitors selects for viruses that have acquired drug resistance mutations in protease due to the high replication rate of HIV-1 and to lack of a proofreading mechanism in its reverse transcriptase. The drug-resistant protease variants decrease their high binding affinity to the inhibitors, while maintaining enough enzyme activity for the virus to propagate.4 Comparison between the crystal structures of wild-type and drug-resistant variant protease’s in complex with inhibitors partially elucidates how specific protease mutations decrease protease-inhibitor binding affinity.5–7 However, it is still a challenge to elucidate the critical components of the binding affinity quantitatively from the structural data. Free-energy simulations, in principle, can aid in elucidating these components of the binding affinities to particular atomic interactions.8–10 The calculation results can be further analyzed, e.g., for free energy decomposition, to provide information about affinity changes due to specific kinds of interaction on an atomic level, which could not be determined by experimental methods. Darunavir (DRV) (Figure 1A) is a recently FDA approved HIV-1 protease inhibitor.11,12 The Gibbs free energy change for DRV-WT binding measured by ITC is −15.2kcal/mol. Drug resistant protease variant ACT (Figure 1B) has two active site mutations V82T and I84V.7 The binding free energy change for DRV-ACT binding is −13.6kcal/mol. Energetic studies by computational methods have found that the vdW interaction has dominate influence in protease-inhibitor recognition.13,14 In this context, the vdW energy contributions were calculated by the MD simulation package AMBER15 for each DRV atom and compared between the WT and ACT protease variant. Understanding how the protease mutates to decrease its binding affinity with a very high affinity inhibitor will contribute to developing better strategies to design protease inhibitors.
Figure 1.
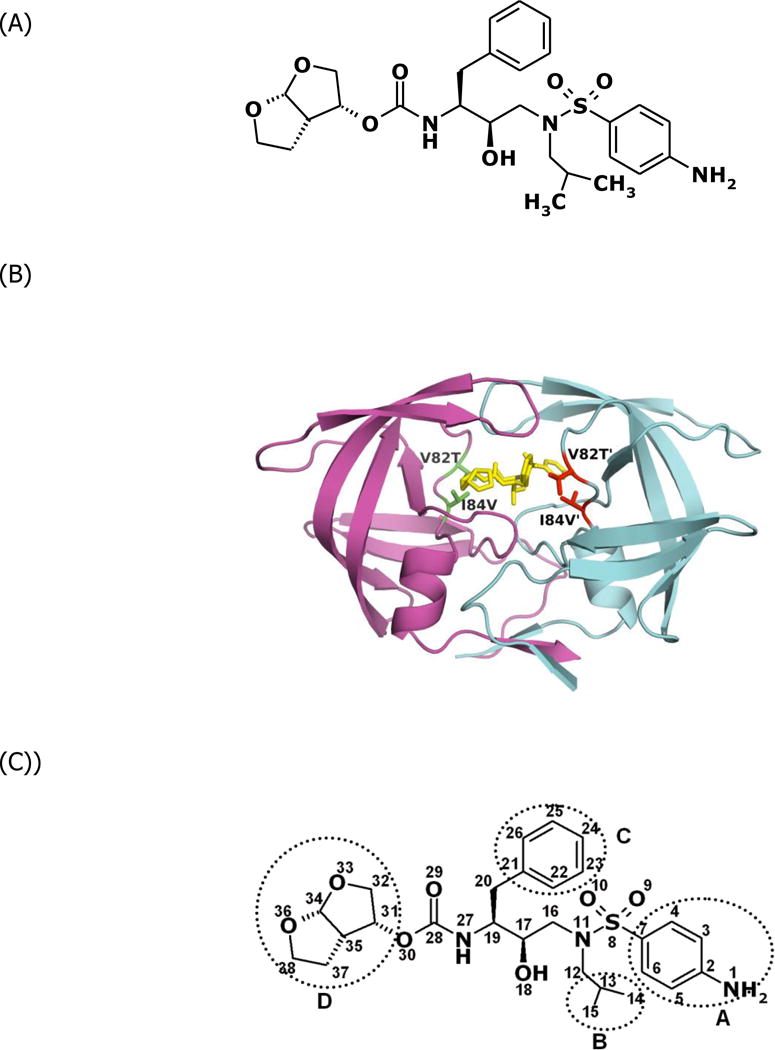
(A) Chemical structure of DRV. (B) Structure of protease variant ACT-DRV complex. DRV is colored yellow. The side chains of the mutated residues Thr82 and Val84 are displayed and colored red or green. (C) The four moieties of DRV.
2. Methods
2.1 Generate topology and coordinates files from the crystal structures for the MD simulations
Create a text file “hivpr_md.leap” with the following contents. (Do not include the texts in the parenthesis.)
source $AMBERHOME/dat/leap/cmd/leaprc.ff03 (see Note 1) source $AMBERHOME/dat/leap/cmd/leaprc.gaff loadamberprep DRV.in (see Note 2) WAT = TP3 SWT = TP3 HOH = TP3 DRVwt=loadpdb 1T3R.pdb DRVact=loadpdb 1T7J.pdb alignaxes DRVwt addions DRVwt Cl- 6 solvatebox DRVwt TIP3PBOX {10 10 10} saveamberparm DRVwt DRVwt.top DRVwt.crd alignaxes DRVact addions DRVact Cl- 6 solvatebox DRVact TIP3PBOX {10 10 10} saveamberparm DRVact DRVact.top DRVact.crd
Enter “/$AMBERHOME/exe/teLeap –f hivpr_md.leap” to create the topology and coordinates files.
2.2 Perform Energy Minimizations for both WT-DRV and ACT-DRV systems
Create a text file “emin” with the following context.
&cntrl imin=1, ntmin=2, ntr=1, maxcyc=4000, ntpr=25, ntwx=50 &end END
Perform the energy minimization by typing the following command line.
$AMBERHOME/exe/sander -O -i emin | -o DRVwt.emin.out -p DRVwt.top -c DRVwt.crd | -ref DRVwt.crd -r DRVwt.emin.rst
2.3 Assign initial velocities of each atom of both WT-DRV and ACT-DRV systems
Create a text file “thermin” with the following context.
therm to 300K with restrain &cntrl imin=0, iwrap=1, irest=0, ntx=1, ntrx=1, ntxo=1, ntpr=100, ntwr=100, ntwx=50, ntwv=0, ntwe=0, ntf=2, ntb=1, cut=8.0, ibelly=0, ntr=1, nstlim=10000, nscm=1000000, t=0.00, dt=0.001, (see Note 3) temp0=300.0, tempi=300.0, ig=1001, (see Note 4) ntt=1, tautp=2.0, ntp=0, nrespa=2, ntc=2, lastrst=5000000, lastist=5000000, &end Restrained the heavy atoms 9.55 FIND C * * * O * * * N * * * S * * * SEARCH RES 1 1000 END END
Assign the velocities by typing the following command line.
$AMBERHOME/exe/sander -O -i thermin | -o DRVwt.therm.out -p DRVwt.top -c DRVwt.emin.rst | -ref DRVwt.emin.rst -r DRVwt.therm.rst -x DRVwt.therm.x
2.4 Perform restrained MD simulations to equilibrate the system
Create a text file “equilin” with the following context.
Equil at 300K &cntrl icfe=2, imin=0, iwrap=1, irest=1, ntx=5, ntrx=1, ntxo=1, ntpr=100, ntwr=100, ntwx=50, ntwv=0, ntwe=0, ntf=2, ntb=2, cut=8.0, ibelly=0, ntr=1, nstlim=50000, nscm=1000000, t=0.00, dt=0.001, temp0=300.0, ntt=1, tautp=2.0, ntp=1, nrespa=1, ntc=2, lastrst=5000000, lastist=5000000, &end END
Type the following command line and hit enter.
$AMBERHOME/exe/sander -O -i equilin -o DRVwt.equil.out | -p DRVwt.top -c DRVwt.therm.rst | -ref DRVwt.therm.rst -r DRVwt.equil.rst -x DRVwt.equil.x
2.5 Performing MD simulations to sample the conformations
Create a text file “mdin” with the following context.
MD at 300K &cntrl imin=0, iwrap=1, ntx=5, irest=1, ntrx=1, ntxo=1, ntpr=10000, ntwr=100000, ntwx=500, ntwv=0, ntwe=0, ntf=2, ntb=2, cut=8.0, igb=0, ibelly=0, ntr=0, nstlim=500000, nscm=1000, t=0.00, dt=0.001, temp0=300.0, tempi=300.0, ig=100000, heat=0.0, ntt=1, tautp=0.1, ntp=1, pres0=1.013, comp=27.5, taup=0.5, ntc=2, tol=0.0001, lastrst=5000000, lastist=5000000, &end END
Type the following command line and hit enter.
$AMBERHOME/exe/sander -O -i mdin | -o DRVwt.1.out -p DRVwt.top -c DRVwt.equil.rst | -ref DRVwt.equil.rst -r DRVwt.1.rst -x DRVwt.1.x
Perform step 2.2 to 2.5 for DRV-ACT
2.6 Create the topology file for free energy decompositions
Open the 1T3R PDB file and define the DRV atoms residue indexes as shown below.
ATOM 3129 C4 D1 200 38.304 35.393 26.577 ATOM 3130 H4 D2 201 39.040 35.702 25.841 ATOM 3131 C3 D3 202 37.237 36.233 26.904 …… ATOM 3201 2H30 D73 272 43.496 27.149 26.713 ATOM 3202 C31 D74 273 45.684 26.771 26.888 ATOM 3203 1H3 D75 274 46.089 26.521 25.908
Remove all atoms other than the protease and inhibitor atoms. Save it as “1T3R.dc.pdb”. Make another PDB file named “1T3R.rec.pdb” from 1T3R.dc.pdb by deleting the inhibitor informations. Make another PDB file named “1T3R.lig.pdb” from 1T3R.dc.pdb by deleting the protease atoms informations.
Create a text file “decom.leap” with the following contents.
source $AMBERHOME/dat/leap/cmd/leaprc.ff03 source $AMBERHOME/dat/leap/cmd/leaprc.gaff loadamberprep DRV.dc.in (see Note 5) DRVwt=loadpdb 1T3R.dc.pdb DRVwtrec=loadpdb 1T3R.rec.pdb DRVwtlig=loadpdb 1T3R.lig.pdb saveamberparm DRVwt DRVwt.dc.top DRVwt.dc.crd saveamberparm DRVwtrec DRVwt.rec.top DRVwt.rec.crd saveamberparm DRVwtlig DRV.dc.top DRV.dc.crd
Enter “/$AMBERHOME/exe/teLeap –f hivpr_md.leap” to create the topology and coordinates files.
2.7 Process the trajectories
Create a text file “DRVwt.coor.in” with the following context.
@GENERAL PREFIX DRVwt PATH ./ COMPLEX 1 RECEPTOR 1 LIGAND 1 COMPT ../DRVwt.dc.top RECPT ../DRVwt.rec.top LIGPT ../DRV.dc.top GC 1 AS 0 DC 0 MM 0 GB 0 PB 0 MS 0 NM 0 @MAKECRD BOX NO NTOTAL 7455 (see Note 6) NSTART 1 NSTOP 1000 NFREQ 1 NUMBER_LIG_GROUPS 1 LSTART 3135 (see Note 7) LSTOP 3209 (see Note 8) NUMBER_REC_GROUPS 1 RSTART 1 RSTOP 3134 (see Note 9) @TRAJECTORY TRAJECTORY ./DRVwt.1.x
Type the following command line and hit enter.
$AMBERHOME/exe/mm_pbsa.pl DRVwt.coor.in >& DRVwt.coor.out
2.8 Calculate the vdW energy change for each atoms of DRV
Create a text file “DRVwt.decom.in” with the following context.
@GENERAL PREFIX DRVwt PATH ./ COMPLEX 1 RECEPTOR 1 LIGAND 1 COMPT ./DRVwt.dc.top RECPT ./DRVwt.rec.top LIGPT ./DRV.dc.top GC 0 AS 0 DC 1 MM 1 GB 1 PB 0 MS 0 NM 0 @DECOMP DCTYPE 1 COMREC 1–198 COMLIG 199–273 COMPRI 1–273 RECRES 1–198 RECPRI 1–198 RECMAP 1–198 LIGRES 1–75 LIGPRI 1–75 LIGMAP 199–273 @MM DIELC 1.0 @GB IGB 2 GBSA 2 SALTCON 0.0 EXTDIEL 80.0 INTDIEL 1.0 SURFTEN 0.0072 SURFOFF 0.00 @MS PROBE 0.0
Type the following command line and hit enter.
$AMBERHOME/exe/mm_pbsa.pl DRVwt.decom.in >& DRVwt.decom.out
A file name “DRVwt_statistics.out” will be created after the calculations is done.
Perform the same operation of step 2.6 and 2.8 on DRV-ACT system.
A file name “DRVact_statistics.out “ will be created after the calculations is done.
Extract the data under “TVDW” colume label, the last 75 lines are the 75 atoms of DRV intereaction energy with the protease. The order of the atoms of DRV will be the same as in the PDB file. (see Note 10)
Table 1.
Loss of van der Waals’ Interaction Energy for different Moieties of DRV and APV
| DRV | 4–Amino Phenyl Group | Isopropyl Group | Benzyl Ring | bis-Tetrahydrofuranyl | |
|---|---|---|---|---|---|
| DRV-ACT | kcal/mol | 1.11 | 0.83 | 0.30 | 0.20 |
| % | 18.9 | 28.0 | 6.5 | 3.2 | |
Footnotes
“$AMBERHOME” is the directory where the AMBER package is.
“DRV.in” provides information and parameters of DRV. See attachment.
“dt=0.001” – the Time interval of the calculation is 1 femto-second.
“ig=1001” – This is the random seed value. Changing this value can generate a parallel MD simulations with different initial conditions of the system.
“DRV.dc.in” is the parameter file of DRV where each atom is defined as a unit. See attachment.
Total number of the system with explicit solvent. Check it in the file “DRVWT.top” and “DRVACT.top”.
The number of the first DRV atoms.
The number of the last DRV atoms.
Last number of the Last protease atoms.
DRV had 37 hydrogen atoms with very limited contribution to the vdW interaction energy. Thus, data were analyzed for the 38 non - hydrogen atoms of DRV. Structurally, DRV can be considered formed by four major moieties: A) 4–aminophenyl group, B) isopropyl group, C) benzyl ring, and D) bis-tetrahydrofuranylurethane (THF) (Figure 1C). The percentage of energy lost of each moiety can be calculated. (Table 1) The bis-THF group and benzyl ring of DRV sustain their vdW interactions with the drug-resistant protease variants and contribute most to the inhibitor-protease binding, while DRV’s 4 – amino phenyl and isopropyl groups are susceptible to changes in the protease’s binding pocket and adopt conformations that lose vdW interaction with drug-resistant variants. (Table 1) The analysis suggest that modifying the 4–aminophenyl and isopropyl groups might help to design new protease inhibitors which likely have higher binding affinities with wild-type protease and drug-resistant variants.
References
- 1.Debouck C. AIDS Research and Human Retroviruses. 1992;8:153–164. doi: 10.1089/aid.1992.8.153. [DOI] [PubMed] [Google Scholar]
- 2.Wlodawer A, Erickson JW. Annual Review of Biochemistry. 1993;62:543–585. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]
- 3.Wood E, Hogg RS, Yip B, Moore D, Harrigan PR, Montaner JS. HIV Med. 2007;8:80–5. doi: 10.1111/j.1468-1293.2007.00430.x. [DOI] [PubMed] [Google Scholar]
- 4.Schinazi RF, Larder BA, Mellors JW. Internatl Antiviral News. 1997;5:129–142. [Google Scholar]
- 5.King NM, Melnick L, Prabu-Jeyabalan M, Nalivaika EA, Yang SS, Gao Y, Nie X, Zepp C, Heefner DL, Schiffer CA. Protein Science. 2002;11:418–429. doi: 10.1110/ps.25502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prabu-Jeyabalan M, Nalivaika EA, King NM, Schiffer CA. Journal of Virology. 2003;77:1306–15. doi: 10.1128/JVI.77.2.1306-1315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Bethune MP, Schiffer CA. Journal of Virology. 2004;78:12012–12021. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang W, Kollman PA. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14937–42. doi: 10.1073/pnas.251265598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huo S, Massova I, Kollman PA. Journal of Computational Chemistry. 2002;23:15–27. doi: 10.1002/jcc.1153. [DOI] [PubMed] [Google Scholar]
- 10.Michielin O, Karplus M. Journal of Molecular Biology. 2002;324:547–569. doi: 10.1016/s0022-2836(02)00880-x. [DOI] [PubMed] [Google Scholar]
- 11.Surleraux DL, de Kock HA, Verschueren WG, Pille GM, Maes LJ, Peeters A, Vendeville S, De Meyer S, Azijn H, Pauwels R, de Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PB. Journal of Medicinal Chemistry. 2005;48:1965–73. doi: 10.1021/jm049454n. [DOI] [PubMed] [Google Scholar]
- 12.Surleraux DL, Tahri A, Verschueren WG, Pille GM, de Kock HA, Jonckers TH, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PB. Journal of Medicinal Chemistry. 2005;48:1813–22. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]
- 13.Hou T, Yu R. Journal of Medicinal Chemistry. 2007;50:1177–88. doi: 10.1021/jm0609162. [DOI] [PubMed] [Google Scholar]
- 14.Stoica I, Sadiq SK, Coveney PV. Journal of the American Chemical Society. 2008;130:2639–48. doi: 10.1021/ja0779250. [DOI] [PubMed] [Google Scholar]
- 15.Case DA, Cheatham TE3rd, Darden T, Gohlke H, Luo R, Merz KMJr, Onufriev A, Simmerling C, Wang B, Woods RJ. Journal of Computational Chemistry. 2005;26:1668–88. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]