

Abstract
A shift in macrophage metabolism from oxidative phosphorylation to aerobic glycolysis is a requirement for activation to effectively combat invading pathogens. Francisella tularensis is a facultative intracellular bacterium that causes an acute, fatal disease called tularemia. Its primary mechanism of virulence is its ability to evade and suppress inflammatory responses while replicating in the cytosol of macrophages. The means by which F. tularensis modulates macrophage activation are not fully elucidated. Here we demonstrate that virulent F. tularensis impairs production of inflammatory cytokines in primary macrophages by preventing their shift to aerobic glycolysis, as evidenced by the down regulation of HIF-1α, and failure to upregulate pfkfb3. We also uncover that Francisella capsule is required for this process. In addition to modulating inflammatory responses, inhibition of glycolysis in host cells is also required for early replication of virulent Francisella. Together our data demonstrate that metabolic reprogramming of host cells by F. tularensis is a key component of both inhibition of host defense mechanisms and replication of the bacterium.
Introduction
Activation of macrophages and dendritic cells requires a complex network of cellular processes to generate pro-inflammatory cytokines and anti-microbial products for control of pathogen replication. One of these processes involves regulated changes in host metabolism. Specifically, activation and maturation of macrophages and dendritic cells following engagement of pattern recognition receptors (PRR) is dependent on a shift in metabolism from oxidative phosphorylation to aerobic glycolysis (1). Induction of aerobic glycolysis generates several products required for host cell activation and maturation (2). First, the glycolytic process evokes rapid production of ATP to fuel the increased energy needs of the activated cell. Glycolysis also initiates metabolic pathways that generate substrates required for activation of transcription factors responsible for promoting expression of genes involved in potentiating glycolysis and pro-inflammatory cytokines.
Francisella tularensis subsp tularensis (Ftt) is a facultative intracellular bacterium that causes a lethal disease known as tularemia. There are approximately 200 cases of tularemia each year in the United States and approximately 500–1000 cases in Europe; thus this pathogen represents a consistent public health problem (3, 4). Additionally, Ftt was developed as a biological weapon by the United States, the former Soviet Union and Japan (5). These programs also developed at least one antibiotic resistant strain. Given the lack of an approved vaccine for use against natural and nefarious exposures and the presence of antibiotic resistant strains there is a need for novel vaccines and therapeutics.
Although Ftt can infect a variety of cell types, the preferred cellular targets of this pathogen in vivo are macrophages and dendritic cells (6). An important mechanism of virulence for Ftt is its ability to evade and suppress induction of innate immune responses in these cells that would normally control replication and dissemination of Ftt (7–9). However, the mechanisms and bacterial products responsible for modulating the host response have not been comprehensively defined. In the last several years the presence of an O-antigen capsule and the genes required for capsule generation in virulent Ftt have been confirmed (10–12). Since then Ftt capsule has been shown to contribute to resistance of Ftt to killing via the alternative pathway of complement activation following exposure to human serum (12). Also, under specific conditions, presence of capsule on Francisella has also been reported to influence bacterial replication in the intracellular compartment (11). Together, these features suggest capsule contributes to the virulence of the bacterium. Indeed, defined mutations in capsule synthesis genes results in modest attenuation of Francisella in vivo (13). Despite these advances, the contribution of capsule in the evasion suppression of pro-inflammatory responses is largely unexplored.
Bacterial capsules are generally thought to contribute to evasion of host defenses by cloaking immunostimulatory structures present on the bacterial surface, providing resistance to complement mediated killing, and limiting phagocytosis by host cells [as reviewed, (14)]. However, there is some data suggesting that capsular material may take a more active role in modulating the immune response by suppressing the ability of host cells to mount protective inflammatory responses (15).
In this report, we demonstrate that Ftt capsule aids in direct suppression of inflammatory responses in vitro and in vivo. Further, we provide evidence that Ftt capsule mediates this suppression by reprogramming host metabolism via inhibition of the shift to aerobic glycolysis required for macrophage activation. Finally, we show that inhibition of host cell glycolysis is essential for the early intracellular replication of Ftt. Together our findings identify a unique intercept of bacterial pathogenesis and modulation of host metabolism.
Materials and Methods
Mice and generation of bone marrow derived macrophages (BMDM)
C57BL/6J wild type mice (WT) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were housed in ABSL-2 and ABSL-3 animal facilities at Rocky Mountain Laboratories (RML) and were provided food and water ad libitum. All research involving animals was conducted in accordance with Animal Care and Use guidelines and animal protocols were approved by the Animal Care and Use Committee at RML. BMDM were generated as previously described (16). As indicated, BMDM were treated with 2-deoxyglucose (2-DG; Sigma-Aldrich, St. Louis, MO) 2h prior to infection or exposure to capsule.
Bacteria
Stock cultures of F. tularensis ssp. tularensis SchuS4 (Ftt) (Dr. Jeannine Peterson, CDC, Fort Collins, CO), SchuS4Δ1238, SchuS4Δ1236, SchuS4Δ1464c (Dr. Bradley Jones, University of Iowa, Iowa City IA) were generated and utilized as previously described (8). SchuS4Δ1238 (Δ1238) and SchuS4Δ1236 (Δ1236) were previously characterized as capsule mutants due to deletions in waa locus encoding enzymes responsible for adding O-antigen subunits to the LPS core or adding sugars to LPS core, respectively (13). SchuS4Δ1464c (Δ1464c) was also previously described and is devoid of capsule due to the mutation in the dTDP-glucose 4,6-dehydratase that enables formation of 6-deoxy sugars that make up the capsular structure (17). All experiments were performed under approved BSL-3 safety protocols at RML.
Purification of Ftt capsule
Capsule was purified from Ftt as previously described with minor modifications (10). Briefly, Ftt grown on modified Mueller hinton (MMH) agar was collected in buffer containing 6mM Tris, 10mM EDTA and 2% [wt/vol] SDS at pH 6.8 and incubated for 24 h at 65°C, followed by addition of 50 µg/ml proteinase K and an additional incubation at 37–42°C for 24 h. SDS was removed by ethanol precipitation, samples were centrifuged at 12,000 × g at 4°C and the resulting pellets were resuspended in 10mM Tris-base/10mM CaCl2 pH 7.4 containing 80 units micrococcal nuclease (Sigma-Aldrich). Samples were incubated at 37°C for 24 h followed by addition of an equal volume of phenol and an additional incubation at 65°C for 30 min. Samples were then cooled on ice, centrifuged 2,000 × g for 10 min at 4°C. The aqueous layer was collected. The phenol layer was back extracted with deionized water. Aqueous layers were combined and phenol removed via ethanol precipitations. Following precipitation the pellet was resuspended in HPLC grade water (Sigma-Aldrich) containing 5% [vol/vol] Triton X-114 and incubated at 4°C for 24 h followed by incubation at 37°C for 1 h. Samples were centrifuged at 2,000 × g for 10 min and the upper aqueous phase containing capsule was collected and lyophilized. Capsule was resuspended in sterile tissue culture grade water (Life Technologies, Grand Island, NY) before use.
Infection of BMDM with Ftt
BMDM were infected with various strains of Ftt as previously described (16). Briefly, bacteria were resuspended at a MOI = 50 in 250 µL complete DMEM (cDMEM, DMEM with glutamine, HEPES, and NEAA added [Life Technologies, Grand Island, NY], plus 10% heat inactivated FBS [Thermo Fisher, Waltham, MA]). Actual inoculum was confirmed by plating on MMH agar. Medium from BMDM was removed and reserved for replacement after infection. Bacteria were added to BMDM, incubated at 37°C for 90 min. Infected BMDM were subsequently incubated with gentamicin (50 µg/mL for 45 min at 37oC) to eliminate extracellular bacteria and washed three times with PBS. As indicated, in some experiments the gentamicin incubation step was eliminated. Where indicated BMDM were infected with bacteria utilizing centrifugation as previously described (18). Cells were washed three times with PBS and cultured in reserved cDMEM. R848 (Enzo Life Sciences, Farmingdale, NY) was added at 5 ng/ml either 90 min or 8 h after infection. Intracellular bacteria were enumerated at the designated time points as previously described (16). Briefly, BMDM were incubated with gentamicin as described above. Cells were washed three times followed by lysis with sterile water. Cell lysates were serially diluted and plated on modified Mueller Hinton (MMH) agar plates for enumeration of colonies. Eight or 16 h after infection cell lysates and culture supernatants were collected for assessment of mRNA, cellular protein and cytokines.
In vitro assessment of purified Ftt capsule
Ftt capsule was diluted in cDMEM, vortexed, and added to macrophages at the indicated concentrations. Water was diluted using the same scheme and was utilized as vehicle control (mock). Eighteen hours after addition of capsule, cells were stimulated with the following TLR agonists: 5 ng/ml Pam3Csk4 (TLR2) or 5 ng/ml R848 (TLR8) (both from Enzo Life Sciences). Eight or 16 h later cell lysates and culture supernatants were collected for assessment of mRNA, cellular protein and cytokines.
In vivo assessment of capsule deficient Francisella and purified capsule
Mice were anesthetized by intraperitoneal injection of 100 µl of 12.5 mg/ml ketamine + 3.8 mg/ml xylazine. Animals were intranasally inoculated with 50 CFU SchuS4, SchuS4Δ1238 or 10 µg purified Ftt capsule in 25µl PBS. Mice receiving PBS alone served as negative controls for infected or capsule treated mice. Sixteen hours later mice were anesthetized as described above and given 50 µg/25 µl R848 in PBS or 25 µl PBS alone (mock treated controls). Four hours later mice were euthanized, tracheas were exposed, cannulated with a disposable 18G catheter and airways were flushed with 0.5 ml PBS. Lavage fluid was collected for assessment of cytokines.
Detection of cell death and cytokines
Cell death was measured as release of lactate dehydrogenase (LDH) into culture supernatants using Cytotox 96 non-radioactive cytotoxicity kit according to the manufacturer’s instructions (Promega, Madison, WI). TNF-α, IL-6, IL-12p40 (all from BD Biosciences, San Jose, CA), CCL5 and CXCL1 (both from R&D Systems, Minneapolis, MN) present in cell culture supernatants or bronchoalveolar lavage fluid were quantitated using commercially available ELISA kits following manufacturer’s instructions.
Western blotting and qRT-PCR
At the indicated time points, medium was removed from BMDM. Cells were lysed in 150 µl 1X cell lysis buffer (Cell Signal Technologies, Danvers, MA)supplemented with PMSF (Sigma-Aldrich). Lysates were added to NuPAGE LDS Sample Buffer (Life Technologies), heated at 95°C for 10 min, homogenized by centrifugation using a QiaShredder (Qiagen, Valencia, CA) and immediately placed on ice prior to loading onto 4–12% SDS NuPAGE gradient gels (Life Technologies). Western blots were generated from SDS-NuPAGE gels as previously described (19). Blots were probed with antibodies to phospho-p44/42, p44/42, HIF-1α or β-actin (13E5), (all from Cell Signaling Technologies). RNA was purified using RNeasy kits (Qiagen), cDNA generated using the Superscript VILO kit (Life Technologies) and real-time quantitative PCR was run using primer/probe sets for pfkfb3 (ID number: Mm00504650_m1) (all from Life Technologies) and an ABI 7900HT (Life Technologies). Input RNA was normalized to HPRT. Fold change of the indicated genes as compared to untreated, uninfected controls was quantified as ΔΔCT.
Detection of Lactate
Lactate present in culture medium was quantitated using Amplex Red Glucose/Glucose Oxidase kit (Life Technologies) following manufacturer’s instructions, substituting lactate and lactate oxidase (both from Sigma-Aldrich, St. Louis, MO) for glucose and glucose oxidase.
Statistics
Statistical differences between two groups were determined using a two-tailed Student’s t test. For comparison between three or more groups, analysis was done by one-way ANOVA followed by Tukey’s multiple comparisons test. Significance using both methods was determined at p<0.05.
Results
Ftt capsule is required for replication and evasion of triggering host inflammatory responses
Previous reports have shown that presence of Ftt capsule impacts host cell death and replication efficiency of the bacterium, in a species dependent manner. For example, in human monocyte derived macrophages Ftt capsule mutants replicated exponentially over the first 16 hours of infection similar to wild type Ftt, followed by induction of dramatic host cell death that was not observed in wild type Ftt infected cells (12). Following infection of mouse BMDM, the capsule mutant bacteria also appeared to induce increased cell death distinct from that observed in wild type Ftt infected cells. However, in BMDM capsule deficient Ftt did not replicate over the first 24 hours (11). The contribution of capsule to evasion of triggering inflammatory responses and how this might contribute to bacterial replication and/or host cell death was not evaluated in either study. Thus, we first determined if we could replicate previous findings with regard to replication and cell death in addition to examining elicitation of cytokine production in BMDM infected with various Ftt capsule mutants.
In agreement with previous work, Ftt capsule mutants were nearly eliminated from the intracellular compartment of BMDM within the first 24 hours of infection. However, replication was partially restored over the last 48–72 hours infection (Figure 1A). Over the first 24 hours of infection none of the capsule mutants tested induced significant cell death. Rather, statistically significant differences in cell death among BMDM infected with capsule mutants compared with mock infected controls were not noted until 72 hours after infection (Figure 1B). In contrast to induction of cell death, capsule was required for evasion of induction of inflammatory responses as indicated by the significant increase in secretion of IL-12p40 and CCL5 among BMDM infected with Ftt capsule mutants compared to cells infected with wild type Ftt and mock infected controls (Figure 1C). There were small, but notable, differences in the ability of each mutant to trigger cytokine production. Δ1238 induced the greatest amount of both IL-12 and CCL5 in the first 24 hours after infection. By 48 hours after infection all capsule mutants were eliciting cytokine secretion (Figure 1C). Similar to our previous findings, wild type Ftt grew exponentially over the 72 hour culture period and did not induce detectable cytokine production until 72 hours after infection. However, this was paired with almost complete eradication of the cellular monolayer (unpublished observation). Thus, the direct contribution of microbial products versus massive cell death in wild type infected Ftt cultures was difficult to discern. In the last 72 hours, statistically significant cell death among all infected cells compared to uninfected cells was noted (Figure 1B).
Figure 1. Differential induction of pro-inflammatory cytokines by capsule deficient Francisella in mouse cells.
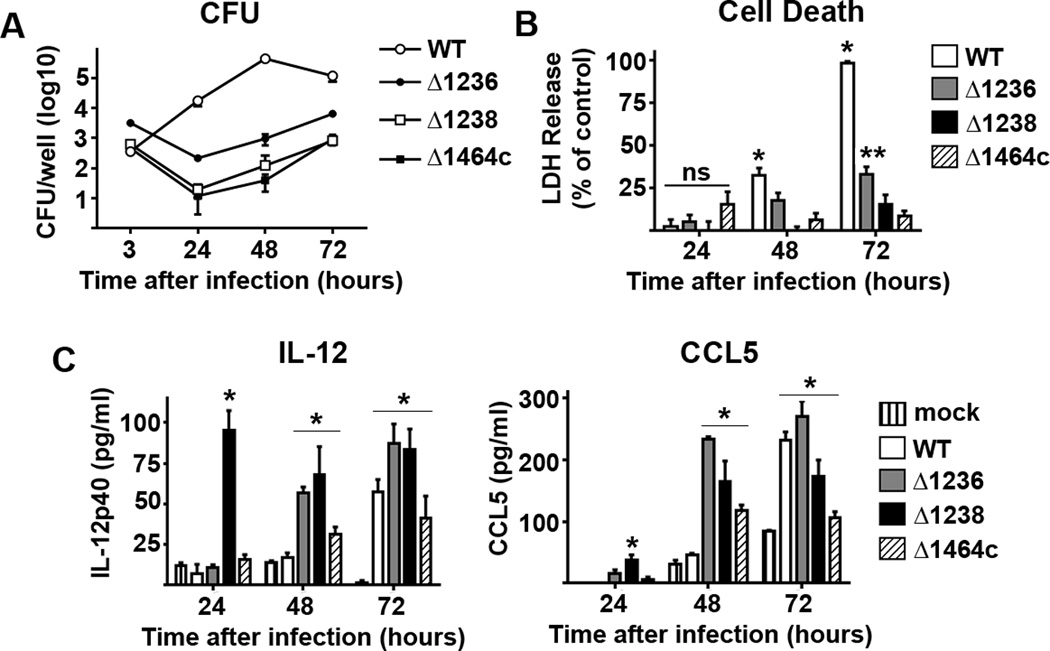
BMDM were infected with MOI = 50 of wild type F. tularensis SchuS4 (WT) or the indicated SchuS4 mutant. Each data point represents the mean of triplicate samples. (A) Viable intracellular bacteria were assessed at the indicated time points. (B) Cell death was quantitated using lactate dehydrogenase (LDH) release. (C) IL-12p40 and CCL5 secreted into culture supernatant was assessed 24 hours after infection. Error bars represent standard deviation (SD). BLD = below level of detection. ns = not significantly different. * =compared to all capsule mutant infected cells. ** = compared to Δ1238 and Δ1464c infected cells. *** = p<0.05 compared to uninfected (−) and WT infected cells. Statistical significance was determined following analysis by ANOVA. Data is representative of 4 experiments of similar design.
The early control followed by recovery of Ftt capsule mutant replication in combination with triggering early cytokine response was similar to findings following in vivo infection with these mutants. Yet, our results did not replicate previously published in vitro work with mouse derived cells (11). In the previous report the method of infection was different and involved centrifugation of BMDM with bacteria. It was possible that this difference in methodology contributed to the disparity in our findings. To confirm this hypothesis we performed infections in BMDM duplicating the method of infection described in the previous report. Using this technique, we observed early control of Ftt capsule mutant replication accompanied by significant host cell death (Supplemental Figure 1). We also observed that capsule mutants triggered cytokine secretion from BMDM similar to that observed using the infection method described in the present report. Therefore, the method of infection of BMDM altered the ability of Ftt capsule mutants to trigger cell death, but not cytokine production. Centrifugation of host cells can alter receptor expression and change the outcome of infection in other models of host-pathogen interaction (20). Our data suggest that a “passive” infection model, as opposed to centrifugation, may more closely mimic the interaction of Ftt with host cells in the in vivo environment, thus this method is used in the remainder of our report.
Ftt capsule required for suppression
The data presented above demonstrated that capsule is required for evasion of detection by macrophages early after infection. We hypothesized that capsule also contributes to the ability of Ftt to suppress induction of inflammatory responses. We first examined the ability of a capsule mutant to suppress cytokine secretion in macrophages early after infection that had also been treated with an unrelated secondary stimuli. Treating newly infected cells with gentamicin followed by washing with PBS resulted in elimination of nearly all of the capsule mutants within 24 hours of infection. Failure of these mutants to inhibit host cell responses could be a factor of too few bacteria present in the cell. Therefore, we modified our protocol to ensure that similar numbers of wild type and capsule mutant bacteria were present at the time of stimulation and that there was not significant cell death among infected macrophages as compared to uninfected controls (Figure 2A). We also selected Δ1238 to test as a representative capsule mutant. As expected, wild type Ftt infected cells treated with R848 or Pam3Csk4 (P3C) secreted significantly less IL-12p40 compared to treated, mock infected cells (Figure 2A). In contrast, despite similar numbers of intracellular bacteria, Δ1238 was unable to impair macrophage secretion of cytokine in response to R848 or P3C (Figure 2A).
Figure 2. Ftt capsule is required for inhibition of pro-inflammatory responses in vitro and in vivo.
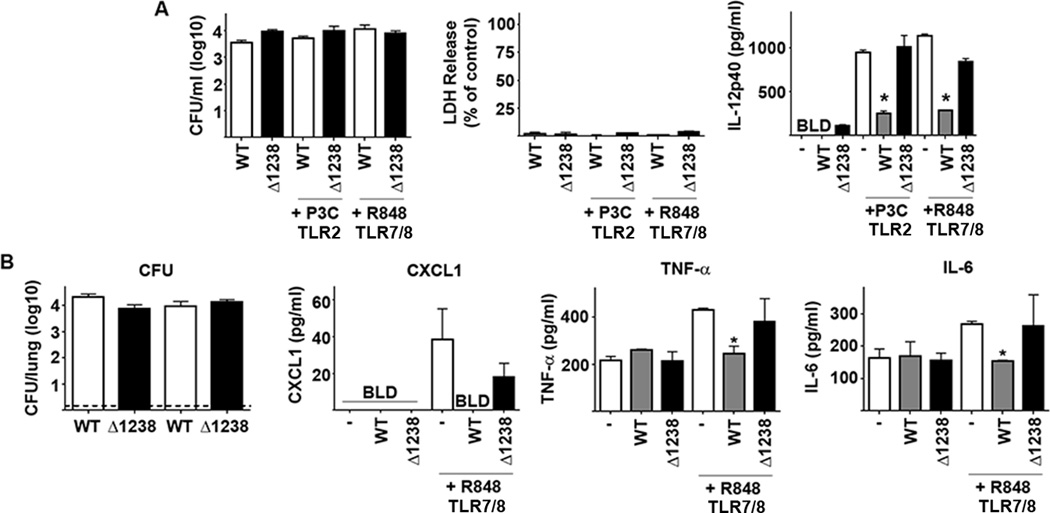
(A) BMDM were infected with MOI = 50 of wild type F. tularensis SchuS4 (WT) or SchuS4Δ1238 mutant (Δ1238). Mock infected and vehicle treated cells (−) served as negative and positive controls. Each data point represents the mean of triplicate samples. Immediately after infection bacteria were removed and cells were treated with Pam3CSK4 (P3C) or R848. Culture supernatants were assessed for IL-12p40 8 hours later. Error bars represent SD. BLD = below level of detection. Statistical significance was determined following analysis by performing student’s t-test comparing infected cells to the vehicle control. * = p<0.05 compared to vehicle treated cells. Data is representative of 3 experiments of similar design. (B) Mice (n=5/group) were intranasally inoculated with approximately 50 CFU SchuS4 (WT) or SchuS4Δ1238 mutant (Δ1238). Twenty-four hours after infection, mice were given 50 µg R848 intranasally and the airways were assessed for cytokine 4 hours later. Lungs were also assessed for bacterial load 4 hours after exposure to R848. Uninfected mice receiving vehicle controls (−) served as negative and positive controls. Error bars represent SD. BLD = below level of detection. * = p<0.05 compared to uninfected/vehicle treated and SchuS4Δ1238 infected controls. Data are representative of 4 experiments of similar design.
We also compared the ability of wild type and capsule mutant Ftt to suppress inflammatory responses in vivo. Similar numbers of wild type Ftt and Δ1238 were recovered from the lungs of infected mice (Figure 2B). As previously observed, wild type Ftt did not trigger inflammation within the first 24 hours of infection and the organism significantly impaired secretion of TNF-α and IL-6 in response to R848 (Figure 2B) (7). Animals infected with wild type Ftt also had less, but not significantly different, CXCL1 in their BAL following exposure to R848. Similar to wild type Ftt, Δ1238 did not induce pro-inflammatory cytokines following a low dose infection (Figure 2B). However, consistent with our in vitro data, the capsule mutant was unable to inhibit secretion of TNF-α, IL-6 and CXCL1 in response to R848 (Figure 2B). Together our data show that presence of capsule on viable bacteria was required for suppression of pro-inflammatory responses in vitro and in vivo.
We next confirmed that Ftt capsule, independent of intact bacteria, triggers a suppressive program in macrophages. We tested purified Ftt capsule for its ability to stimulate and/or suppress pro-inflammatory responses in macrophages. Capsule did not induce secretion of IL-12p40 from macrophages (Figure 3A). However, capsule treated cells secreted significantly less cytokine in response to R848 and Pam3Csk4 (Figure 3A). We then determined if purified capsule could inhibit production of cytokines in the airways in vivo. Similar to our in vitro findings capsule did not trigger production of pro-inflammatory cytokines in the airways (Figure 3B), nor did capsule induce an influx of inflammatory cells or depletion of alveolar macrophages (unpublished observations). Four hours after treatment with R848, no influx of inflammatory cells was observed in mock treated or capsule treated animals (unpublished observations), nor did capsule impair secretion of TNF-α or IL-6 into the airways following administration of R848. However, we did observe significantly less CXCL-1 in BAL fluid among capsule treated animals compared to mock controls that received R848 (Figure 3B). Thus, Ftt capsule recapitulates the ability of intact Ftt to suppress host cell inflammatory cytokine production in vitro and partially mimics this response in vivo.
Figure 3. Purified Ftt capsule inhibits pro-inflammatory responses in vitro and in vivo.
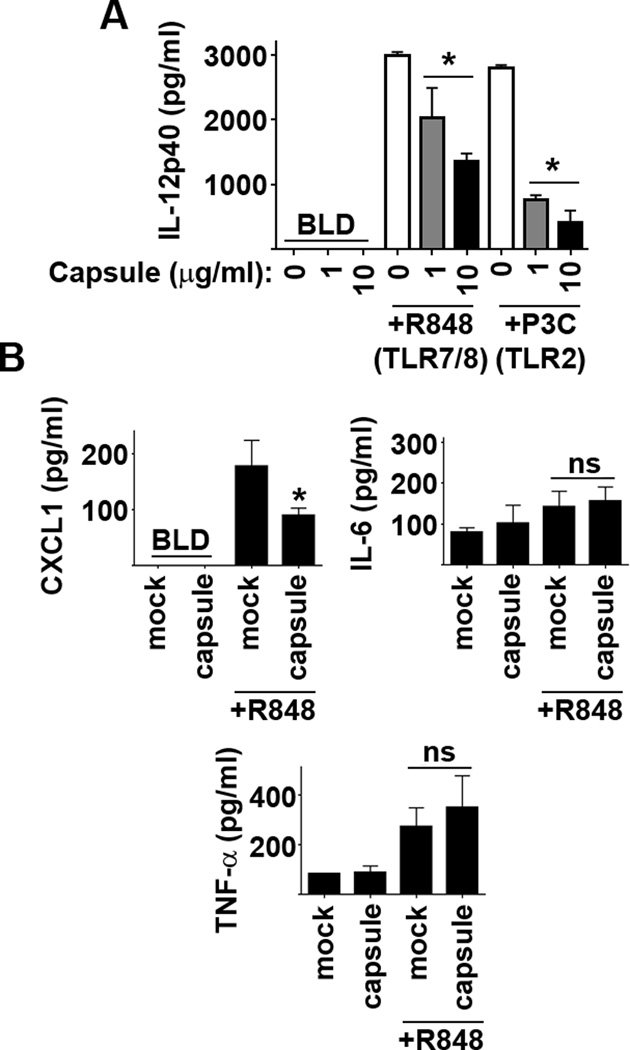
(A) BMDM were treated with the indicated concentration of purified SchuS4 capsule overnight followed by addition of TLR agonists. Vehicle (−) treated cells served as negative controls. Each data point represents the mean of triplicate samples. Culture supernatants were assessed for IL-12p40 18 hours later. Error bars represent SD. BLD = below level of detection. Statistical significance was determined following analysis by performing student’s t-test comparing infected cells to the vehicle control. * = p<0.05 compared to vehicle treated cells. Data is representative of 3 experiments of similar design. (B) Mice (n=5/group) were inoculated intranasally with 10µg purified SchuS4 capsule. Eighteen hours later mice were given 50 µg R848 intranasally and the airways were assessed for cytokine 4 hours later. Mice receiving vehicle controls (−) served as negative and positive controls. Error bars represent SD. BLD = below level of detection. Statistical significance was determined following analysis by ANOVA. * = p<0.05 compared to uninfected/vehicle treated controls. Data are representative of 4 experiments of similar design.
Purified capsule modulates host cell metabolism
Activation of macrophages, and secretion of many pro-inflammatory cytokines, is dependent on a shift in metabolism from oxidative phosphorylation to aerobic glycolysis [as reviewed, (1)]. We hypothesized that Ftt capsule may inhibit this shift as a mechanism to impair cytokine production by macrophages. Therefore, we examined the ability of Ftt capsule to modulate induction of host cell glycolysis in three ways. Lactate is a secreted product of aerobic glycolysis and is increased in the supernatant following activation of cells with TLR ligands (21). In agreement with these previous findings, R848 induced an increase in lactate secretion among macrophages and treatment with Ftt capsule significantly impaired this response (Figure 4A). A shift to aerobic glycolysis among host cells can also be monitored by the stabilization of the transcription factor Hypoxia Inducible Factor1-α (HIF1-α) and the genes it targets, i.e. pfkfb3 (21). Similar to our findings with lactate secretion, R848 increased stabilized HIF-1α and increased expression of pfkfb3 while Ftt capsule significantly inhibited each of these responses (Figure 4B and D). Translation of the HIF-1α gene and activation of HIF-1α are controlled, in part, by activation of the MAPK p44/42 (22). Thus, we also determined if capsule inhibited activation of p44/42 triggered by R848. Stimulation of BMM with R848 induced rapid phosphorylation of p44/42 and pretreatment of BMM with capsule inhibited this activation (Figure 4C).
Figure 4. Purified Ftt capsule impairs glycolysis in macrophages.
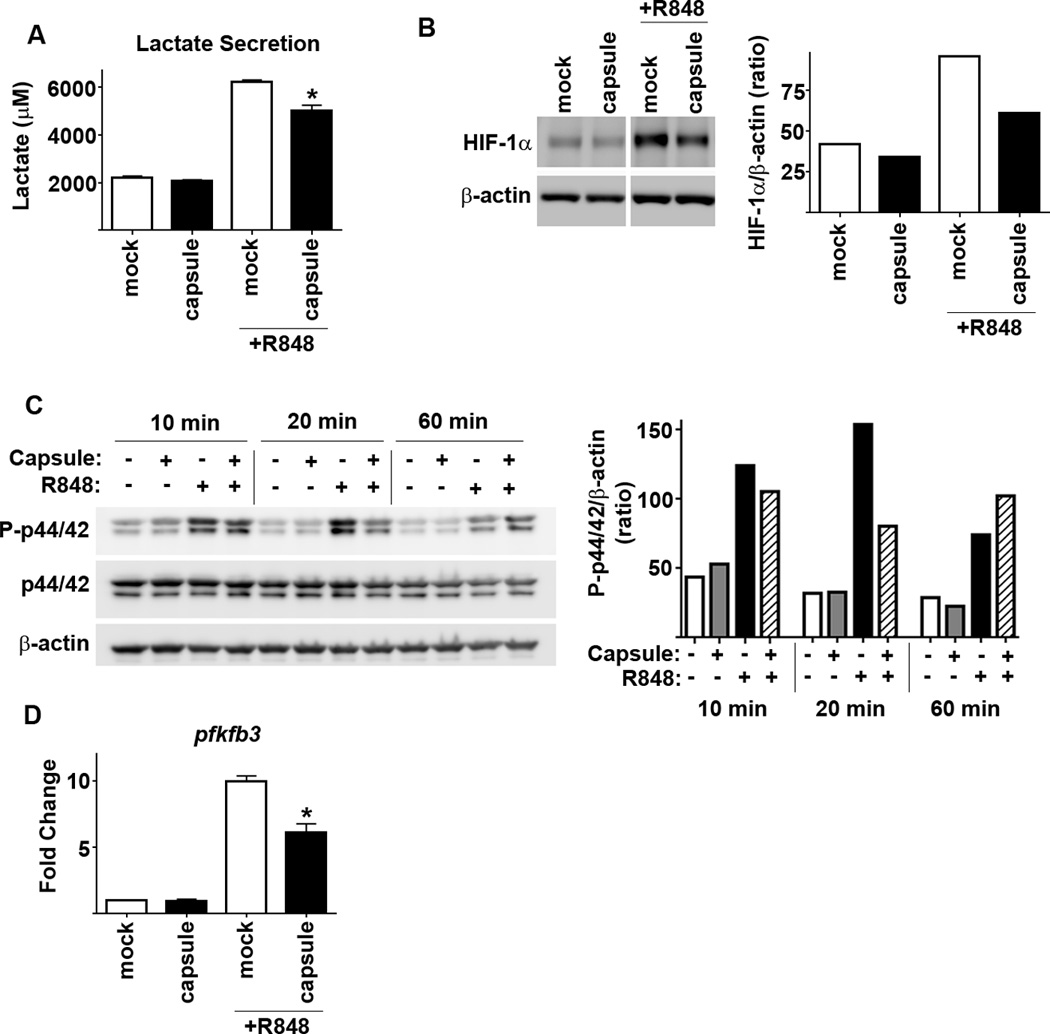
BMDM were treated with the indicated concentration of purified SchuS4 capsule overnight followed by addition of R848. Vehicle (−) treated cells served as negative controls. Each data point represents the mean of triplicate samples. (A) Culture supernatants were assessed for lactate 18 hours later. (B and D) Protein levels of HIF-1αand transcripts pfkfb3 mRNA and were quantitated 8 hours after addition of R848. (C) Protein levels of P-p44/42, p44/42 and were quantitated at the indicated time points after addition of R848. Protein levels of HIF-1α, P-p44/42, p44/42 and actin were obtained from pooling lysates from 2 individual wells/groups. Error bars represent SD. BLD = below level of detection. Statistical significance was determined following analysis by performing student’s t-test comparing infected cells to the vehicle control. * = p<0.05 compared to vehicle treated cells. Data are representative of 3 experiments of similar design.
To confirm that modulation of host cell metabolism by Ftt capsule was not a peculiarity of purified capsule, we also compared the ability of wild type Ftt and Δ1238 to induce and/or inhibit host cell glycolysis. Since the bacteria are also metabolically active we monitored the host cell specific markers of aerobic glycolysis including stabilization of HIF-1α, activation of p44/42 and transcription of pfkfb3. Consistent with its ability to evade triggering host cell processes, wild type Ftt did not significantly affect stabilization of HIF-1α or increase expression of pfkfb3 (Figure 5A and C). However, Ftt infection resulted in dephosphorylation of p44/42 in host cells (Figure 5B). In contrast, Δ1238 stabilized HIF-1α, did not inhibit phosphorylation of p44/42, and induced expression of pfkfb3 (Figure 5A–C). Following treatment with R848, wild type Ftt infected macrophages had significantly less HIF-1α and Ftt impaired R848 mediated phosphorylation (Figure 5D and E). Consistent with decreased HIF-1α, Ftt infected cells also had significantly fewer transcripts encoding pfkfb3 following R848 treatment compared to uninfected controls (Figure 5F). In contrast, Δ1238 infected cells had significantly increased expression of pfkfb3 upon stimulation with R848 compared to uninfected controls (Figure 5F), suggesting that Δ1238 was promoting, or unable to inhibit, the development of an activated glycolytic program among infected cells. Therefore, wild type Ftt modulates host cell metabolism by impairing a shift to aerobic glycolysis and capsule contributes to this process.
Figure 5. Capsule is required for evasion and suppression of induction of the host glycolytic program by Francisella.
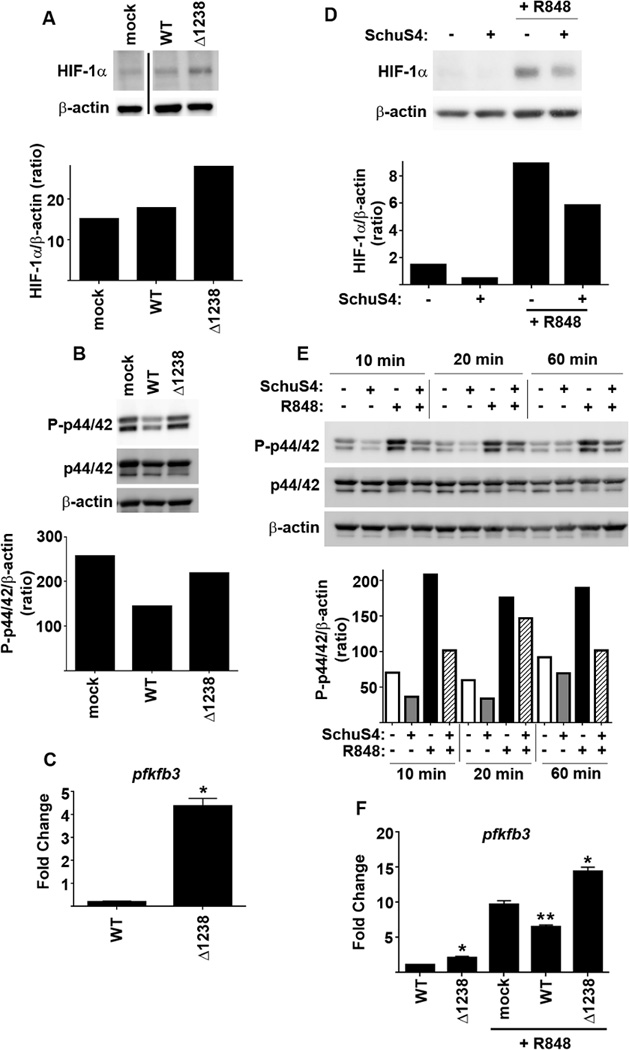
BMDM were infected with MOI = 50 of wild type F. tularensis SchuS4 (WT) or SchuS4Δ1238 mutant (Δ1238). Mock infected and vehicle treated cells (−) served as negative and positive controls. Eight hours after infection (A) protein levels of HIF-1α, (B) P-p44/42 and (C) pfkfb3 mRNA transcripts were quantitated. Cells were infected as described above and were treated with R848 either immediately (D) or 8 hours after infection (E–F). (D) Sixteen hours later protein levels of HIF-1α were quantitated. (E)P-p44/42 was assessed at the indicated time points after treatment with R848. (F) Four hours after R848 treatment pfkfb3 mRNA transcripts were quantitated. Protein levels of HIF-1α, P-p44/42, p44/42 and actin were obtained from pooling lysates from 2 individual wells/groups. Each data point assessing pfkfb3 mRNA transcripts represents the mean of triplicate samples. Error bars represent SD. BLD = below level of detection. Statistical significance was determined following analysis by ANOVA. * = p<0.05 compared to WT infected and mock treated cells. ** = p<0.05 compared to mock and Δ1238 infected cells treated with R848. Data are representative of 3 experiments of similar design.
Inhibition of aerobic glycolysis is required for optimal replication of Francisella
Induction of aerobic glycolysis alters both the intracellular pool of metabolites and induces pathways that could contribute to control of bacterial replication. The killing of Δ1238 during the first 24 hours of infection could be attributed to bacterial driven induction of aerobic glycolysis. Thus, we tested whether inhibition of aerobic glycolysis would restore replication of Δ1238. Since Ftt capsule independently inhibited host cell glycolysis we first determined if addition of capsule to Δ1238 infected cells would impact bacterial survival and production of IL-12p40. Addition of exogenous capsule resulted in significantly higher numbers of Δ1238 bacteria 24 hours after infection compared to mock treated controls (Figure 6A). Similarly, in the presence of capsule Δ1238 triggered significantly less IL-12p40 compared to mock treated, Δ1238 infected cells (Figure 6A). While the addition of capsule attenuated killing of Δ1238 over the first 24 hours of infection, it did not enhance replication of the bacteria. It was possible that inhibition of aerobic glycolysis mediated by the concentration of capsule was not sufficient to reverse inhibition of Δ1238 replication. Therefore, we repeated the experiments in the presence of a specific and potent inhibitor of aerobic glycolysis, 2-deoxyglucose (23). Addition of 2-DG had no effect on the ability of wild type Ftt to replicate nor was there any difference in cytokine production among wild type Ftt infected BMDM compared to vehicle controls (Figure 6B). In marked contrast, BMDM treated with 2-DG had statistically significantly more Δ1238 compared to mock treated controls (Figure 6B). Treatment of BMDM with 2-DG also resulted in less secretion of IL-12p40 by Δ1238 infected cells (Figure 6B).
Figure 6. Inhibition of glycolysis restores growth of capsule deficient Francisella.
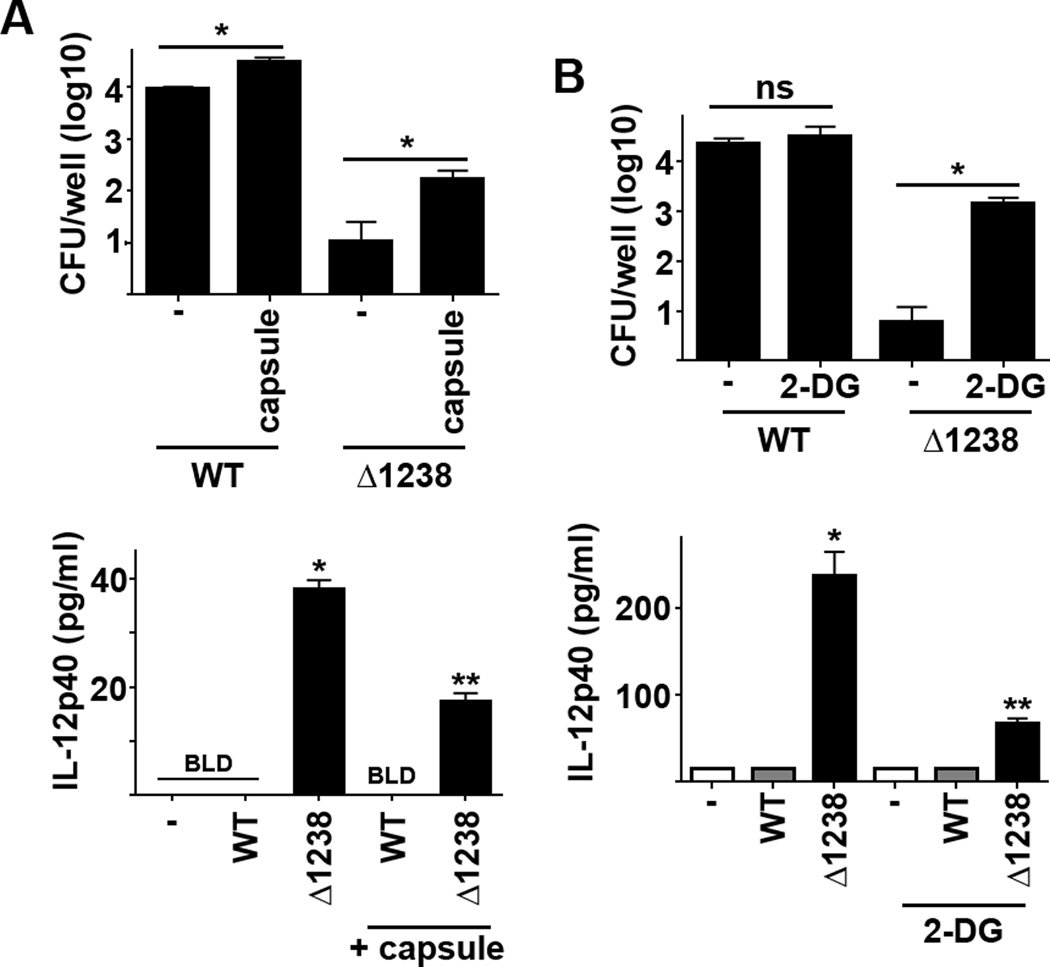
Two hours prior to infection BMDM were treated with (A) Ftt capsule (10 µg/ml) or (B) 2-DG (10 mM) or PBS (−). BMDM were infected with MOI = 50 of wild type F. tularensis SchuS4 (WT) or SchuS4Δ1238 mutant (Δ1238). Twenty-four hours later intracellular bacteria were enumerated and culture supernatants were assessed for IL-12p40. Each data point represents the mean of triplicate samples. Error bars represent SD. BLD = below level of detection. Statistical significance was determined following analysis by ANOVA. * = p<0.05 compared to vehicle treated cells. ** = p<0.05 compared to untreated Δ1238 infected cells. Data are representative of 3 experiments of similar design.
Discussion
Virulent F. tularensis causes a lethal disease following infection of host cells with as few as 1–2 bacteria per cell and 10–15 bacteria in the entire inoculum. Given this low infectious dose, it suggests that successful infection is greatly dependent on the ability of the bacteria to both evade and suppress anti-microbial responses in host cells from the moment of contact through the course of infection. The intersect of bacterial replication, manipulation of innate immunity and host metabolism have not been fully explored with regard to tularemia. Our current report provides evidence that Ftt capsule plays a key role in the early evasion and suppression of pro-inflammatory responses in vitro and in vivo and that this process, in part, involves modulating host cell metabolism.
Initially, it was not surprising that strains of Ftt lacking capsule were unable to evade detection by host cells. It is appreciated that one function of bacterial capsules is to shield immunogenic structures present on the surface of the microorganisms (14). Indeed, attenuated strains of Francisella lacking capsule triggered even higher pro-inflammatory responses than encapsulated wild type strains (24, 25). It is likely in our model Ftt capsule acts to shield immunogenic proteins present on the surface and it is the recognition of these proteins by host cells infected with capsule mutants that triggers the early transition to aerobic glycolysis. In contrast to the activity of capsules shielding structures on the surface of bacteria, there is less evidence for bacterial capsules directly dampening the host response as we observed here with purified Ftt capsule. The best example of a bacterial capsule triggering anti-inflammatory responses is the polysaccharide A (PSA) capsule derived from Bacteroides fragilis. However, the anti-inflammatory mechanism of this glycan lies not within direct impairment of macrophages or dendritic cells, but rather its ability to induce expansion of IL-10 producing CD4+ T cells following its presentation to T cells by dendritic cells (26). Thus, the ability of Ftt capsule to direct an anti-inflammatory program among macrophages in the absence of other cell types is a unique feature of virulence embodied by these bacteria.
After establishing that capsule played a role in inhibiting inflammatory responses in macrophages, we next determined the mechanism by which capsule executed this suppressive program. As a polysaccharide, Ftt capsule could have initiated anti-inflammatory responses in host cells following engagement of glycan binding receptors present on the cell surface. We analyzed the potential role of a wide variety of receptors known to interact with microbial carbohydrates including mannose receptor, scavenger receptor A, CD11b, Mincle and Dectin-1. However, none of these receptors were required for capsule mediated inhibition of pro-inflammatory responses in macrophages. We have previously established that Ftt lipids utilize TLR2 to antagonize general inflammatory responses in host cells (27). However, we confirmed that TLR2 was not required for capsule mediated inhibition of pro-inflammatory responses in macrophages (unpublished observations). Alternatively, Ftt capsule may non-specifically bind surface exposed TLRs inhibiting the ability of TLR agonists to interact with their cognate receptor. However, we routinely observed Ftt mediated inhibition of TLR7/8 signaling. These receptors are only found associated with acidic compartments within the host cytosol. The agonists for TLR7/8 are small nucleic acids. Thus, if capsule were covering these TLR receptors it would have to gain access to the cytosol and acidic compartments and mimic nucleic acids ability to bind to these receptors. In our opinion, it does not seem likely that a carbohydrate such as Ftt capsule would be able to fulfill both of these requirements for inhibiting TLR7/8 mediated signaling.
As an intracellular bacterium that replicates in the cytosol of the host cell, by default the Ftt capsule could engage any number of other host pathways that guide macrophage activation and production of pro-inflammatory cytokines. Transition in host cell metabolism is a key feature for induction of inflammation. Specifically, induction of aerobic glycolysis is a requirement for macrophages and dendritic cells to efficiently activate production of pro-inflammatory cytokines and anti-microbial molecules (28–30). Additionally, shifts in host cell metabolism also alter the composition of intracellular metabolites and other pathways such as autophagy that may impact survival of intracellular bacteria (31). Thus, we hypothesized that capsule mediated interference with induction of host cell aerobic glycolysis could explain both the inability of capsule deficient mutants to evade and suppress inflammatory responses as well as their survival. We established that both capsule and wild type Ftt impair induction of host aerobic glycolysis and provided evidence that a capsule mutant is incapable of this process. Importantly, supplementation of capsule to macrophages infected with capsule deficient Ftt partially restored bacterial replication and dampened the production of cytokine triggered by infection. It was not clear the mechanism by which Ftt capsule modulates the transition to aerobic glycolysis in host cells. The AKT/mTOR pathway has been implicated in the induction of aerobic glycolysis in cancer cells. It has been suggested that Ftt impairs mTOR activity as measured by phosphorylation of the mTOR target, S6 ribosomal protein (P-S6) (32). Thus, we examined activation of mTOR among cells infected with wild type versus capsule mutant Ftt. We did not observe significant changes in mTOR activity among wild type or capsule mutant infected BMM (unpublished observations). This suggested that suppression of aerobic glycolysis by wild type Ftt may be independent of the mTOR pathway. Alternatively to directly targeting host metabolic pathways, other bacterial capsules have been documented to act as mimics for host carbohydrates (33). It is possible that Ftt capsule is acting in that capacity herein. Specifically, Ftt capsule may engage specific host enzymes required to support aerobic glycolysis and as a consequence out compete the natural ligands.
As stated above, a shift in host cell aerobic glycolysis impacts the presence of anti-microbial effector molecules such as reactive oxygen species (ROS) (34). Thus, the early replication defect observed among capsule deficient mutants may be a result of increased presence of ROS. We examined the role of NADPH derived ROS utilizing gp91/nos2 deficient macrophages and found that absence of this molecule did not restore the ability of capsule mutants to replicate during the first 24 hours (unpublished observations). Mitochondrial ROS (mROS) is an additional source of ROS in host cells which is increased during aerobic glycolysis (35). We have previously established that enhanced susceptibility of attenuated F. novicida to mROS contributes to the ability of these organisms to promote inflammatory responses in macrophages (36). However, inhibition of mROS also reverses aerobic glycolysis (35). Therefore it is not currently possible to separate the role for mROS independent of other features of aerobic glycolysis that may affect Ftt replication.
In cancer cells and fibroblasts associated with tumors undergoing aerobic glycolysis, autophagy is increased. Autophagy is a critical part of maintenance of the general physiology of host cells, delivering spent or unnecessary cytosolic content to lysosomes for degradation. Autophagy also plays a role in control of intracellular pathogens (37). A recent report examining two capsule mutants of Ftt suggested that the impaired replication observed in mouse cells was due to failure of these mutants to evade being taken up into autophagic vacuoles (11). Although they did not observe an increase in autophagic vacuoles following infection with capsule mutants, it is possible that the method used to quantitate these compartments (microscopy) was not sensitive enough to detect differences triggered by capsule mutants versus wild type Ftt. Therefore, it is possible that the early defect in survival of capsule mutants in mouse macrophages is due to increased autophagy triggered by aerobic glycolysis. Alternatively, as the metabolic content of host cells shifts during aerobic glycolysis, host cell metabolites critical for Ftt replication may be depleted resulting in impaired replication of Ftt.
Regardless of the mechanism by which induction of aerobic glycolysis contributes to limiting Ftt replication, this process is transient and reflects the modest attenuation of Ftt capsule mutants in vivo. It is appreciated that sustained aerobic glycolysis can be lethal for macrophages and dendritic cells, thus we propose that the recovery phase of Ftt mutants observed at 48 and 72 hours after infection reflects another shift in host cell metabolism toward one that is more favorable for replication of Ftt. Moreover, we suggest that a similar phenomenon occurs in vivo. Specifically, Ftt capsule mutants trigger aerobic glycolysis upon invading macrophages resulting in a subset of the bacteria to be killed by the host cell. However, as the metabolism of the host cell shifts back to a resting oxidative phosphorylation the surviving bacteria are free to replicate which ultimately leads to a lethal outcome. Together our data suggest that identification of the specific mechanism by which induction of aerobic glycolysis controls Ftt replication will yield new strategies and targets for novel therapeutics aimed to limit Ftt and resolve infection.
Supplementary Material
Acknowledgments
The authors thank Dr. Chris Bosio for his helpful comments and review of this manuscript.
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
References
- 1.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease, C., and Prevention. Tularemia--United States, 1990–2000. MMWR. Morbidity and mortality weekly report. 2002;51:181–184. [PubMed] [Google Scholar]
- 4.Hestvik G, Warns-Petit E, Smith LA, Fox NJ, Uhlhorn H, Artois M, Hannant D, Hutchings MR, Mattsson R, Yon L, Gavier-Widen D. The status of tularemia in Europe in a one-health context: a review. Epidemiol Infect. 2015;143:2137–2160. doi: 10.1017/S0950268814002398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris S. Japanese biological warfare research on humans: a case study of microbiology and ethics. Ann N Y Acad Sci. 1992;666:21–52. doi: 10.1111/j.1749-6632.1992.tb38021.x. [DOI] [PubMed] [Google Scholar]
- 6.Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, Frelinger JA, Kawula TH. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect Immun. 2008;76:5843–5852. doi: 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosio CM, Bielefeldt-Ohmann H, Belisle JT. Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J Immunol. 2007;178:4538–4547. doi: 10.4049/jimmunol.178.7.4538. [DOI] [PubMed] [Google Scholar]
- 8.Chase JC, Celli J, Bosio CM. Direct and indirect impairment of human dendritic cell function by virulent Francisella tularensis Schu S4. Infect Immun. 2009;77:180–195. doi: 10.1128/IAI.00879-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayes CG, Burans JP, Ksiazek TG, Del Rosario RA, Miranda ME, Manaloto CR, Barrientos AB, Robles CG, Dayrit MM, Peters CJ. Outbreak of fatal illness among captive macaques in the Philippines caused by an Ebola-related filovirus. Am J Trop Med Hyg. 1992;46:664–671. doi: 10.4269/ajtmh.1992.46.664. [DOI] [PubMed] [Google Scholar]
- 10.Apicella MA, Post DM, Fowler AC, Jones BD, Rasmussen JA, Hunt JR, Imagawa S, Choudhury B, Inzana TJ, Maier TM, Frank DW, Zahrt TC, Chaloner K, Jennings MP, McLendon MK, Gibson BW. Identification, characterization and immunogenicity of an O-antigen capsular polysaccharide of Francisella tularensis. PLoS One. 2010;5:e11060. doi: 10.1371/journal.pone.0011060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Case ED, Chong A, Wehrly TD, Hansen B, Child R, Hwang S, Virgin HW, Celli J. The Francisella O-antigen mediates survival in the macrophage cytosol via autophagy avoidance. Cell Microbiol. 2014;16:862–877. doi: 10.1111/cmi.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindemann SR, Peng K, Long ME, Hunt JR, Apicella MA, Monack DM, Allen LA, Jones BD. Francisella tularensis Schu S4 O-antigen and capsule biosynthesis gene mutants induce early cell death in human macrophages. Infect Immun. 2011;79:581–594. doi: 10.1128/IAI.00863-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasmussen JA, Post DM, Gibson BW, Lindemann SR, Apicella MA, Meyerholz DK, Jones BD. Francisella tularensis Schu S4 lipopolysaccharide core sugar and O-antigen mutants are attenuated in a mouse model of tularemia. Infect Immun. 2014;82:1523–1539. doi: 10.1128/IAI.01640-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merino S, Tomas JA. Bacterial Capsules and Evasion of Immune Responses. eLife Science. 2015:1–10. [Google Scholar]
- 15.Sharma A, Qadri A. Vi polysaccharide of Salmonella typhi targets the prohibitin family of molecules in intestinal epithelial cells and suppresses early inflammatory responses. Proc Natl Acad Sci U S A. 2004;101:17492–17497. doi: 10.1073/pnas.0407536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffin AJ, Crane DD, Wehrly TD, Scott DP, Bosio CM. Alternative activation of macrophages and induction of arginase are not components of pathogenesis mediated by francisella species. PLoS One. 2013;8:e82096. doi: 10.1371/journal.pone.0082096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen JA, Fletcher JR, Long ME, Allen LA, Jones BD. Characterization of Francisella tularensis Schu S4 mutants identified from a transposon library screened for O-antigen and capsule deficiencies. Frontiers in microbiology. 2015;6:338. doi: 10.3389/fmicb.2015.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chong A, Wehrly TD, Nair V, Fischer ER, Barker JR, Klose KE, Celli J. The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect Immun. 2008;76:5488–5499. doi: 10.1128/IAI.00682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bauler TJ, Chase JC, Bosio CM. IFN-beta mediates suppression of IL-12p40 in human dendritic cells following infection with virulent Francisella tularensis. J Immunol. 2011;187:1845–1855. doi: 10.4049/jimmunol.1100377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo J, Wang W, Yu D, Wu Y. Spinoculation triggers dynamic actin and cofilin activity that facilitates HIV-1 infection of transformed and resting CD4 T cells. J Virol. 2011;85:9824–9833. doi: 10.1128/JVI.05170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, Cascante M, Bosca L. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185:605–614. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 22.Traves PG, de Atauri P, Marin S, Pimentel-Santillana M, Rodriguez-Prados JC, Marin de Mas I, Selivanov VA, Martin-Sanz P, Bosca L, Cascante M. Relevance of the MEK/ERK signaling pathway in the metabolism of activated macrophages: a metabolomic approach. J Immunol. 2012;188:1402–1410. doi: 10.4049/jimmunol.1101781. [DOI] [PubMed] [Google Scholar]
- 23.Barban S, Schulze HO. The effects of 2-deoxyglucose on the growth and metabolism of cultured human cells. J Biol Chem. 1961;236:1887–1890. [PubMed] [Google Scholar]
- 24.Okan NA, Chalabaev S, Kim TH, Fink A, Ross RA, Kasper DL. Kdo hydrolase is required for Francisella tularensis virulence and evasion of TLR2-mediated innate immunity. mBio. 2013;4:e00638-00612. doi: 10.1128/mBio.00638-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh A, Rahman T, Malik M, Hickey AJ, Leifer CA, Hazlett KR, Sellati TJ. Discordant results obtained with Francisella tularensis during in vitro and in vivo immunological studies are attributable to compromised bacterial structural integrity. PLoS One. 2013;8:e58513. doi: 10.1371/journal.pone.0058513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crane DD, Ireland R, Alinger JB, Small P, Bosio CM. Lipids derived from virulent Francisella tularensis broadly inhibit pulmonary inflammation via toll-like receptor 2 and peroxisome proliferator-activated receptor alpha. Clin Vaccine Immunol. 2013;20:1531–1540. doi: 10.1128/CVI.00319-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, Artyomov MN, Jones RG, Pearce EL, Pearce EJ. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15:323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vander Heiden M, Xavier RJ, O'Neill LA. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell metabolism. 2015;21:65–80. doi: 10.1016/j.cmet.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O'Neill LA. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duan L, Perez RE, Davaadelger B, Dedkova EN, Blatter LA, Maki CG. p53-regulated autophagy is controlled by glycolysis and determines cell fate. Oncotarget. 2015;6:23135–23156. doi: 10.18632/oncotarget.5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steele S, Brunton J, Ziehr B, Taft-Benz S, Moorman N, Kawula T. Francisella tularensis harvests nutrients derived via ATG5-independent autophagy to support intracellular growth. PLoS Pathog. 2013;9:e1003562. doi: 10.1371/journal.ppat.1003562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cress BF, Englaender JA, He W, Kasper D, Linhardt RJ, Koffas MA. Masquerading microbial pathogens: capsular polysaccharides mimic host-tissue molecules. FEMS Microbiol Rev. 2014;38:660–697. doi: 10.1111/1574-6976.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 35.Nazarewicz RR, Dikalova A, Bikineyeva A, Ivanov S, Kirilyuk IA, Grigor'ev IA, Dikalov SI. Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxidants & redox signaling. 2013;19:344–349. doi: 10.1089/ars.2013.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crane DD, Bauler TJ, Wehrly TD, Bosio CM. Mitochondrial ROS potentiates indirect activation of the AIM2 inflammasome. Frontiers in microbiology. 2014;5:438. doi: 10.3389/fmicb.2014.00438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jo EK, Yuk JM, Shin DM, Sasakawa C. Roles of autophagy in elimination of intracellular bacterial pathogens. Frontiers in immunology. 2013;4:97. doi: 10.3389/fimmu.2013.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.