

Abstract
Influenza A virus gene segment 7 encodes two proteins: the M1 protein translated from unspliced mRNA and the M2 protein produced by mRNA splicing and largely encoded by the M1 +1 reading frame. To better understand the generation of defective ribosomal products (DRiPs) relevant to MHC class I antigen presentation, we engineered influenza A virus gene segment 7 to encode the model H-2 Kb class I peptide ligand SIINFEKL at the M2 protein C-terminus. Remarkably, after treating virus-infected cells with the RNA splicing inhibitor spliceostatin A to prevent M2 mRNA generation, Kb-SIINFEKL complexes were still presented on the cell surface at levels of up to 60% of untreated cells. Three key findings indicate that SIINFEKL is produced by cytoplasmic translation of unspliced M1 mRNA initiating at CUG codons within the +1 reading frame:
Synonymous mutation of CUG codons in the M2-reading frame reduced Kb-SIINFEKL generation.
Kb-SIINFEKL generation was not affected by drug-mediated inhibition of AUG-initiated M1 synthesis.
Kb-SIINFEKL was generated in vitro and in vivo from mRNA synthesized in the cytoplasm by vaccinia virus, and hence cannot be spliced.
These findings define a viral DRiP generated by cytoplasmic non-canonical translation and demonstrate the participation of CUG-codon-based translation initiation in pathogen immunosurveillance.
Introduction
CD8+ T cells play central roles in immunosurveillance of transplants, tumors, and intracellular microbes, including viruses. Antiviral CD8+ T cells recognize virus-encoded oligopeptides bound to MHC class I molecules and presented on the infected-cell surface. Peptides are typically generated by proteasomal cleavage of viral proteins in the cytoplasm or nucleus. They are then transported by the TAP peptide transporter into the endoplasmic reticulum where they are trimmed and loaded onto waiting class I molecules in a exquisitely choreographed molecular ballet (1). After peptide binding, class I molecules are exported from the ER through the Golgi complex to the cell surface for immunosurveillance.
The entire process of peptide creation, loading and MHC complex delivery to the cell surface can occur with surprising speed. Antiviral T cells can recognize viral peptides within 45 min of cellular infection, despite the stability of the corresponding source full-length viral gene product (2). This observation spawned the defective ribosomal product (DRiP) hypothesis of peptide generation, which posits that the class I system exploits a distinct pool of metabolically unstable translation products for immunosurveillance (3).
Considerable experimental evidence from numerous approaches indicate that DRiPs are a major contributor to immunosurveillance (4), including recent mass spectrometry studies that elegantly demonstrate the disparate kinetics between peptide generation and degradation of the full-length source protein (5, 6). Myriad translational mechanisms can generate DRiPs (4, 7–9), including: (i) degradation of misfolded or mistargeted full-length proteins; (ii) overproduction of a polypeptide relative to the expression of its normal interaction partner(s); (iii) truncation due to mistranslation (i.e. frame shifting, alternative initiation on CUG codons or downstream AUG); (iv) non-canonical translation in the nucleus of immature and mature mRNA (10, 11).
Despite the wide variety of studies that support DRiPs as a major source of class I peptide ligands, the mechanisms involved in the generation of DRiPs are poorly defined outside of experimental systems in which DRiPs are artificially created. A problem inherent to characterizing DRiPs is the high ratio of viral proteins synthesized by infected cells (typically in the range of 105 to 107 copies per cell) vs. the number of complexes presented on the cell surface (typically in the range of 101 to 103 copies per cell). The roughly 10,000-fold ratio of native protein to class I peptide complex makes biochemical analysis difficult. Since complexes can be generated from viral gene products with an efficiency at least as high as 2.5%, antigenically relevant DRiPs can represent only a small fraction of the synthesis of a given gene product. For example, at an efficiency of 2.5%, 1000 complexes would derive from 40,000 substrates, which represent just 4% of the total pool of 106 native viral proteins synthesized. These circumstances favor experimental strategies based on genetic or chemical manipulation of source antigen expression.
Here, we study the generation of a specific model peptide (SIINFEKL) from DRiPs encoded by influenza A virus (IAV) mRNA in a context that allows us to dissect the contributions of standard vs. alternative reading frames and initiation codons. SIINFEKL forms highly stable complexes with the mouse Kb class I molecule, enabling detection by the 25-D1.16 mAb (12) or by OT-I transgenic T cells (13) via T cell activation assays in vitro and in vivo. Our findings demonstrate the contribution of non-canonical CUG-codon-based translation initiation to antiviral CD8+ T cell immunosurveillance.
Materials and Methods
Cell culture, reagents and antibodies
L929 cells (ATCC) and HeLa cells from American Type Culture Collection stably transfected with a cDNA encoding the MHC class I Kb molecule were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 7.5% fetal bovine serum (FBS) at 37 °C with 9% CO2. 293T and MDCK cells from American Type Culture Collection were propagated in DMEM supplemented with 10% FBS at 37 °C with 9% CO2.
Spliceostatin A was synthesized as described (14). Cycloheximide (Sigma), anti-β-actin antibody (Sigma), MG132 (Calbiochem) were obtained from vendors. Anti-SIINFEKL C-terminal antibody (1F10.2.2) was obtained from Kenneth Rock (U Mass Med). Anti-IAV mAbs were described previously (15): anti-HA, H36-26; anti-NA, NA2-1C1; anti-M1, M2-1C6; anti-M2, O19 (16). The anti-NA rabbit polyclonal Ab used for western blotting was described (17). For flow cytometry, FITC anti-mouse H-2Kb (clone#AF6-88.5) was from BD Biosciences. MAb 25D1.16 (anti-Kb-SIINFEKL) and M2-1C6 were labeled with Alexa Fluor 647 (Life Technologies). NA2-1C1 was labeled with Pacific Blue (Life Technologies). H36-26 and O19 were labeled with Alexa Fluor 488 (Life Technologies).
Recombinant IAV PR8 virus construction and infection
PR8-M2-SIIN, PR8-M2(24)-SIIN, PR8-M2(45)-SIIN, PR8-M2(47)-SIIN, PR8-M2-C888T-SIIN, PR8-M2-M72L-SIIN, PR8-M2-G1001A-SIIN and PR8-M2-C888T-G1001A-SIIN were generated by 8 plasmid transfection into a mixture of 293T and MDCK cells as described (18). The rescue plasmids pDZ-PR8(M2-SIIN), pDZ-PR8(M2-(24)SIIN), pDZ-PR8(M2-(45)SIIN), pDZ-PR8(M2-(47)SIIN), pDZ-PR8(M2-C888T-SIIN), pDZ-PR8 (M2-M72L-SIIN), pDZ-PR8(M2-G1001A-SIIN) and pDZ-PR8(M2-C888T-G1001A-SIIN) were cloned by PCR mutagenesis of PR8 segment 7. Rescued viruses were propagated in 10-day embryonated chicken eggs and sequenced to confirm their identity. IAV titers were determined by standard 50% tissue culture infective dose assay using MDCK cells. For PR8 infection, cells were infected at an MOI of 3–10 for 1 h at 37 °C with mixing in DMEM without FBS. Then cells were washed and treated with inhibitors as specified for indicated times in growth medium. For overnight infection, cells were infected at an MOI of 0.2–0.5 to minimize cell death.
Flow cytometry
For measuring cell surface proteins, L-Kb cells were incubated at 4°C for 30 min with fluorescently tagged antibodies at various times post-infectin (p.i.) After three washes with HBSS/BSA, cells were analyzed using a BD LSR Π flow cytometry (BD Biosciences) and Flowjo software (Tree Star). To measure intracellular M1, cells were fixed in 3% paraformaldehyde with 10 mM HEPES in PBS, washed with PBS and incubated with Alexa Fluor 647-conjugated M2-1C6 in PBS containing 1% bovine calf serum and 0.5% saponin.
Mice
Pathogen-free C57BL/6 were acquired from Taconic. OT-I TCR transgenic mice were bred in-house and crossed to mice ubiquitously expressing dsRed (Jackson Laboratories stock #5441) to create OT-I dsRed mice. 6–12-wk-old adult mice were used in all experiments. All mice were housed under specific pathogen–free conditions (including MNV, MPV, and MHV) and maintained on standard rodent chow and water supplied ad libitum.
OT-I experiments
OT-I CD8+ T cells were purified using an AutoMacs and a negative selection kit (Miltenyi Biotech) from OT-I dsRed mice to ~95% purity. Approximately 5 x 105 cells were adoptively transferred iv. into C57Bl/6 recipients. Mice were infected i.p. with 1 x 106 pfu of the indicated virus. Five days p.i., spleens were removed and restimulated for 4 hours with either 100 nM irrelevant or SIINFEKL peptide. Brefeldin A (10 μg/ml, Sigma-Aldrich) was added during restimulation. After incubation at 37°C, cells were stained for CD8, fixed for 20 min with 1% paraformaldehyde, washed, and then stained with anti-IFN-γ antibody (clone XMG1.2, eBioscience) in 0.5% saponin overnight at 4°C.
Western blotting
Cells were lysed in 50 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS supplemented with protease inhibitors (Roche). Lysates were separated on 4–12% PAGE gels and transferred to nitrocellulose. Membranes were probed with primary antibodies, followed by anti-rabbit or anti-mouse secondary Abs coupled to the IR dyes 680 or 800CW. Membranes were analyzed using an Odyssey infrared imager (LI-COR).
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (GraphPad, San Diego, CA). Error bars in graphs show standard deviations.
Ethics Statement
All animal experiments were conducted in accordance with the USA Animal Welfare Act and the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All procedures were approved by the NIAID Animal Care and Use Committee (protocol LVD 37E).
Results
Generation and initial characterization of PR8 M2-SIIN
We generated a recombinant PR8 influenza A virus (IAV) encoding SIINFEKL appended in-frame to the C-terminus of the viral M2 protein (PR8 M2-SIIN) (schematic shown in Fig. 1). Due to the nuclear localization of IAV mRNA synthesis, the virus can exploit cellular splicing machinery to increase its repertoire of gene products. M2 is a type I integral membrane protein abundantly expressed on infected cell surfaces and is one of two influenza proteins (the other being NEP) generated by splicing (19). The M2 mRNA is generated by splicing of the M1 mRNA to create a protein consisting of the amino terminal 17 amino acids of M1 fused to 80 amino acids coded by the +1 reading frame of M1, extending past the M1 stop codon (Fig. 1A).
Fig. 1. Characterization of PR8-M2-SIIN infection.

(A) Genetic structure of M1 and M2 RNA with post-splice site sequence of M2. SIINFEKL encoding nucleotides were genetically inserted (green arrows) after the COOH terminus (used for most experiments in the study) or residues 24,45, and 47 (used in experiments shown in Figure 4 to gain insight to the manner of SIINFEKL generation). Red residues indicate potential alternative M1 +1 frame downstream initiation codons: LeuCUG or Met.
(B) Flow cytometric analysis of cell surface levels of M2 (red) and Kb-SIIN (black) in L-Kb cells after PR8 or PR8-M2-SIIN infection for up to 4 h.
(C) Immunoblot analysis of M1, M2 or PB1-F2 in lysates of L-Kb cells treated with cycloheximide (CHX) (25 μg/ml) for indicated hours after 12 h infection with PR8-M2-SIIN
(D) L-Kb cells were infected with PR8-M2-SIIN, then SSA (100 ng/ml) or vehicle control was immediately added. Levels of cell surface M2 or intracellular M1 detected by cognate mAbs were determined at indicated times p.i. by flow cytometry, with relative amounts plotted compared to maximum expression for vehicle control (set at 100%).
(E) Immunoblot of NA, M1 and M2 in L-Kb cells infected with PR8-M2-SIIN for 20 h in the presence of SSA (100 ng/ml) or vehicle control. β-actin signal serves as a loading control.
Data are representative of at least three experiments.
PR8 M2-SIIN replicated robustly in eggs or MDCK cells, consistent with a prior study that M2 function is not significantly affected by short C-terminal extensions (20). We confirmed M2-SIIN expression using flow cytometry of infected L-Kb cells (L cells permanently transfected to express the Kb) after staining with Abs recognizing Kb-SIINFEKL complexes (25-D-1.16) or against M2 protein and comparing to cells infected with wild-type IAV PR8 (Fig. S1A and S1C). Immunoblotting cell lysates 12 hrs post-infection (p.i.) with a SIINFEKL-specific mAb also revealed M2 SIINFEKL expression (21) (Fig. S1B).
To determine the contribution of DRiPs to Kb-SIINFEKL generation from M2-SIIN, we measured the temporal kinetics of Kb-SIINFEKL expression using the 25-D1.16 mAb (Fig. 1B). This revealed a strong kinetic connection between the cell surface expression of M2 and Kb-SIINFEKL. As expected, presentation was completely inhibited by incubating cells with the protein synthesis inhibitor cycloheximide (CHX) at the time of infection to prevent viral protein synthesis (Fig. S1C). This demonstrated that Kb-SIINFEKL complexes were generated from nascent viral proteins and not the small amounts of M2 present on incoming virions (~ 5 copies per virion (22)). Immunoblotting infected cells treated with CHX showed that the M2-SIIN fusion protein is highly stable (Fig. 1C), similar to M1 (top row) but distinct from the IAV protein PB1-F2 (third row), which possesses a major rapidly degraded cohort (23). If Kb-SIINFEKL complexes were generated from standard turnover of “retirees,” i.e. native proteins being degraded after reaching their predetermined life spans, presentation should begin only as degraded M2-SIIN protein accumulates. Instead, maximal Kb-SIINFEKL formation closely tracked with M2 synthesis (Fig. 1B), clearly indicating that Kb-SIINFEKL complexes are predominantly generated from a rapidly degraded cohort of DRiP antigenic precursors.
Spliceostatin A prevents M2 expression but still allows Kb-SIINFEKL generation
Since M1 mRNA splicing generates M2 mRNA, SIINFEKL can potentially be produced from either M2 mRNA in the proper reading frame, or M1 mRNA in the +1 reading frame downstream of the normal stop codon. To examine the contributions of M1 vs. M2 mRNA to Kb-SIINFEKL expression, we treated cells with spliceostatin A (SSA), a small, cell permeable natural product that blocks mRNA splicing by binding to splicing factor 3b (14, 24). SSA was highly effective in preventing splicing of M1 mRNA, as indicated by 95% or greater inhibition of M2 expression and a concomitant increase in M1 expression (Fig. 1D, E).
We next examined the effect of SSA treatment on antigen (Ag) presentation (Fig. 2). As a control for SSA specificity on M2 synthesis inhibition, we infected cells with a commonly used recombinant PR8 IAV with SIINFEKL inserted into the stalk region of neuraminidase (NA) (NA-SIIN), an IAV protein that is not translated from a spliced mRNA (25). Treating NA-SIIN infected cells with SSA did not inhibit NA expression as determined by immunoblotting (Fig. 1E), and enhanced cell surface NA expression as determined by flow cytometry (Fig. 2D).
Fig. 2. Kb-SIIN generation persists despite near complete SSA-inhibition of M2 expression.

(A) Flow cytometric analysis of cell surface levels of M2 (red) and Kb-SIINFEKL (black) in L-Kb cells at indicated time after infection with PR8 M2-SIIN in the presence of SSA (squares) or vehicle control. 100% = 5 h vehicle levels.
(B) 25D1.16/M2 ratio calculated from experiment in panel A.
(C) Graph showing ratios of M2 or SIINFEKL expression as determined by flow cytometry 20 h p.i. with PR8 M2-SIIN of HeLa-Kb cells.
(D, E) As in panel A and B, but using PR8 NA-SIIN to infect L-Kb cells.
All experiments were performed at least three times.
We next examined the effect of SSA on the generation of Kb-SIINFEKL complexes from M2-SIIN. Despite near total inhibition of M2 synthesis in SSA-treated L-Kb (Fig. 2A–C) or HeLa-Kb (Fig. 2D–E) cells (permanent HeLa cell transfectants expressing Kb), Kb-SIINFEKL was expressed between 25% and 60% of levels in untreated cells. In L-Kb cells, the net affect of SSA was to increase the ratio of Kb-SIINFEKL complexes to M2 approximately 10-fold (Fig. 2B–C). In contrast, SSA had little effect on Kb-SIINFEKL complex generation in cells infected with the control NA-SIIN, decreasing the ratio of Kb-SIINFEKL complex/NA due to the enhanced expression of NA (Fig. 2E).
These findings demonstrate that in the presence of SSA, Kb-SIINFEKL generation occurs independently of M2 biosynthesis. Based on this finding, we hypothesized that when SSA blocks M2 mRNA biogenesis, SIINFEKL is largely generated from the +1 reading frame of the M1 mRNA via frame shifting or alternative initiation.
SIINFEKL is generated from cytoplasmic translation of M1 mRNA
Antigenic peptides can be generated from translation within the nucleus (10, 11). Previously, we demonstrated that preventing nuclear export of IAV-encoded NA-SIIN mRNA by treating cells with the RNA polymerase II inhibitor 5,6-dichloro-1-beta-D-ribofuranosyl benzimidazole (DRB) inhibited NA biosynthesis but had little impact on Kb-SIINFEKL complex generation, suggesting that SINFEKL peptides were produced from nuclear translation of NA-SIIN mRNA (10). Could aberrant translation of M1 mRNA in the nucleus account for the generation of Kb-SIINFEKL complexes in SSA-treated cells?
To explore this possibility, we characterized the nuclear vs. cytoplasmic localization of M2 and M1 mRNA under various conditions (Fig. 3). Quantitative PCR confirmed that SSA nearly completely prevented M2 mRNA generation, concomitantly increasing both nuclear (detergent insoluble) and cytoplasmic (detergent soluble) M1 mRNA (Fig. 3A), accounting for the effects of SSA on M1 and M2 protein levels.
Fig. 3. SIINFEKL derives from cytoplasmic translation in SSA-treated cells.
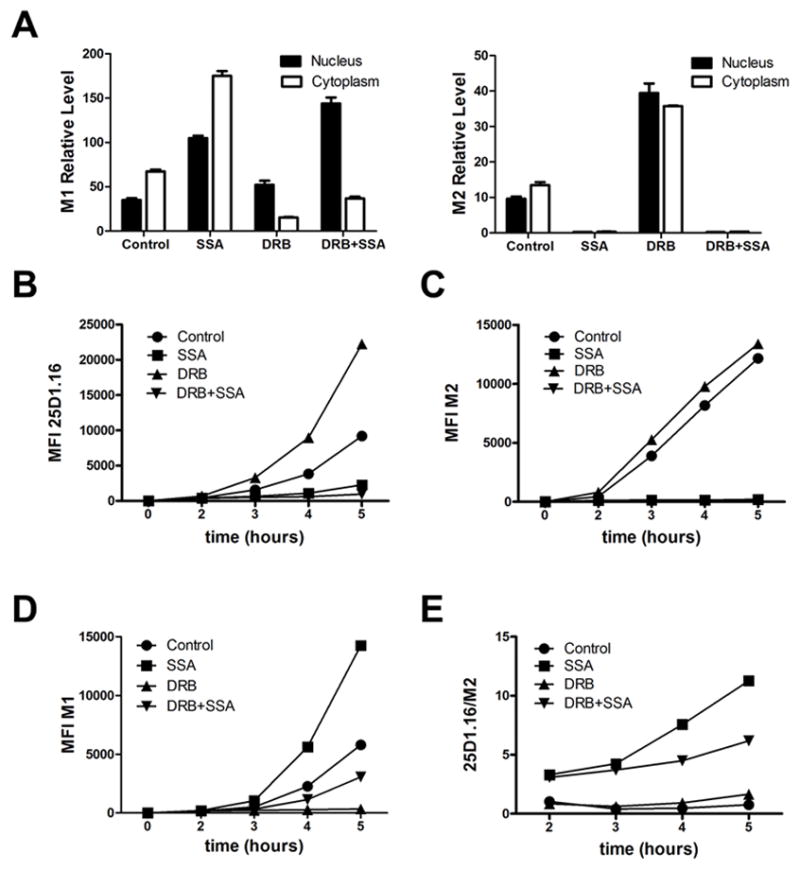
L-Kb cells infected with PR8-M2-SIIN in the presence of SSA (100 ng/ml), DRB (150 μM) or vehicle control for various times.
(A) M1 and M2 mRNA levels present 4 h p.i. in cytosol vs. nucleus determined by real-time PCR, normalizing levels of M2 and M1 mRNA to GAPDH mRNA to control for differential recovery of mRNA from samples. (B–D) Flow cytometric analysis of L-Kb cell surface levels of Kb-SIIN determined by 25D-1.16 staining (B), M2 (C), and intracellular M1 levels (D) at the indicated time p.i. (E) Kb-SIIN/M2 ratio calculated from experiment shown in C and D.
Data are representative of three independent experiments.
As reported by Amorim et al. (26), DRB nearly completely inhibited M1 expression as determined by intracellular flow cytometry (Fig. 3D), which correlated with reduced cytoplasmic M1 mRNA levels (Fig. 3A, left panel). Concomitantly, DRB enhanced M2 mRNA levels in both nucleus and cytoplasm by approximately 4-fold (Fig. 3A, right panel). DRB treatment increased Kb-SIINFEKL expression to a similar extent (Fig. 3B). Curiously, M2 cell surface expression was only slightly increased (Fig. 3C).
DRB did not interfere with the ability of SSA to block M2 mRNA generation and M2 synthesis (Fig. 3A–C). Importantly, Kb-SIINFEKL expression paralleled the decreased amount of M1 protein synthesized, tracking with the diminished cytoplasmic levels of M1 RNA. This is consistent with the conclusion that under SSA blockade of M2 mRNA biogenesis, Kb-SIINFEKL complexes are generated from cytoplasmic translation of M1 RNA in the +1 reading frame.
Kb-SIINFEKL generation from M1 mRNA is highly dependent on SIINFEKL location in SSA-treated Cells
To better understand the generation of Kb-SIINFEKL complexes from M1-M2 mRNAs in SSA-treated cells, we inserted the SIINFEKL coding sequence into different regions of the mRNA in the M2 reading frame. Placing SIINFEKL in frame after M2 residues 24, 45, or 47 (Fig. 1A) maintained M2 function, allowing robust virus growth with strong M2 surface expression (Fig. 4). Additionally, insertion in these positions supported the robust generation of Kb-SIINFEKL complexes, although the ratio of M2 to Kb-SIINFEKL varied, likely due to expected the influence of flanking sequences on peptide liberation (27, 28). Indeed, locating SIINFEKL after positions 24 and 45 was a more efficient source of Kb-SIINFEKL complexes than C-terminal SIINFEKL (Fig. 4).
Fig. 4. Generation of Kb-SIINFEKL complexes from unspliced M1 mRNA is highly dependent on SIINFEKL position in the M2 reading frame.

Flow cytometric analysis of M2 and Kb-SIINFEKL expression on live L-Kb cells infected for the indicated time with PR8 viruses with SIINFEKL inserted after the indicated M2-residue (see insertions depicted in Figure 1A). For comparison, M2-SIIN is shown (orange), with the standard carboxy-terminal M2 insertion of SIINFEKL. Data are representative of three experiments.
Remarkably, after treating infected cells with SSA, little to no cell surface Kb-SIINFEKL complexes were generated from M2 with SIINFEKL at any of the upstream locations, despite enhanced generation in the absence of SSA. This implies that in SSA treated-cells, translation initiation in the +1 reading frame of the M1 mRNA downstream of M2 residue 47 can generate SIINFEKL when the peptide is encoded at the M2 C-terminus.
Kb-SIINFEKL complexes are generated from CUG-Initiation of M1 mRNA in SSA-treated Cells
Viral peptides can be produced from non-canonical downstream AUG initiation sites in both standard and alternative reading frames (29–32). Furthermore, the elegant studies of Shastri and colleagues demonstrate the contribution of class I peptide-ligand generation from CUG-codon initiation (33–35). This work presaged recent ribosome profiling papers that clearly show the general importance of CUG-mediated initiation to mammalian cell translation initiation (36, 37).
Because of the importance of non-standard translation in peptide generation, we next investigated the role of non-canonical +1 initiation from M1 mRNA in generating Kb-SIINFEKL in SSA-treated cells. Examination of the +1 reading frame of the downstream M1 RNA sequence revealed one AUG (encoding Met72) and two CUG in-frame codons (encoding Leu59 and Leu96) (see Fig. 1A). We therefore generated IAV viruses with synonymously altered CUG codons (C888T or G1001A). For Met72, we altered the codon to encode Leu (UUG, M72L).
Infection with the mutated viruses revealed that alteration of the AUG or individual CUG codons did not inhibit SSA-resistant Kb-SIINFEKL complex generation relative to presentation in the absence of the drug (Fig. 5A). Altering both CUG residues, however, nearly abrogated Kb-SIINFEKL complex generation in SSA-treated cells (Figure 5A, lower right panel), despite maintaining the expected increase in M1 expression levels (Fig. 5B and S2)
Fig. 5. CUG-initiated translation contributes to +1 frame SIINFEKL generation from M1 mRNA.

(A) Flow cytometric analysis of cell surface M2 and Kb-SIINFEKL expression on L-Kb cells infected in the presence of SSA or vehicle control with IAVs expressing M2-SIIN with the coding alterations indicated. (B) Flow cytometric analysis of intracellular M1 levels In C888T-G1001A infection. (C) Graph showing Kb-SIIN and M2 values at 5 h p.i. in the experiments shown in panel A. Cell surface M2 expression for each IAV (red bars); Kb-SIIN levels in the present (black bars) or absence (white bars) of SSA.
Data are representative of three experiments.
In the absence of SSA, each of the CUG mutations reduced the number of Kb-SIINFEKL complexes relative to M2-SIIN cell surface expression, with the greatest reduction occurring with the double CUG mutant (Fig. 5C). These findings suggest that even when M2 is generated by an intact splicing mechanism, CUG initiation provides a significant fraction of the Kb-SIINFEKL complexes generated.
To corroborate the participation of CUG-based initiation in peptide generation, we infected cells with M2-M72L-SIIN to maximize the relative contribution of CUG codons to +1 initiation, preventing peptide generation from M2 mRNA by treating cells with SSA at the time of infection. At 3 h p.i., we added NSC119893, a specific inhibitor of Met-based translation initiation (33) (Fig. 6). Although NSC119893 reduced M1 synthesis by ~50% in SSA-treated cells (Fig. 6C), consistent with partial inhibition of canonical Met initiation, it had no effect on Kb-SIINFEKL generation in SSA-treated cells (Fig. 6A). By contrast, NSC119893 + SSA inhibited Kb-SIINFEKL generation from NA-SIIN (Fig. 6B) by 1.5- to 3-fold, while inhibiting NA cell-surface expression by 10–30%.
Fig. 6. Inhibition of AUG-initiated translation reduces M1 expression but has little effect on +1 frame SIINFEKL generation from M1 mRNA.
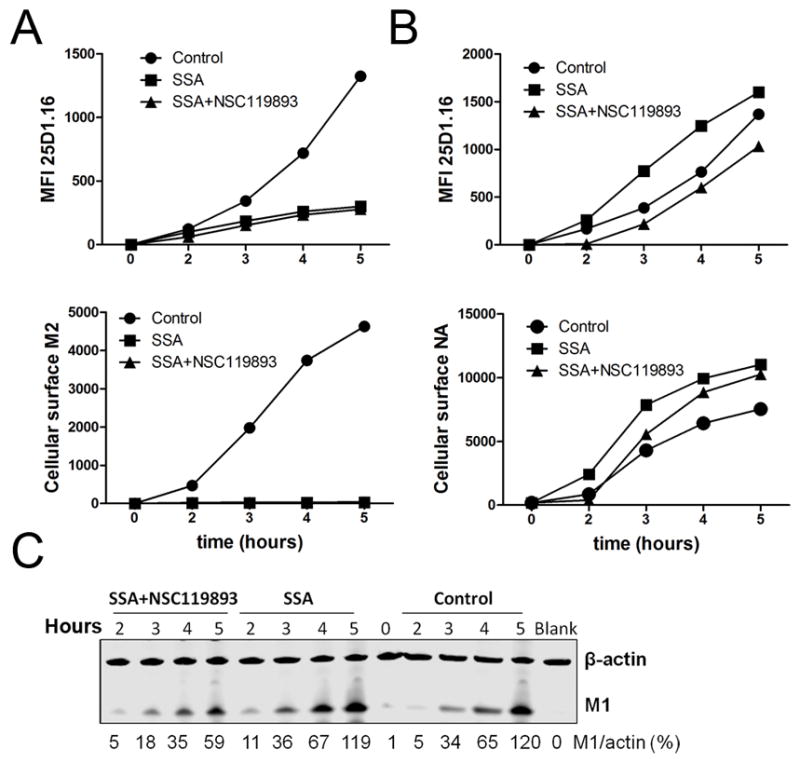
Flow cytometric analysis of L-Kb cell surface M2, NA, or Kb-SIINFEKL expression after infection for the indicated time with PR8-M2-SIIN (A) or PR8-NA-SIIN (B) in the presence of SSA, SSA + NSC119893 or vehicle control. (C) Immunoblot for M1 in cells used in experiment shown in panel A. β-actin was used as a loading control.
Data are representative of three experiments.
Taken together, these findings support the conclusion that peptides are generated via CUG initiation from in the +1 frame of the IAV M1 mRNA.
Evidence for alternative reading frame SIINFEKL from M1 mRNA in vitro and in vivo
To more directly demonstrate that SIINFEKL can be generated by cytoplasmic translation from the M1 mRNA in the +1 reading frame, we inserted a cDNA encoding the IAV segment 7 from PR8 M2-SIIN (encoding the unspliced M1/M2 mRNA) into vaccinia virus (VV) to create VV-M1-SIINARF (Fig. S3). As a strictly cytoplasmic virus, VV-encoded mRNAs are not spliced, and are translated exclusively in the cytoplasm in viral factories (38, 39). As a positive control for VV-mediated Kb-SIINFEKL expression, we generated a recombinant VV (rVV) expressing the spliced M2-SIIN mRNA (SIIN located at the M2 COOH-terminus).
We first tested the ability of rVVs to generate Kb-SIINFEKL complexes after infection of L-Kb cells. VV-M2-SIIN generated a robust signal detectable by 25D1.16 staining within 3 hr p.i. (Fig. 7A,C). However, VV-M1-SIINARF failed to generate a significant signal, even by 5 hr p.i. We were able, however to detect a specific Kb-SIINFEKL signal (relative to cells infected with a rVV expressing M1 without SIINFEKL) if we pre-treated cells with IFN-γ to increase expression of the class I processing machinery and thereby enhance antigen presentation (Fig. 7B,D).
Fig. 7. Generation of Kb-SIINFEKL in cells and mice from VV-mRNA +1 translation.

Flow cytometric analysis of L-Kb cell surface Kb-SIINFEKL expression (A–B) or intracellular M1 (C–D) after infection with recombinant vaccinia virus expressing M2-SIIN, M1-SIINARF or M1 lacking SIIN (control). In B and D, cells were treated for 48 h with IFN-γ prior to infection to up-regulate the antigen processing machinery and increase antigen presentation. (E) OT-I CD8+ cells per spleen 60 h.p.i. with control (black), M1-SIINARF (red), or M2-SIIN (blue) rVV. (F) Undivided OT-I CD8+ cells per spleen 60 h.p.i as indicated by failure to dilute CFSE signal. (G) OT-I CD8+ cells per spleen at 5 d.p.i. (H) Interferon-γ+ OT-I CD8+ T cells determined by intracellular cytokine staining. Bars represent 3–4 mice/group. Error bars = SEM. Statistics = students two-tailed t test.
All experiments were performed at least three times.
Although the number of Kb-SIINFEKL complexes generated from M1-SIINARF may seem small, 25D1-16 staining is much less sensitive (threshold of hundreds of complexes) than T cell recognition (threshold of tens of complexes or less). To confirm SIINFEKL generation from M1-SIINARF and demonstrate its in vivo relevance, we examined the capacity of rVVs to activate CFSE-labeled OT-I TCR transgenic CD8+ T cells (specific for Kb-SIINFEKL) adoptively transferred into B6 mice. Two and a half days after i.p. infection with VV-M2-SIIN (positive control), splenic OT-I CD8+ T cell numbers were significantly increased over mice infected with a control rVV lacking SIINFEKL (3.2 x 106 vs 7.8 x 104), indicating Ag-induced T cell division (Fig. 7E). Infection with VV-M1-SIINARF likewise produced increased numbers of OT-I cells (3.7 x 105). Furthermore, the fraction of undivided OT-I cells (Fig. 7F) was clearly lower in mice infected with VV-M1-SIINARF vs. than control rVV, although to a lesser extent than VV-M2-SIIN infected mice (83.4% undivided in control-VV-infected mice; 23.4% for VV-M1-SIINARF; 1.0% for VV-M2-SIIN).
To confirm OT-I activation by VV-M1-SIINARF, we examined T cell effector function 5 d.p.i. (Fig. 7G–H). As expected from the 2.5 day time point, we recovered far greater numbers of splenic OT-I cells from mice infected with either VV-M2-SIIN (1.1 x 107) or VV-M1-SIINARF (5.9 x 105) relative to the control rVV (2.1 x 104). Additionally, infection with either VV-M1-SIINARF or VV-M2-SIIN greatly increased the number OT-I cells synthesizing IFN-γ, an indicator of fully activated effectors (control=1.7 x 104; M2-SIIN=1.6x107; M1-SIINARF =5.2 x 105).
Together these data demonstrate that SIINFEKL is generated from cytoplasmic mRNA in vitro and in vivo despite its location in a +1 reading frame after the stop codon of the standard M2 open reading frame (Fig. S3). The study provides in vivo relevance for translation initiation on CUG for the purpose of viral immunosurveillance.
Discussion
We have investigated the sources of viral encoded class I peptide ligands by inserting the model peptide SIINFEKL into the IAV segment 7 gene at the C-terminus of M2. Flow cytometry of infected cells revealed that Kb-SIINFEKL complexes were generated in a close kinetic relationship with M2-SIIN synthesis, despite the high metabolic stability of M2-SIIN. Such tight linkage between viral protein synthesis and peptide generation is the rule and not the exception, as shown by a mounting number of studies (reviewed in Anton and Yewdell (4)). This is particularly well shown by applying mass spectrometry to kinetic measurements of viral protein vs. peptide generation, which enables the simultaneous characterization of dozens of gene products (5, 40), revealing the dominant contribution of DRiPs to peptide generation from viral gene products.
We show that SSA, which targets cellular splicing machinery, blocked M2 synthesis. Remarkably, despite SSA mediated-inhibition of M2 synthesis, Kb-SIINFEKL complexes continued to be generated at a robust rate. Co-treating cells with DRB and SSA revealed that Kb-SIINFEKL generation paralleled cytoplasmic levels of M1 mRNA, suggesting that Kb-SIINFEKL was generated from unspliced M1 mRNA in the +1 reading frame. Using VV-M1-SIINARF we conclusively show that SIINFEKL was synthesized from the M1 mRNA by cytoplasmic translation, as VV mRNAs are not spliced, and translation of VV-infected cells is limited to cytoplasmic viral factories (39).
Relocating SIINFEKL to various positions within the M2 coding sequence revealed the plasticity for M2 to accept this peptide at the amino- (position 24) or carboxy- (position 45 or 47) terminus of its transmembrane helix (which approximately encompasses residues 22–43 (41)). This plasticity is consistent with a previous study in which IAV genes were screened for their ability to function after random transposon insertional mutagenesis (42). M2’s mutational flexibility allowed us to determine that while each of these locations supported robust SIINFEKL presentation in untreated cells, SSA reduced presentation to near background levels, implying that peptide location is critical in generating antigenic peptides from the M1 mRNA.
An explanation for this finding is the absence of potential alternative AUG or CUG start codons in the +1 (M2) reading frame upstream of position 47. M2 encodes 2 Met residues, at positions 1 and 72, and two CUG codons, located downstream of position 47 at residues 59 and 96. CUG was described as an initiation codon for mammalian (43) and viral (44) proteins more than 25 years ago. A substantial body of work from Shastri and colleagues established that CUG initiation can generate small open reading frames in transgenes for immunosurveillance (8).
Simultaneously modifying CUG codons encoding Leu59 and Leu96 greatly reduced peptide generation in SSA-treated M2-SIIN infected cells, strongly indicating that CUG-based initiation is important for SIINFEKL synthesis. Supporting this conclusion, NSC119893, a specific inhibitor of standard AUG Met-initiated translation, had little effect on Kb-SIINFEKL generation from M1 mRNA in SSA-treated cells while inhibiting M1 synthesis ~ 50%. While we cannot eliminate a contribution of frame shifting to SIINFEKL generation from M1 mRNA, these findings suggest that peptides are largely derived from CUG-based initiation.
Together, our findings support alternative initiation as a source of IAV DRiPs. Ironically, our initial search for IAV alternative reading frame DRiPs (45) led to the discovery of PB1-F2, generated by AUG initiation in the +1 reading frame of the PB1 gene, downstream of the standard PB1-initiating AUG. (23, 45). Since then, additional bona fide IAV gene products are know to arise from downstream Met initiation in the standard reading frame (PB1-N40 (46)) and by frame shifting (PAX (47)). While not impossible, it is unlikely that the translation we detect at the 3′ end of the M1 mRNA produces a functional gene product or otherwise functions to increase viral fitness in some manner. Translation initiation at Leu59 would create a polypeptide of 40 amino acids, which would make it the shortest described IAV polypeptide, but still possibly functional. M42, a splice variant of M2, is 54 residues in length, (48), and a functional 45-residue protein has been defined in Luteroviradae (49). Our findings imply that translation can be also be initiated at Leu96, which would create a 2 amino acid gene product before the stop codon at position 98--surely too short to be functional. Philosophically, it is basically impossible to disprove that a given genetic element or gene product is functional in some evolutionary context. Practically, having an open mind about potential function is a good idea. “ Junk” peptides (like junk DNA) can only get more important as knowledge about a system grows.
In studying the generation of SIINFEKL we artificially altered the IAV genome, which could potentially influence the occurrence of alternative translation initiation. This seems unlikely to have greatly influenced our results that strongly implicate initiation on CUG encoding Leu59, nearly 100 nucleotides distant from the SIINFEKL coding sequence. How widespread is alternative non-functional initiation to creating viral DRiPs? We previously reported that 14% of newly synthesized NP is truncated from the N-terminus in IAV infected cells, possibly due to downstream initiation (30). It seems likely that many more viral peptides are generated by non-traditional translation of gene products that are inconsequential to viral transmission (the driving force in viral evolution), except as serving as targets for host immunity.
Supplementary Material
Acknowledgments
We thank Tong-Ming Fu, Merck Research Laboratories, and Kenneth Rock, U Mass Medical School for their generous gifts of antibodies, and Tom Kristie, NIAID for critical comments on the manuscript.
Abbreviations used in text
- ARF
alternative reading frame
- DRB
RNA polymerase II inhibitor 5,6-dichloro-1-beta-D-ribofuranosyl benzimidazole
- DRiP
defective ribosome product
- IAV
influenza A virus
- p.i
post-infection
- SSA
spliceostatin A
References
- 1.Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annual review of immunology. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esquivel F, Yewdell JW, Bennink JR. RMA/S cells present endogenously synthesized cytosolic proteins to class I-restricted cytotoxic T lymphocytes. Journal of Experimental Medicine. 1992;175:163–168. doi: 10.1084/jem.175.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yewdell JW, Anton LC, Bennink JR. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J Immunol. 1996;157:1823–1826. [PubMed] [Google Scholar]
- 4.Anton LC, Yewdell JW. Translating DRiPs: MHC class I immunosurveillance of pathogens and tumors. Journal of leukocyte biology. 2014;95:551–562. doi: 10.1189/jlb.1113599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Croft NP, Smith SA, Wong YC, Tan CT, Dudek NL, Flesch IEA, Lin LCW, Tscharke DC, Purcell AW. Kinetics of Antigen Expression and Epitope Presentation during Virus Infection. PLoS pathogens. 2013;9:e1003129. doi: 10.1371/journal.ppat.1003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourdetsky D, Schmelzer CE, Admon A. The nature and extent of contributions by defective ribosome products to the HLA peptidome. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E1591–1599. doi: 10.1073/pnas.1321902111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenlohr LC, Huang L, Golovina TN. Rethinking peptide supply to MHC class I molecules. Nature Reviews Immunology. 2007;7:403–410. doi: 10.1038/nri2077. [DOI] [PubMed] [Google Scholar]
- 8.Starck SR, Shastri N. Non-conventional sources of peptides presented by MHC class I. Cell Mol Life Sci. 2011;68:1471–1479. doi: 10.1007/s00018-011-0655-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Granados DP, Laumont CM, Thibault P, Perreault C. The nature of self for T cells-a systems-level perspective. Current opinion in immunology. 2015;34:1–8. doi: 10.1016/j.coi.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Dolan BP, Knowlton JJ, David A, Bennink JR, Yewdell JW. RNA polymerase II inhibitors dissociate antigenic peptide generation from normal viral protein synthesis: a role for nuclear translation in defective ribosomal product synthesis? J Immunol. 2010;185:6728–6733. doi: 10.4049/jimmunol.1002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apcher S, Millot G, Daskalogianni C, Scherl A, Manoury B, Fahraeus R. Translation of pre-spliced RNAs in the nuclear compartment generates peptides for the MHC class I pathway. Proceedings of the National Academy of Sciences of the United States of America. 2013 doi: 10.1073/pnas.1309956110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6:715–726. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 13.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh AK, Chen ZH, Effenberger KA, Jurica MS. Enantioselective total syntheses of FR901464 and spliceostatin A and evaluation of splicing activity of key derivatives. The Journal of organic chemistry. 2014;79:5697–5709. doi: 10.1021/jo500800k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brooke CB, Ince WL, Wrammert J, Ahmed R, Wilson PC, Bennink JR, Yewdell JW. Most influenza a virions fail to express at least one essential viral protein. Journal of virology. 2013;87:3155–3162. doi: 10.1128/JVI.02284-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu TM, Freed DC, Horton MS, Fan J, Citron MP, Joyce JG, Garsky VM, Casimiro DR, Zhao Q, Shiver JW, Liang X. Characterizations of four monoclonal antibodies against M2 protein ectodomain of influenza A virus. Virology. 2009;385:218–226. doi: 10.1016/j.virol.2008.11.035. [DOI] [PubMed] [Google Scholar]
- 17.Dolan BP, Li L, Takeda K, Bennink JR, Yewdell JW. Defective ribosomal products are the major source of antigenic peptides endogenously generated from influenza A virus neuraminidase. The journal of immunology. 2010;184:1419–1424. doi: 10.4049/jimmunol.0901907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martínez-Sobrido L, García-Sastre A. Generation of recombinant influenza virus from plasmid DNA. Journal of visualized experiments: JoVE. 2010 doi: 10.3791/2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamb RA, Choppin PW. The gene structure and replication of influenza virus. Annual Reviews in Biochemistry. 1983;52:467–506. doi: 10.1146/annurev.bi.52.070183.002343. [DOI] [PubMed] [Google Scholar]
- 20.Wu WH, Pekosz A. Extending the cytoplasmic tail of the influenza a virus M2 protein leads to reduced virus replication in vivo but not in vitro. Journal of virology. 2008;82:1059–1063. doi: 10.1128/JVI.01499-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hilton CJ, Dahl AM, Rock KL. Anti-peptide antibody blocks peptide binding to MHC class I molecules in the endoplasmic reticulum. Journal of Immunology. 2001;166:3952–3956. doi: 10.4049/jimmunol.166.6.3952. [DOI] [PubMed] [Google Scholar]
- 22.Hutchinson EC, Charles PD, Hester SS, Thomas B, Trudgian D, Martinez-Alonso M, Fodor E. Conserved and host-specific features of influenza virion architecture. Nature communications. 2014;5:4816. doi: 10.1038/ncomms5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O’Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- 24.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, Tani T, Horinouchi S, Yoshida M. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 25.Jenkins MR, Webby R, Doherty PC, Turner SJ. Addition of a prominent epitope affects influenza A virus-specific CD8+ T cell immunodominance hierarchies when antigen is limiting. J Immunol. 2006;177:2917–2925. doi: 10.4049/jimmunol.177.5.2917. [DOI] [PubMed] [Google Scholar]
- 26.Amorim MJ, Read EK, Dalton RM, Medcalf L, Digard P. Nuclear export of influenza A virus mRNAs requires ongoing RNA polymerase II activity. Traffic. 2007;8:1–11. doi: 10.1111/j.1600-0854.2006.00507.x. [DOI] [PubMed] [Google Scholar]
- 27.del Val M, Schlicht HJ, Ruppert T, Reddehase MJ, Koszinowski UH. Efficient processing of an antigenic sequence for presentation by MHC class I molecules depends on its neighboring residues in the protein. Cell. 1991;66:1145–1153. doi: 10.1016/0092-8674(91)90037-y. [DOI] [PubMed] [Google Scholar]
- 28.Eisenlohr LC, Yewdell JW, Bennink JR. Flanking sequences influence the presentation of an endogenously synthesized peptide to cytotoxic T lymphocytes. The Journal of experimental medicine. 1992;175:481–487. doi: 10.1084/jem.175.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bullock TN, Eisenlohr LC. Ribosomal scanning past the primary initiation codon as a mechanism for expression of CTL epitopes encoded in alternative reading frames. The Journal of experimental medicine. 1996;184:1319–1329. doi: 10.1084/jem.184.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berglund P, Finzi D, Bennink JR, Yewdell JW. Viral alteration of cellular translational machinery increases defective ribosomal products. Journal of virology. 2007;81:7220–7229. doi: 10.1128/JVI.00137-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bullock TN, Patterson AE, Franlin LL, Notidis E, Eisenlohr LC. Initiation codon scanthrough versus termination codon readthrough demonstrates strong potential for major histocompatibility complex class I-restricted cryptic epitope expression. J Exp Med. 1997;186:1051–1058. doi: 10.1084/jem.186.7.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cardinaud S, Moris A, Fevrier M, Rohrlich PS, Weiss L, Langlade-Demoyen P, Lemonnier FA, Schwartz O, Habel A. Identification of cryptic MHC I-restricted epitopes encoded by HIV-1 alternative reading frames. The Journal of experimental medicine. 2004;199:1053–1063. doi: 10.1084/jem.20031869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Starck SR, Jiang V, Pavon-Eternod M, Prasad S, McCarthy B, Pan T, Shastri N. Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science. 2012;336:1719–1723. doi: 10.1126/science.1220270. [DOI] [PubMed] [Google Scholar]
- 34.Schwab SR, Shugart JA, Horng T, Malarkannan S, Shastri N. Unanticipated antigens: Translation initiation at CUG with leucine. PLoS Biol. 2004;2:1774–1784. doi: 10.1371/journal.pbio.0020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malarkannan S, Horng T, Shih PP, Schwab S, Shastri N. Presentation of out-of-frame peptide/MHC class I complexes by a novel translation initiation mechanism. Immunity. 1999;10:681–690. doi: 10.1016/s1074-7613(00)80067-9. [DOI] [PubMed] [Google Scholar]
- 36.Ingolia Nicholas T, Lareau Liana F, Weissman Jonathan S. Ribosome Profiling of Mouse Embryonic Stem Cells Reveals the Complexity and Dynamics of Mammalian Proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. Decoding human cytomegalovirus. Science. 2012;338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katsafanas GC, Moss B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe. 2007;2:221–228. doi: 10.1016/j.chom.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.David A, Dolan BP, Hickman HD, Knowlton JJ, Clavarino G, Pierre P, Bennink JR, Yewdell JW. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. The Journal of cell biology. 2012;197:45–57. doi: 10.1083/jcb.201112145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Croft NP, Purcell AW, Tscharke DC. Quantifying epitope presentation using mass spectrometry. Molecular immunology. 2015 doi: 10.1016/j.molimm.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Forrest LR, Peter Tieleman D, Sansom MSP. Defining the Transmembrane Helix of M2 Protein from Influenza A by Molecular Dynamics Simulations in a Lipid Bilayer. Biophysical Journal. 1999;76:1886–1896. doi: 10.1016/s0006-3495(99)77347-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heaton NS, Sachs D, Chen CJ, Hai R, Palese P. Genome-wide mutagenesis of influenza virus reveals unique plasticity of the hemagglutinin and NS1 proteins. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:20248–20253. doi: 10.1073/pnas.1320524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hann SR, King MW, Bentley DL, Anderson CW, Eisenman RN. A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt’s lymphomas. Cell. 1988;52:185–195. doi: 10.1016/0092-8674(88)90507-7. [DOI] [PubMed] [Google Scholar]
- 44.Mehdi H, Ono E, Gupta KC. Initiation of translation at CUG, GUG, and ACG codons in mammalian cells. Gene. 1990;91:173–178. doi: 10.1016/0378-1119(90)90085-6. [DOI] [PubMed] [Google Scholar]
- 45.Chen W, Bennink JR, Yewdell JW. Systematic search fails to detect immunogenic MHC class-I-restricted determinants encoded by influenza A virus noncoding sequences. Virology. 2003;305:50–54. doi: 10.1006/viro.2002.1744. [DOI] [PubMed] [Google Scholar]
- 46.Wise HM, Barbezange C, Jagger BW, Dalton RM, Gog JR, Curran MD, Taubenberger JK, Anderson EC, Digard P. Overlapping signals for translational regulation and packaging of influenza A virus segment 2. Nucleic acids research. 2011;39:7775–7790. doi: 10.1093/nar/gkr487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science. 2012;337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wise HM, Hutchinson EC, Jagger BW, Stuart AD, Kang ZH, Robb N, Schwartzman LM, Kash JC, Fodor E, Firth AE, Gog JR, Taubenberger JK, Digard P. Identification of a Novel Splice Variant Form of the Influenza A Virus M2 Ion Channel with an Antigenically Distinct Ectodomain. PLoS pathogens. 2012;8:e1002998. doi: 10.1371/journal.ppat.1002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smirnova E, Firth AE, Miller WA, Scheidecker D, Brault V, Reinbold C, Rakotondrafara AM, Chung BY, Ziegler-Graff V. Discovery of a Small Non-AUG-Initiated ORF in Poleroviruses and Luteoviruses That Is Required for Long-Distance Movement. PLoS pathogens. 2015;11:e1004868. doi: 10.1371/journal.ppat.1004868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.