

Abstract
One of the most prominent features of the human brain is the fabulous size of the cerebral cortex and its intricate folding. Cortical folding takes place during embryonic development and is important to optimize the functional organization and wiring of the brain, as well as to allow fitting a large cortex in a limited cranial volume. Pathological alterations in size or folding of the human cortex lead to severe intellectual disability and intractable epilepsy. Hence, cortical expansion and folding are viewed as key processes in mammalian brain development and evolution, ultimately leading to increased intellectual performance and, eventually, to the emergence of human cognition. Here, we provide an overview and discuss some of the most significant advances in our understanding of cortical expansion and folding over the last decades. These include discoveries in multiple and diverse disciplines, from cellular and molecular mechanisms regulating cortical development and neurogenesis, genetic mechanisms defining the patterns of cortical folds, the biomechanics of cortical growth and buckling, lessons from human disease, and how genetic evolution steered cortical size and folding during mammalian evolution.
Keywords: evolution, ferret, gyrencephaly, humans, neocortex
Subject Categories: Neuroscience
Introduction
In 1990, Wally Welker published a landmark book chapter updating on all that was known about the physiological, experimental, and pathological causes of cortical folding and misfolding, including comparative and phylogenetic considerations (Welker, 1990). Twenty‐five years later, we have been invited to present an update of current knowledge on this matter. Welker's review was 134 pages long and cited 665 references, and much progress has been made since then. Clearly, if our review on cortical folding has to fit in this issue of The EMBO Journal, something has to be done. Our solution is to limit the size of this review by focusing and briefly touching on what we feel have been the most significant advances in this period leading to our current understanding of the biology of this problem.
One of the most prominent features of the human brain is the fabulous size of the cerebral cortex and its folding, visible as bulges and grooves on its external surface. Most animals with a large brain have a folded cortex, whereas most animals with a small brain have a smooth cortex, without folds. The cerebral cortex is a laminar tissue where neurons lie on the upper part, and the lower or inner part contains most of the wire connecting neurons between brain areas. In big brains, this sheet of neural tissue covering the outside of the brain is disproportionately larger than the deep brain structures it covers, and instead of adopting a balloon‐like conformation it folds onto itself, minimizing total brain and cranial volume. In addition to minimizing brain volume, cortical folding is of key importance for the optimization of brain wiring and functional organization (Klyachko & Stevens, 2003), and alterations in cortical size or folding lead to severe intellectual disability and intractable epilepsy in humans (Walsh, 1999; Barkovich et al, 2012).
The process of cortical folding takes place during brain development, and thus, it is essentially a developmental problem. In this review, we will start describing exciting discoveries made over the last fifteen years on the central role of progenitor cell proliferation and survival in cortical size, the discovery of novel germinal zones and progenitor cell types, which are greatly overrepresented in gyrated brains, and their key function in cortical folding. This will be followed by our understanding of the genetic mechanisms regulating the patterns of cortical folding and the biomechanics of this process, what we have learned from human disease, and finally will close with a view of how genetic evolution may have steered cortical size and folding during mammalian evolution.
Cellular mechanisms leading to cerebral cortex expansion and folding
In order to understand the developmental mechanisms involved in cortical expansion and folding, we will frame these within the basic principles of cerebral cortex development as found in the most common animal model, the mouse.
Telencephalic neuroepithelium
In the early embryo (in mouse c. 9 days postconception, E9), the expansion of the rostral‐most domain of the neural tube gives rise to the two telencephalic vesicles. The dorsal half of these vesicles is then molecularly specified as the primordium of the cerebral cortex. At this stage, the cortical primordium is solely composed of a monolayer of neural stem neuroepithelial cells (NECs; Bayer & Altman, 1991). NECs are highly polarized and attached to each other by adherens and tight junctions at the level of the apical domain (inner surface of the telencephalic vesicle) and which move their cell nucleus between the apical and the basal sides of the neuroepithelium in coordination with the cell cycle: basal‐directed movement during G1, basal position during S‐phase, apical‐directed movement during G2, and mitosis at the apical surface. This cyclic movement is known as interkinetic nuclear migration and is completely asynchronous between NECs, conferring the neuroepithelium a pseudostratified appearance (Sauer, 1935; Bayer & Altman, 1991; Taverna & Huttner, 2010). NECs only undergo symmetric self‐amplificative divisions, whereby each division generates two daughter NECs, hence exponentially increasing their number (Miyata et al, 2010; Fig 1).
Figure 1. Stem cells in the developing cerebral cortex of gyrencephalic brains and their molecular regulation.
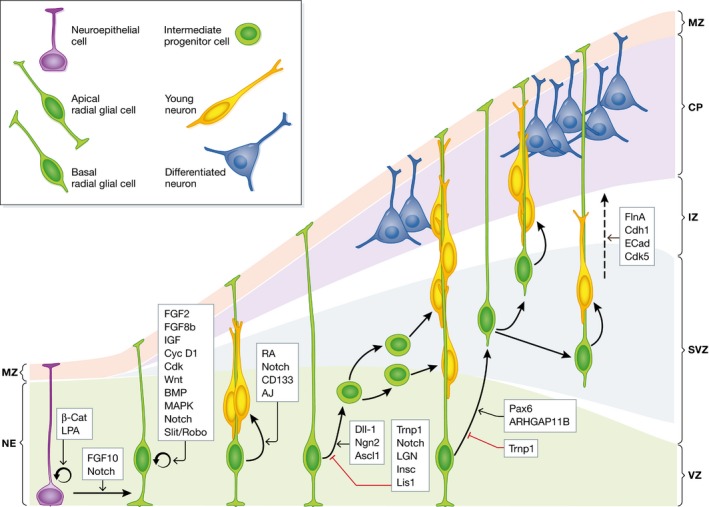
Schema depicting the main types of progenitor cells and their lineage relationships in the developing cerebral cortex. Arrows indicate lineage relationships demonstrated by time‐lapse imaging and/or by retroviral lineage tracing. During the expansion phase, most neuroepithelial cells divide symmetrically to self‐amplify to generate apical radial glial cells. During the neurogenic phase, most aRGCs divide asymmetrically to generate neurons, either directly or indirectly through intermediate progenitor cells or basal radial glial cells. Molecules or pathways regulating some of these steps are indicated. MZ, marginal zone; CP, cortical plate; IZ, intermediate zone; SVZ, subventricular zone; VZ, ventricular zone.
Because NECs are the founder progenitor cells of the cerebral cortex, their pool size determines the numbers of their derived neurogenic progenitor cells and the final number of cortical neurons, and hence, it has a fundamental impact on the size of the mature cerebral cortex. Accordingly, already at this early stage, the size of the telencephalic vesicles is much larger in the human than in mouse embryo, reflecting significant differences in size of the neuroepithelium as a consequence of different NEC abundance (Sidman & Rakic, 1973; Rakic, 1995). NEC abundance may be increased by extending the time period of their self‐amplification and delaying the onset of neurogenesis, as observed in primates compared to rodents (Rakic, 1995, 2009; Kornack & Rakic, 1998). The importance of NEC amplification on cortical size has been experimentally demonstrated in mouse, where NEC abundance in the embryonic cortex may be increased by either promoting their re‐entry into cell cycle or preventing programmed cell death (Chenn & Walsh, 2002; Kingsbury et al, 2003). In both cases, increased NEC abundance leads to expansion in surface area and folding of the neuroepithelium.
Proliferation and neurogenesis
Immediately prior to the onset of neurogenesis, NECs start losing tight junctions and begin acquiring features typical of glial cells, including the expression of brain lipid‐binding protein (BLBP), vimentin, and Pax6, thus becoming apical radial glial cells (aRGCs; see Taverna et al (2014) for a detailed review on the cell biology of this process). Like NECs, aRGCs undergo interkinetic nuclear migration, divide at the apical surface of the developing cortex and, at this early stage (c. E10 in mouse), also undergo self‐amplifying divisions. However, aRGCs gradually start dividing asymmetrically to generate one aRGC plus a different cell. These new cells accumulate at the basal side of the cortical primordium, while the cell bodies of aRGCs remain in the apical side, forming the ventricular zone (VZ; Fig 1). With the accumulation of cells above the VZ, the basal process of aRGCs elongates while remaining attached to the basal lamina and is now termed radial glial fiber. Asymmetric aRGC divisions generate one aRGC plus either one neuron or one intermediate progenitor cell (IPC; Malatesta et al, 2000; Noctor et al, 2001, 2004; Haubensak et al, 2004; Miyata et al, 2004; Fig 1). IPCs are secondary progenitor cells without apical–basal polarity, do not undergo interkinetic nuclear migration, reside and divide in a location immediately basal to the VZ, the subventricular zone (SVZ), and contrary to aRGCs, they all express the transcription factor Tbr2 (Englund et al, 2005). In mouse, the vast majority of IPCs divide once to produce 2 neurons (neurogenic, self‐consuming divisions), and hence, they are viewed as a strategy to amplify the production of cortical neurons. However, because each IPC self‐consumes at mitosis, their relative abundance compared to aRGCs is quite low (Kowalczyk et al, 2009). IPCs in the cerebral cortex generate most cortical excitatory neurons (Attardo et al, 2008; Kowalczyk et al, 2009), whereas inhibitory interneurons are generated extra‐cortically (Anderson et al, 1997). As neurogenesis progresses, there are a lower requirement for aRGC expansion/renewal and a greater need for neuron production, so there is a gradual predominance of asymmetric aRGC divisions producing IPCs (Noctor et al, 2004; Kowalczyk et al, 2009).
In addition to aRGCs and IPCs, the embryonic mouse cortex includes other much less abundant types of progenitor cells. Populating the VZ, we find apical intermediate progenitors (aIPs), which divide at the apical surface to produce neurons (Stancik et al, 2010; Tyler et al, 2015), and subapical progenitors (SAPs), which divide within the VZ, but away from the apical surface to generate IPCs (Pilz et al, 2013). Populating the SVZ, we find basal radial glial cells (bRGCs), which share many similarities with aRGCs including a basal process extended radially and contacting the basal lamina of the telencephalon, and expression of the transcription factor Pax6, but whose cell body is located and divides at basal positions in the SVZ (Shitamukai et al, 2011; Wang et al, 2011; Fig 1). As opposed to aRGCs, bRGCs in mouse do not self‐amplify nor produce IPCs, but are highly neurogenic (Wang et al, 2011).
In gyrencephalic species like humans, monkey, or ferret, the abundance of aRGCs is much greater than in species with a smooth cortex like mouse, producing a more extended VZ. This is a direct consequence of the higher abundance of founder NECs at earlier stages (see above), further promoted by an increased self‐amplification of aRGCs in these species. In addition to having an extended VZ, the most remarkable distinction of gyrencephalic species is having a thickened SVZ populated by an outstanding abundance of basal progenitors, especially at later stages of neurogenesis when these greatly outnumber apical progenitors (Smart et al, 2002; Lukaszewicz et al, 2005; Reillo et al, 2011; Reillo & Borrell, 2012). This abundance of basal progenitors is accompanied by the splitting of the SVZ in inner (ISVZ) and outer (OSVZ) subdivisions, not found in mouse (Smart et al, 2002; Fietz et al, 2010; Hansen et al, 2010; Reillo et al, 2011; Reillo & Borrell, 2012).
The OSVZ contains a wide diversity of progenitor cell types with high amplificative potential, and the combination of these two factors is considered key for cortical expansion and folding (Lui et al, 2011; Betizeau et al, 2013; Borrell & Gotz, 2014). Contrary to the lissencephalic mouse, in gyrencephalic species few basal progenitors are IPCs, but most are bRGCs (Hansen et al, 2010; Reillo et al, 2011; Reillo & Borrell, 2012; Betizeau et al, 2013). A seminal videomicroscopy study demonstrated that in macaque (a gyrated primate), bRGCs come in different modalities, which frequently transition between them and with IPCs, and all of these types of progenitors may self‐amplify prior to generating neurons (Betizeau et al, 2013). Importantly, in gyrencephalic cortices like macaque and ferret, neurogenesis takes place during a period of time much longer than in rodents (up to tenfold; Takahashi et al, 1993; Kornack & Rakic, 1998; Lukaszewicz et al, 2005; Reillo & Borrell, 2012), allowing more rounds of cell division and increasing neuronal output (Dehay & Kennedy, 2007; Florio & Huttner, 2014). On these grounds, we and others have proposed that the OSVZ, with its wealth of neurogenic basal progenitors, plays central roles in the dramatically increased neurogenesis and folding in the cerebral cortex of higher mammals (Fig 2A; Fietz & Huttner, 2011; Lui et al, 2011; Borrell & Reillo, 2012; Borrell & Calegari, 2014; Borrell & Gotz, 2014; Florio & Huttner, 2014). This idea is well supported by experimental work in the gyrencephalic ferret, which demonstrates that forced overproliferation of OSVZ progenitors increases cortical surface area and folding, whereas blockade of their proliferation has the opposite effect (Reillo et al, 2011; Nonaka‐Kinoshita et al, 2013). The central relevance of the OSVZ in cortical expansion and folding resides not only on its prominent contribution to increase neuron production, but also specifically on its high content of bRGCs, as explained in the next section.
Figure 2. Patterns of cell division in the embryonic cerebral cortex and mechanisms for tangential versus radial expansion.
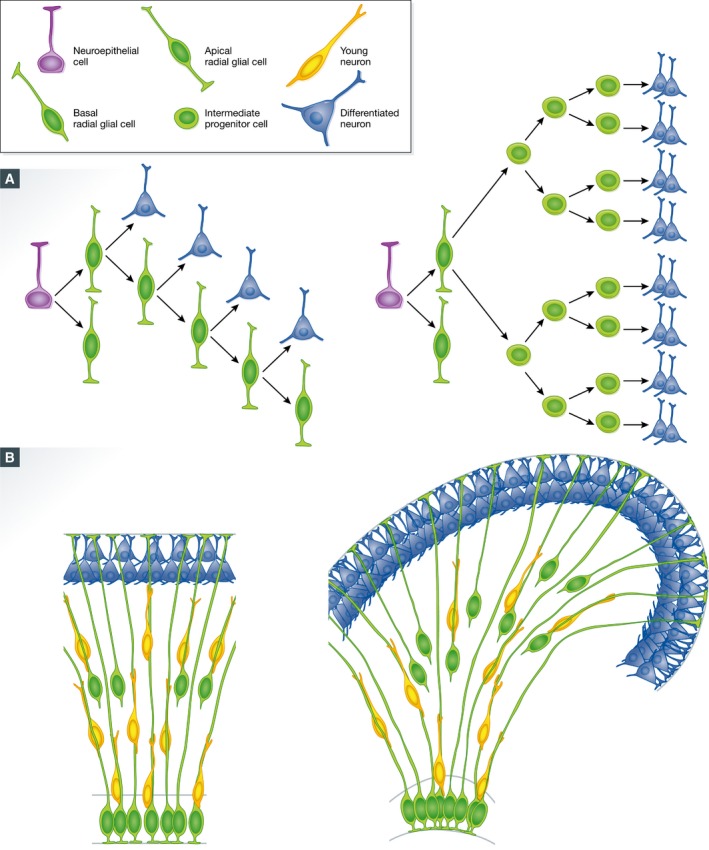
(A) Two patterns of cortical progenitor cell division with opposite neurogenic outcome: The total number of neurons produced is proportional to the generation of intermediate progenitor cells, very small in species with a smooth cortex (left) and large in species with a folded cortex (right). (B) Difference in the general arrangement of the radial fiber scaffold in the cerebral cortex undergoing tangential (left) versus radial expansion (right). In species with a smooth cortex like mouse (left), radial glial fibers are parallel (green) and there is no net lateral dispersion of radially migrating neurons (yellow) with respect to the positions of their progenitor aRGCs (green). In gyrencephalic species, the radial fiber scaffold becomes divergent due to the intercalation of radial fibers from bRGCs. As a result, radially migrating neurons follow divergent trajectories which cause their lateral dispersion; this increases cortical surface area and ultimately promotes folding (modified from Borrell & Reillo, 2012).
Radial migration
Newborn cortical excitatory neurons must travel (migrate) from their layer of birth to the vicinity of the cortical surface, where they will coalesce into nascent neuronal layers. This process is named radial migration (Rakic, 1972). The scaffold of radial glial fibers, which span perpendicular between the ventricular and the pial surface of the cortex, provides the necessary substrate and guide for these neurons during their radial migration, much like the rail tracks for a train. This cell–cell interaction is under tight molecular regulation, and its disruption leads to severe defects of neuronal positioning and layer formation in the cortex (Sidman & Rakic, 1973; Rakic et al, 1974; Rakic, 1978; Anton et al, 1997, 1999; Elias et al, 2007). Due to this dependence of radially migrating neurons on radial glial fibers, the trajectory of these fibers largely defines the migratory route and final location along the cortical surface of new neurons (Rakic, 1995). Consequently, sibling neurons born from one progenitor normally occupy neighboring positions in the mature cerebral cortex (Fig 2B; Soriano et al, 1995; Gupta et al, 2003; O'Leary & Borngasser, 2006; Gao et al, 2014).
In contrast to the mouse cerebral cortex, a characteristic feature of folded cortices during development is their much greater surface area on the pial than on the ventricular side (Sidman & Rakic, 1973; Kriegstein et al, 2006; Rakic, 2009). This expansion of the pial surface relates to the increased numbers of cortical neurons, largely based on the expansion of basal progenitors. Several hypotheses have been proposed to explain how basal progenitors may promote this difference between pial and ventricular surface during development (Kriegstein et al, 2006; Fietz & Huttner, 2011). However, a critical issue overseen in early hypotheses is that whereas the addition of basal progenitors explains increased neurogenesis without expanding the ventricular surface area, the amount of radial fibers from aRGCs also does not expand, which is problematic. If the radial fiber scaffold does not increase as neurogenesis increases, then radial fibers (the rail tracks for migrating neurons) become a limiting factor to be shared by the augmented population of radially migrating neurons. As a result, neurons pile up in thickened cortical layers without a significant lateral separation, and thus, surface area does not increase and folds do not form. Intriguingly, this is reminiscent of human lissencephaly, where cortical layers are several‐fold thicker than normal and folds fail to form (see section below). However, this is not the case in gyrencephalic brains, where the outer surface area of the cerebral cortex increases several orders of magnitude more than thickness (i.e., the human cortex has 1,000 times more surface area than in mouse, but is only 10 times thicker) and without a similar increase in VZ surface area.
Thus, the radial fiber scaffold must be modified so that the increased numbers of radially migrating neurons do not pile in thick layers, but are distributed along the cortex. An elegant solution to this problem is to create a divergence in the array of radial fibers with additional, intercalated fibers. Under this circumstance, neurons migrating radially and in intimate association with radial fibers find additional paths for migration, which has two advantages: (i) overcrowding of the migratory paths is released so radial migration is not delayed and (ii) radially migrating neurons are delivered to distant positions along the cortical surface (Fig 2B). The source of additional radial fibers to create this divergence is bRGCs (Reillo et al, 2011). Basal RGCs have a radial fiber extended to the pial surface, their cell soma is located in ISVZ or OSVZ, basal from aRGCs, and they are not anchored to the apical VZ surface. Hence, bRGCs are in the optimal position to provide additional radial fibers to create the divergence and fanning out of the radial fiber scaffold, without increasing VZ surface area, as reported in ferret (Fig 2B; Reillo et al, 2011). Importantly, in spite of the divergence of radial fibers, thanks to bRGCs their density remains high through the cortical thickness, and many are even intercrossed (Rakic et al, 1974; Reillo et al, 2011), so neurons can disperse laterally while maintaining radial glia‐dependent migration, as recently demonstrated in ferret (Gertz & Kriegstein, 2015). In the developing ferret cortex, the fanning of the radial fiber scaffold is significant in prospective gyrus, but not in prospective sulcus regions, which further supports that this is a driving force in cortical surface area expansion and folding (Lui et al, 2011; Reillo et al, 2011; Borrell & Reillo, 2012). Importantly, this “radial divergence model” has been validated by several laboratories, where bRGC abundance and proliferation have been experimentally manipulated in ferret and mouse. Partial blockade of OSVZ progenitor proliferation in the developing ferret cortex without a significant neuronal loss leads to a reduction in size of cortical folds (Reillo et al, 2011) and even lissencephaly (Poluch & Juliano, 2015). Conversely, forced overproliferation of OSVZ progenitor cells in ferret significantly increases cortical surface area and folding (Nonaka‐Kinoshita et al, 2013). Finally, even in the naturally smooth mouse cortex, genetic manipulations forcing an increased abundance of bRGCs during embryonic development lead to the formation of cortical folds (Stahl et al, 2013; Florio et al, 2015).
Differentiation
Once neurons finish radial migration and detach from the radial fiber, they begin terminal differentiation. This process has a fundamental impact in the final size of the cerebral cortex, essentially by increasing the size of cell somas and the volume of the neuropile: growth and branching of apical and basal dendrites; extension, navigation, and branching of the axon; formation of spines and boutons for synaptic connectivity. Packing density of cortical neurons, cell body size, and extent of their dendritic and axonal arbors are remarkably different between mammals, correlating with brain size (Purves, 1988) and also contributing to cortical expansion (Reillo et al, 2011).
Molecular regulation of cellular mechanisms
Over the last two decades, hundreds of studies have deciphered some of the key molecular mechanisms regulating cerebral cortex development in mouse. Although this invaluable knowledge is frequently extensible to humans and other species at the level of basic cellular mechanisms, much less is really known about the molecular regulation of events key for cortical expansion and folding. In this section, we provide an overview of some of this knowledge, and how it may be extensive to cortical folding.
NEC amplification versus transition to RGC
At early stages of cortical development, several signaling cascades regulate the maintenance of NEC self‐amplification. One of the most potent promoters of NEC self‐amplification is the β‐catenin pathway, such that its constitutive activation in mouse embryos leads to the overexpansion and folding of the cortical neuroepithelium (Chenn & Walsh, 2002). Expansion and significant folding of the mouse cortical neuroepithelium is also achieved by limiting developmental apoptosis, by means of lysophosphatidic acid signaling (Kingsbury et al, 2003). Conversely, the fibroblast growth factor (FGF) pathway, by means of FGF10, drives NECs into expressing radial glial cell markers and thus promotes their transition toward aRGC fate, terminating the phase of NEC amplification (Sahara & O'Leary, 2009). Intriguingly, FGF signaling promotes retention of the aRGC fate, as it also inhibits the subsequent transition of aRGCs toward basal progenitors (Kang et al, 2009). The transition from NECs to aRGCs is also strongly promoted by the activation of the Notch signaling pathway (Gaiano et al, 2000; Hatakeyama et al, 2004; Fig 1).
Progenitor amplification
In addition to their role in blocking the maturation of cortical progenitors, FGF ligands promote their proliferation and inhibit neurogenesis by regulating the duration of the cell cycle. This role of FGF signaling is critical for cortical growth, as its loss at early stages accelerates neuron production and loss of RGCs, resulting in reduced cortical surface area (Rash et al, 2011). Conversely, overactivation of FGF signaling by infusion of FGF2 or FGF8b causes the overproliferation of cortical progenitors and cortical expansion in surface area (Rash et al, 2013; Fig 1). FGFs influence cortical progenitors via regulation of cell cycle proteins. FGF2 and insulin‐like growth factor (IGF) 1 upregulate the expression of cyclin D1 and downregulate the expression of p27(kip1), a cyclin‐dependent kinase (Cdk) inhibitor, thereby shortening the G1 phase of the cell cycle and promoting self‐amplificative divisions (Raballo et al, 2000; Lukaszewicz et al, 2002; Mairet‐Coello et al, 2009). Activation of this FGF signaling cascade drives cortical expansion, which is accompanied by the incipient folding of the otherwise smooth mouse cortex (Rash et al, 2013), and in the already gyrencephalic ferret cortex, it causes extra folding (Masuda et al, 2015). Not surprisingly, cortical progenitor populations may be expanded directly by overexpressing Cdk4 and cyclin D1 (Lange et al, 2009). However, in the mouse cortex, this promotes cortical growth and megalencephaly, but not folding (Nonaka‐Kinoshita et al, 2013), which highlights the molecular and cellular complexity of the process of cortical folding (Borrell & Calegari, 2014).
Other important signaling pathways regulating cortical progenitor proliferation and self‐renewal include the Wnt, BMP, MAPK, and Notch pathways. Unfortunately, none of these pathways have been found able to induce bona fide cortical folding in mouse, and there are no experimental data in naturally gyrencephalic species. One of the best‐known pathways regulating the balance between progenitor proliferation and neurogenesis is Notch. In addition to promoting the transition from NECs to aRGCs at early developmental stages (Gaiano et al, 2000; Hatakeyama et al, 2004; Martynoga et al, 2012), activation of the Notch pathway at later stages inhibits the generation of IPCs from aRGCs (Mizutani et al, 2007; Ohata et al, 2011; Martynoga et al, 2012). Conversely, the onset of Dll1 expression (the main ligand of Notch1) coincides with the expression of the pro‐neural proteins Ngn2 and Ascl1, which are major transcriptional regulators of neurogenesis and also directly regulate Dll1 expression (Castro et al, 2006; Martynoga et al, 2012). The Notch pathway can be additionally activated by Slit/Robo signaling, with similar consequences: impairment of neurogenesis at early stages by favoring the self‐renewal of aRGCs (Borrell et al, 2012). Importantly, Notch signaling seems to be required for the self‐renewal of OSVZ progenitors in the human cerebral cortex (Hansen et al, 2010).
Regarding Wnt, activation of this pathway promotes proliferation and self‐renewal of aRGCs at early developmental stages (Machon et al, 2003; Woodhead et al, 2006; Zhou et al, 2006), while at later stages it promotes the maturation of aRGCs into IPCs (Viti et al, 2003; Hirabayashi et al, 2004) and it may even promote neurogenesis (Munji et al, 2011). Thus, the effects of Wnt signaling in cortical development are complex and time‐regulated during development. The BMP pathway has similarly elusive and complex inputs into the regulation of cortical neurogenesis. At early developmental stages, BMP signals induce neurogenesis (Li et al, 1998; Mabie et al, 1999), while at later stages, they block neurogenesis to promote astrocyte differentiation (Gross et al, 1996). The Ras‐MAPK‐ERK pathway controls the mitogen‐stimulated proliferation of cortical progenitors and its negative regulators, thus ensuring progenitor self‐renewal and preventing premature differentiation (Phoenix & Temple, 2010). Finally, IGF‐2 secreted into the cerebrospinal fluid by the choroid plexus is another potent mitogen promoting proliferation of VZ cortical progenitors (Lehtinen & Walsh, 2011; Lehtinen et al, 2011).
Recently, retinoic acid (RA) signaling has also been identified as important in regulating the balance between cortical progenitor self‐renewal and neurogenesis, where RA secreted by the meningeal membranes promotes neurogenesis while limiting aRGC amplification (Siegenthaler et al, 2009). Contrary to the classical signaling pathways mentioned above, the RA pathway holds promise as an important regulator of cortical expansion and folding, as its genetic blockade induces remarkable folding of the mouse cortex (Siegenthaler et al, 2009). The downstream transducers of RA signaling in this context are not known, but the orphan nuclear hormone receptor CoupTF1 and the pro‐neural transcription factors Ngn1 and 2 may be involved (Ribes et al, 2008; Harrison‐Uy et al, 2013).
Progenitor cell lineage
Whereas cortical folding is positively correlated with increased brain size and greater numbers of neurons, mounting evidence from comparative neuroanatomy and human pathology strongly supports that increased neuron numbers are not sufficient, but cortical folding requires additional mechanisms of developmental regulation (Welker, 1990; Borrell & Reillo, 2012). As explained in the previous section, the tangential dispersion of radially migrating neurons seems key in the expansion of cortical surface area that leads to folding, and this depends on the relative abundance of bRGCs (Reillo et al, 2011; Pilz et al, 2013). Hence, lineage regulation of the different types of cortical progenitors, particularly the production and maintenance of bRGCs, is critical in this process (Borrell & Gotz, 2014).
In the cerebral cortex of mouse, ferret, and humans, bRGCs are generated from aRGCs in the VZ (Reillo et al, 2011; Shitamukai et al, 2011; Wang et al, 2011). This process is seemingly associated with, and controlled by, the mitotic spindle orientation of aRGCs (Shitamukai et al, 2011; LaMonica et al, 2013), which is known to have a strong influence on the acquisition of asymmetric cell fates by cortical progenitors (Postiglione et al, 2011; Xie et al, 2013). Progenitors in the early neuroepithelium divide mostly in perpendicular orientations while oblique cleavage planes augment as neurogenesis becomes predominant, and disruption of these orientations at early stages leads to depletion of the progenitor pool (Mitchison & Kirschner, 1984; Chenn & McConnell, 1995; Yingling et al, 2008). Orientation of the mitotic spindle in the mammalian cerebral cortex is regulated by a number of factors, including LGN, Insc, and Lis1 (Yingling et al, 2008; Postiglione et al, 2011; Shitamukai et al, 2011; Fig 1).
In addition to the cleavage plane orientation, there are other cellular mechanisms regulating cortical progenitor cell fate, particularly symmetric versus asymmetric fates, which include the maintenance of the apical–basal polarity by Notch signaling, Par3, Par6, prominin‐1 (CD133), and other proteins related to the apical adherens junctions (Hatakeyama et al, 2004; Gotz & Huttner, 2005; Costa et al, 2008; Bultje et al, 2009; Kriegstein & Alvarez‐Buylla, 2009; Imayoshi et al, 2010). A decrease in apical junction proteins like Par3 or Par6 switch the mode of aRGC division from self‐renewing to neurogenic, while their overexpression promotes aRGC self‐renewal (Costa et al, 2008).
A landmark finding on the molecular regulation of cortical folding via control of progenitor cell lineage was the identification of Trnp1, a key player in this process (Stahl et al, 2013). Trnp1 is a DNA‐associated protein initially identified as being strongly expressed in self‐amplifying aRGCs (Pinto et al, 2008). Starting at high levels in the early embryo, Trnp1 expression decreases as aRGCs gradually stop self‐amplifying to produce IPCs and neurons. Overexpression of Trnp1 promotes aRGC self‐renewal at the expense of IPCs and neurogenesis. Conversely, knockdown of Trnp1 function increases IPCs and, in addition, it dramatically increases the abundance of bRGCs, otherwise very scarce in the mouse embryo. Most importantly, these changes are associated with a significant expansion of the cortical surface area and the formation of structures resembling bona fide cortical folds, including modification of the radial fiber scaffold and the divergent distribution of radially migrating neurons (Stahl et al, 2013). Analyses of Trnp1 expression in the developing cortex of ferret and human embryos are consistent with these functional results, as Trnp1 levels are specifically low in cortical regions prospectively undergoing tangential expansion and folding (Stahl et al, 2013; de Juan Romero et al, 2015). Thus, Trnp1 was the first gene able to induce bona fide folding of the mouse cerebral cortex.
A second gene causing folding of the mouse cortex has been recently found: ARHGAP11B (Florio et al, 2015). This is a truncated paralog of ARHGAP11A that arose on the human evolutionary lineage after the divergence from the chimpanzee (Antonacci et al, 2014), and hence does not exist in mouse. Similar to the loss of function of Trnp1, experimental expression of ARHGAP11B in mouse embryos drives cortical aRGCs into massively producing basal progenitors, many of which are bRGCs, and this ultimately translates into folding of the mouse cerebral cortex. The seminal relevance of this study is the discovery of a gene with a likely central role in human brain evolution and uniqueness, contributing to further promote cortical expansion and folding in the hominid lineage. High levels of the transcription factor Pax6 have also been reported to promote the generation of bRGCs in mouse, but intriguingly this does not cause cortical folding (Wong et al, 2015).
Progenitor cell heterogeneity: single‐cell transcriptomics
One of the classical limitations of studies searching for genes important in cortical development, expansion and folding was to analyze progenitor cell pools instead of individual cells. The advent of single‐cell transcriptomics has revolutionized this field by allowing uncovering an extraordinary molecular heterogeneity of progenitor cell types in the developing cerebral cortex (Pollen et al, 2014). Importantly, this technology has revealed that aRGCs and bRGCs are molecularly more heterogeneous in the developing folded cortex of humans and ferret (3 classes of aRGC, 2 classes of bRGC) than in the smooth cortex of mouse (2 classes of aRGC, 1 class of bRGC; Camp et al, 2015; Johnson et al, 2015; Pollen et al, 2015). Although we are just beginning to profit from the power of this technology, single‐cell transcriptomics is already highlighting the potential relevance of specific signaling cascades and molecular programs in the formation and maintenance of bRGCs in general and the OSVZ in particular. For example, in agreement with previous population‐wide transcriptomic analyses and functional manipulations, the extracellular matrix and its components are being highlighted as central in stimulating cortical progenitor proliferation and self‐renewal (Fietz et al, 2010, 2012; Stenzel et al, 2014; Pollen et al, 2015).
Genetic patterning of cortical folds
What defines the location and shape of cortical folds and fissures as they form in the developing cortex? The traditional view has been that cortical folds form randomly, essentially based on the idea that cortical growth exceeds cranial volume, and thus, cortical folding occurs purely as a mechanical consequence of cranial constraint (Welker, 1990). However, as discussed in a later section, evidence indicates that both notions are wrong. If any portion of the cerebrum fails to develop or grow, due to either pathological or experimental causes, the skull tends to conform to the size and shape of the remaining neural tissue, which in fact still folds, thus demonstrating that limited cranial volume does not force cortical folding (Welker, 1990). Further evidence demonstrating that cortical folds do not form randomly includes that (i) the pattern of folds and fissures is highly stereotyped and well conserved between individuals of a species, particularly in those with small gyrated brains (i.e., ferret, cat); (ii) folding patterns of phylogenetically related species follow remarkably similar trends (Welker, 1990; Borrell & Reillo, 2012); (iii) even in species with large cortices and very complex folding patterns, like humans, the deepest and earliest fissures to develop do so at strikingly conserved positions, particularly in monozygotic twins where the overall folding pattern is significantly well conserved (Lohmann et al, 1999, 2008). Taken together, cortical folding patterns appear subject to strong genetic regulation.
In species with a simple pattern of folds, like ferret and cat, the stereotyped location of cortical folds and fissures is preceded and mirrored by regional variations in progenitor cell proliferation, in all three germinal layers, but most prominently in the OSVZ (Reillo et al, 2011). Local manipulations of OSVZ proliferation in ferret have a significant impact on the size and shape of cortical folds, without altering cortical area identity nor lamination (Reillo et al, 2011; Ghosh & Jessberger, 2013; Nonaka‐Kinoshita et al, 2013). To identify genes whose expression covaries with the stereotypic patterning of progenitor proliferation and cortical folds, microarray technology was recently used to compare the transcriptome of progenitors in prospective folds versus fissures of the developing ferret cortex (de Juan Romero et al, 2015). This analysis demonstrates the existence of thousands of genes differentially expressed (DEGs) between these regions, mostly in OSVZ and VZ. Identified DEGs include genes key in cortical development and folding like Trnp1 or Ccnd1, as well as genes mutated in human malformations of cortical development. Many DEGs are expressed in modules along the OSVZ and other germinal layers of the gyrencephalic ferret and also the human embryo cortex, but not in the lissencephalic mouse cortex (Sansom & Livesey, 2009; Elsen et al, 2013; de Juan Romero et al, 2015). Most remarkably, expression modules along the OSVZ map faithfully the eventual location of cortical folds and fissures (de Juan Romero et al, 2015). This strongly supports a role for the OSVZ and some of those DEGs on cortical patterning, particularly in its stereotyped folding (Kriegstein et al, 2006; Lui et al, 2011; Reillo et al, 2011; Albert & Huttner, 2015; Fig 3).
Figure 3. Patterns of gene expression map the prospective location of cortical folds in the developing ferret brain.
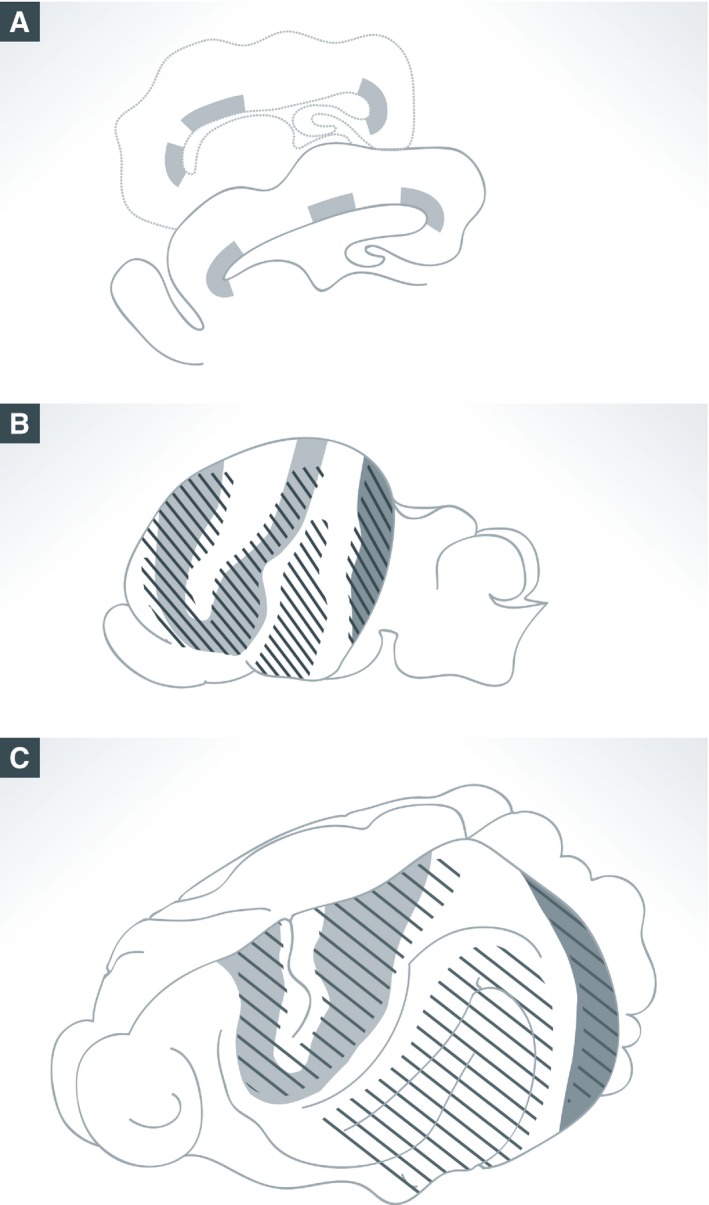
(A) Schema of sagittal sections of ferret brains at postnatal day P6 showing the modular pattern of mRNA expression for Eomes at the outer subventricular zone (shaded areas). (B, C) Representation of the ferret brain surface at postnatal day P2 (B) and adult (C) overlapped with the map of Eomes expression modules (shaded) and prospective gyri (striped pattern), showing the spatial correlation between Eomes expression and gyri (adapted from de Juan Romero et al, 2015).
Multiple genetic maps seem to overlap across germinal layers, as modular expression patterns also exist for DEGs in VZ and ISVZ of ferret and humans (de Juan Romero et al, 2015). Given that these germinal zones are major sites of neurogenesis and neural fate determination, these gene expression patterns may contribute significantly to further define cortical folds and/or functional areas of the cerebral cortex (Lui et al, 2011; Reillo et al, 2011; Taverna et al, 2014; Dehay et al, 2015). Indeed, many DEGs between the prospective gyrus and sulcus are known to regulate progenitor proliferation, neurogenesis, or fate specification, including key signaling pathways such as Notch, Shh, MAPK, and Wnt, which directly regulate cortical growth (see above). In the case of cortical folds, modular patterns of expression for a combination of genes, possibly different depending on the specific gyrus or sulcus, may impose differential tissue growth between modules, eventually leading to the evagination of the cortex and formation of folds (Smart & McSherry, 1986; Fig 3).
Small enhancer elements have been recently identified to drive reporter gene expression in discrete modules, or protodomains, also in the embryonic mouse cerebral cortex (Visel et al, 2013; Pattabiraman et al, 2014). These enhancers have been proposed to integrate broad transcriptional information, including expression of several transcription factors regulating cortical patterning, to define gene expression in those protodomains (Nord et al, 2013; Pattabiraman et al, 2014). Variations in such small enhancer elements may be indeed at the core of cortical patterning (and folding) during development and evolution (Borrell & Gotz, 2014; see below). However, the definition of discrete cortical subdivisions requires the regional control over the expression of protein‐coding genes or their interfering RNAs (i.e., cell cycle regulators, cell fate determinants, neuron terminal selectors) in protodomains along the embryonic germinal layers, as demonstrated in ferret and humans, but not in mouse (Dehay & Kennedy, 2007; Molyneaux et al, 2007; Hobert, 2011; de Juan Romero et al, 2015). Walsh and colleagues recently identified a key regulatory element for the expression of human GPR56 which varies significantly across mammals and, when introduced in transgenic mouse embryos, drives different patterns of expression (Bae et al, 2014). Most importantly, GPR56 expression levels regulate cortical progenitor proliferation, and mutation of this regulatory element disrupts human cortex folding around the Sylvian fissure, demonstrating the importance of the expression pattern of this gene in defining the pattern of folds. Similarly, mutations in the regulatory region of human EOMES lead to significant alterations of cortical size and folding (microcephaly with polymicrogyria; Baala et al, 2007). In agreement with the notion of a protomap of cortical folding, both GPR56 and EOMES are expressed in modular patterns in the developing ferret and human cortex (de Juan Romero et al, 2015).
Biomechanics of cortical folding
Cranial pressure
One of the first hypotheses on the biomechanics of cortical folding proposed that cranial volume limits cortical size, so that as the cortical tissue grows in surface area the skull offers resistance to its outward expansion, forcing the neural tissue to fold onto itself (Le Gros Clark, 1945). The concept that brain morphology adapts to fit in a limited volume has been recently supported by experiments in chick embryos, where an experimentally expanded optic tectum folds to maintain cranial size (McGowan et al, 2012). However, studies focused specifically on the mammalian cerebral cortex have shown that cortical folding takes place even in the absence of compressive constrain from the skull (Welker, 1990). For example, the removal of non‐cortical brain tissue in sheep embryos alleviated any possible cranial pressure onto the growing cortex, and yet this did not suppress or simplify the formation of cortical folds (Barron, 1950; Muckli et al, 2009). On the contrary, development of the skull appears to be strongly influenced by brain growth, as cranial volume is significantly enlarged in pathologies where brain and cortical size are abnormally large, such as hydrocephalus or megalencephaly (Barkovich et al, 2012).
Axonal tension
The first attempt to mathematically model the biomechanical basis of cortical folding dates back four decades (Richman et al, 1975). This model proposed that cortical folding occurs as a result of differential growth between upper and lower cortical layers, which generates stress that is sufficient to induce cortical surface buckling. Thirty years later, Kriegstein and colleagues (Kriegstein et al, 2006) proposed that basal progenitors increase significantly neurogenesis at later stages, precisely when upper layers form, and that this enables the differential growth between layers and ultimately drives cortical folding. Whereas the Richman model showed that this principle is sufficient to convert a flat surface into wavy, it relies on such a difference in stiffness between neuronal layers that it seems unrealistic (Bayly et al, 2014).
Two decades passed before an alternative model was proposed to explain the biomechanics of cortical folding: the tension‐based theory (Van Essen, 1997). This theory by Van Essen was conceptually based on D'Arcy Thompson's analysis on how tension and pressure can interact with structural asymmetries to determine the shape of biological structures (Thompson, 1917). Van Essen argued that cortico‐cortical axons are under strong tension, exerting significant pulling forces capable of deforming the cortical mantle, and that these cortico‐cortical connections are not symmetric or homogeneously distributed, but some areas are more strongly connected together than with others. Under this scenario, he proposed that those areas connected with a larger amount of axons withstand a greater pulling force, and thus come close together to form a fold, whereas areas poorly interconnected are relayed to the opposite banks of a sulcus. This hypothesis attracted the enthusiasm of many, not only for its simplicity but also for its coherence. Indeed, two simple but fundamental observations made this a very attractive model: (i) The cortical sheet is physically tethered in only one axis, initially by radial processes and followed by connections between cortex and subcortical nuclei (De Carlos & O'Leary, 1992). Tension along these processes may provide a cohesive force against intraventricular hydrostatic pressure, ensuring that the cortical mantle remains tightly wrapped around the subcortical interior. (ii) Specific and topographically organized cortico‐cortical projections are established early in development while convolutions are forming (Coogan & Van Essen, 1996; Hilgetag & Barbas, 2006). Van Essen proposed that if developing CNS axons in vivo generate even a modest fraction of the specific tension measured in vitro (Dennerll et al, 1988), then populations of axons pulling together should have ample strength to cause folding of the highly pliable embryonic cortical sheet (Van Essen, 1997). Importantly, this hypothesis also provided a basis for individual variability in brain morphology.
Remarkably, the tension‐based theory was widely accepted for more than a decade without rigorous experimental testing. But 14 years later, Taber, Bayly, and colleagues performed a series of very simple and elegant experiments that frontally challenged the foundations of this theory (Xu et al, 2010). Their idea was that if axonal tension between opposite sides of a gyrus pulls them together, then these should come apart if these axons are cut and tension is released. This was tested in living brain slices from developing ferrets of various ages throughout the period of gyrus formation. These experiments showed that axons are indeed under considerable tension in the developing brain, but most of this tension is found along axon bundles in deep white matter tracts, not within the core of individual gyri and thus too far to play a major role in initiating, sustaining, or maintaining cortical folding (Armstrong et al, 1991; Xu et al, 2010). In addition, other computational models show that cortical folding may occur in the complete absence of cortico‐cortical fibers, only as an effect of buckling instability (see below; Toro & Burnod, 2005).
Tissue buckling
Folding of the cerebral cortex, like any other tissue, is limited by its physical–mechanical properties of rigidity and pliability, which define the relationship between cortical thickness and surface area. As a general principle, given a cortical surface area, the periodicity of folding is inversely correlated with gray matter thickness (Hofman, 1985; Toro & Burnod, 2005; Pillay & Manger, 2007). This applies to interspecies differences (i.e., ungulates have a thinner and more folded cortex than primates, whereas in the manatee, the cortex is thicker and rather smooth; Welker, 1990) and also to human pathology, where lissencephalic patients with reduced folding display an abnormally thick cortex, and polymicrogyric cortices are abnormally thin (Olson & Walsh, 2002; Barkovich et al, 2012).
A very recent study proposes a general law for this inverse correlation between thickness and folding, based on measurements from 62 different species and describing a mathematical relationship between total cortical surface area, thickness, and exposed area (Mota & Herculano‐Houzel, 2015). The basic idea is that cortical folding is similar to crumpling a ball of paper, where the periodicity of individual folds will be high in a ball of thin smoking paper (small folds) and very low if using thick cardboard paper (large folds). According to this study, the combination of total cortical area and thickness grows with exposed cortical area to the power of 1.25, in a similar fashion as the area of a circle increases with its radius raised to the power of 2. This relationship is also proposed to represent the topological configuration involving the minimal energy, and therefore, cortical folding may settle into the configuration of least energy (Mota & Herculano‐Houzel, 2015). But there are several fundamental problems with the paper ball analogy: (i) It describes the folding of a structure that no longer grows, whereas the cortex folds while it continues to develop and grow; (ii) it is based on physical models only valid for single‐molecule thin materials; (iii) the theory considers that the whole brain folds, while folding only involves the cortical gray matter, not the rest of the brain (Mota & Herculano‐Houzel, 2015). Finally, folding of the cerebral cortex does not occur randomly, as crumpling of a paper ball, but in a rather highly stereotyped process, defining patterns that are characteristic and distinct for each species. Prior to their appearance, folding patterns are delineated by regional variations of progenitor cell proliferation (Reillo et al, 2011), and by modules of differential gene expression along germinal layers, as recently demonstrated via transcriptomic expression analyses (de Juan Romero et al, 2015). These fundamental heterogeneities of the developing cortex immediately prior to folding are completely ignored by the Mota and Herculano‐Houzel model. Additional problems with this model, at the mathematical level, have been recently highlighted (De Lussanet, 2016).
Using a finite element computational model, Taber and colleagues suggested that a critical factor leading to the formation of outward folds is differential cortical growth (Xu et al, 2010), which precisely matches our experimental data and histogenic model of regional cortical expansion and folding in ferret (Reillo et al, 2011). This hypothesis where the local difference in tissue growth, particularly lateral/tangential expansion, is a key factor driving cortical folding (Reillo et al, 2011; Borrell & Reillo, 2012; Borrell & Gotz, 2014) is progressively gaining acceptance in the field (Ronan et al, 2014; Ronan & Fletcher, 2015; Striedter et al, 2015). Although these new findings contradict Van Essen's tension‐based hypothesis of cortical folding, and axonal tension may not directly be the driving force of cortical folding, alternative computational models do propose that axonal tension may play an important role in tissue buckling (Chada et al, 1997). In this case, however, models propose that upon incipient folding, the mechanical stress created induces cell proliferation and differential growth, which further accentuates the folded appearance of the cortex and defines the shape of cortical folds (Toro & Burnod, 2005).
A major breakthrough in our understanding of the biomechanics underlying cortical folding came with a recent study by Tallinen and colleagues (Tallinen et al, 2014). Many of the ingredients of this model were already present in the mentioned models of Richman, Toro, and Taber (Richman et al, 1975; Toro & Burnod, 2005; Xu et al, 2010), but this new study is the first to propose a coherent framework with physical simulations, computational simulations, and a very compelling mathematical analysis of the problem. The key point of this model is that two different materials are sticking together while growing homogeneously, if one grows faster than the other, the system becomes unstable and will change shape by folding, which is an emergent property of any such mechanical system (Tallinen et al, 2014). Most interestingly, solely based on the slightly different physical properties of the two materials (i.e., upper versus lower cortical layers), homogeneous continuous growth is sufficient to lead the system to develop a dramatic change in shape, forming a heterogeneous pattern of stress that in turn can influence the biology of the tissue (i.e., cell proliferation, apoptosis, cell fate, and even axon guidance). Therefore, tissue buckling and cortical folding may ultimately result from the mutual influence between physical properties, biomechanics, and differential tissue growth.
Defects of cortical folding: human disease
The size and folding of the cerebral cortex have a fundamental impact on brain function. Significant changes (excess or defect) in these parameters are the most common cause of severe intellectual disability and intractable epilepsy (Guerrini et al, 2008; Andrade, 2009). Defects in human cortical development have been recognized as being originated by the disruption of some of the cellular and molecular mechanisms described above in this review (Barkovich et al, 2012), hence critically impacting on key developmental events. In this section, we briefly present these human malformations grouped according to the main affection: brain size, cortical folding, or the formation of ectopias (groups of cells in an abnormal location). It is important to note that cortical malformations usually appear as compound phenotypes, only very rarely as a single defect (Fig 4).
Figure 4. Human cortical malformations and their phenotypic manifestations.
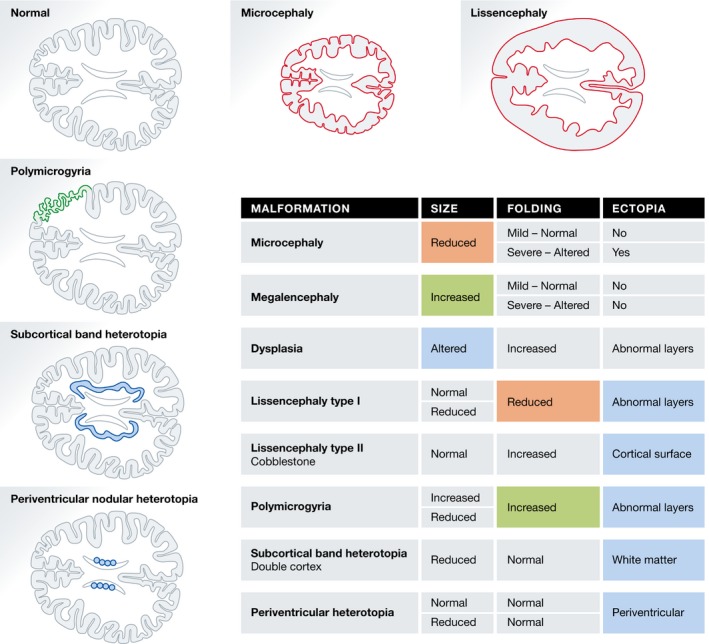
Schematic of horizontal sections through the cerebral cortex of a normal human brain compared to those of patients with cortical malformation: microcephaly, lissencephaly type I, polymicrogyria, subcortical band heterotopia (double cortex), and periventricular nodular heterotopia. The table summarizes the phenotypic manifestations associated with each malformation regarding brain size, cortical folding, and the formation of ectopias. The most representative effects are highlighted and color‐coded: Features negatively affected by the pathology are in red, features augmented in green, and particularities are in blue. Uncolored cells indicate additional alterations that may be associated with the primary defect.
Brain size
As explained in the first section, the pool size of founder and neurogenic cortical progenitors plays a central role in defining cortical size. Consequently, alterations in proliferation and/or survival of neural progenitors lead to abnormal brain size, either excessive (megalencephaly), defective (microcephaly), or imbalanced (dysplasia; Barkovich et al, 2012; Fig 4).
Microcephaly
Microcephaly is a rare developmental disorder in which affected individuals display a significantly reduced brain size compared to controls (Bond et al, 2002; Gilmore & Walsh, 2013). This condition may be mild (only brain size is affected) or severe (small brain size and altered cortical folding; Bilguvar et al, 2010; Yu et al, 2010; Adachi et al, 2011). Microcephaly has been associated with mutations in genes important for a wide variety of cellular processes: DNA repair efficiency; cell cycle length; mitotic spindle positioning; and centrosome maturation, duplication, and position (Table 1). For example, the most common causes of primary microcephaly are mutations in microcephalin (MCPH1), which lengthens the cell cycle and alters chromosome alignment during mitosis (Jackson et al, 2002; Woods et al, 2005; Gruber et al, 2011), and ASPM, which is important to maintain the orientation of the mitotic cleavage plane (Kumar et al, 2004; Shen et al, 2005; Fish et al, 2006; Gul et al, 2006; Table 1). For an extensive review on the role of different genes identified in microcephalic patients on the emergence of this condition, please refer to Bizzotto and Francis (2015).
Table 1.
Types of human cortical malformation, molecular mechanisms altered, and genes associated
| Malformation | Molecular mechanism | Genes | References | |
|---|---|---|---|---|
| Size | Microcephaly | DNA repair efficiency | MCPH1, PNKP, PNCT | Woods et al (2005); Griffith et al (2008); Sheen et al (2010); Gruber et al (2011) |
| Cell cycle length | ASPM, STIL, AKT3 | Boland et al (2007); Desir et al (2008); Kumar et al (2009); Passemard et al (2009) | ||
| Mitotic spindle positioning | ASPM, STIL, WDR62, NDE1, TCOF1, DYNC1H1, TUBG1, KIF5C, KIF2A | Feng & Walsh (2004); Bilguvar et al (2010); Nicholas et al (2010); Yu et al (2010); Sakai et al (2012); Poirier et al (2013) | ||
| Centrosome maturation, duplication, and position | NDE1, CDK5RAP2, CENPJ, ASPM, CMPH1, WDR62, STIL, CEP152, CEP63 | Abrieu et al (2000); Alkuraya et al (2011); Bhat et al (2011); Bond et al (2005); Graser et al (2007); Bakircioglu et al (2011); Marthiens et al (2013); Mirzaa et al (2014); Nicholas et al (2010); Sir et al (2011); Thornton & Woods (2009); Yao et al (2000) | ||
| Megalencephaly | Cell growth | PI3K‐AKT signaling AKT3, PIK3R2, PIK3CA | DiLiberti (1998); Lee et al (2012) #7409; Mirzaa et al (2013); Poduri et al (2013); Riviere et al (2012) | |
| Dysplasia | Cell cycle and growth, ribosome biogenesis, mRNA translation | mTOR pathway activation (tuberous sclerosis complex 1–tuberous sclerosis complex 2) | Crino et al (2006); Barkovich et al (2012) | |
| Folding | Lissencephaly type I | Radial migration | LIS1, DCX, TUBB3, TUBA1A, RELN | D'Arcangelo et al (1995); Sapir et al (1997); Pilz et al (1998); Caspi et al (2000); Dulabon et al (2000); Hong et al (2000); Rice & Curran (2001); Fallet‐Bianco et al (2008); Morris‐Rosendahl et al (2008); Kumar et al (2010) |
| Cortical lamination | RELN | D'Arcangelo et al (1995); Dulabon et al (2000); Hong et al (2000); Rice & Curran (2001) | ||
| Polymicrogyria | Cell adhesion, regulation of phosphorylation, cell motility, synaptogenesis, angiogenesis | SPRX2 | Roll et al (2006) | |
| Gene regulator | GPR56 | Piao et al (2002, 2004, 2005); Bae et al (2014) | ||
| Cytoskeleton regulation | TUBB2B, TUBB3, TUBA1A, TUBA8, KBP | Abdollahi et al (2009); Jaglin & Chelly (2009); Jansen et al (2011); Tischfield et al (2011); Poirier et al (2013); Valence et al (2013); Squier & Jansen (2014) | ||
| Neurite outgrowth | KBP | Valence et al (2013) | ||
| DNA repair efficiency | NHEJ1 | Cantagrel et al (2007) | ||
| Microdeletions in 22q11 | Robin et al (2006) | |||
| Suggested: centrosomal role | WDR62 | Yu et al (2010) | ||
| Ectopia | SBH/double cortex | Cytoskeleton regulation/neuronal migration defects | DCX, LIS1, TUBA1A, TUBG1, EML1 | Gleeson et al (1998); Francis et al (1999); Sicca et al (2003); Keays (2007); Mineyko et al (2010); Kielar et al (2014) |
| Lissencephaly type II (cobblestone) | Pial surface stability | POMT1; POMT2; FKTN, FKRP, LARGE, POMGNT1, LAMB1 | Brockington et al (2001); Yoshida et al (2001); Beltran‐Valero de Bernabe et al (2002); Longman et al (2003); van Reeuwijk et al (2005b); Roscioli et al (2012); Willer et al (2012); Kariminejad et al (2013) | |
| Periventricular heterotopia | Actin cytoskeleton | FLNA | Fox et al (1998); Sheen et al (2001); Parrini et al (2006); Ferland et al (2009) | |
| Vesicle trafficking | ARFGEF2 | Sheen et al (2004); Ferland et al (2009) | ||
| Neuronal migration | C6orf70 | Conti et al (2013) | ||
| Molecular adhesion | FAT4 | Cappello et al (2013) | ||
| Molecular adhesion | DCHS1 | Cappello et al (2013) | ||
| (unknown) | Microdeletions in 22q11 | Kiehl et al (2009) |
Megalencephaly
Megalencephaly is characterized by an abnormal enlargement of the brain, which has been related to an excessive production of progenitor cells and cortical neurons due to a decreased apoptosis, or a shortening of cell cycle and increased cell cycle re‐entry (Dehay & Kennedy, 2007; Hansen et al, 2010; Wang et al, 2011; Barkovich et al, 2012). As in other malformations, severe forms of megalencephaly may occur together with altered patterns of cortical folding. Usually, the increased abundance of progenitor cells and neurons results in polymicrogyria or excessive cortical folding (Barkovich et al, 2005). In fact, megalencephaly normally occurs in syndromes, in combination with other alterations of development, such as MPPH (macrocephaly, polymicrogyria, polydactyly, hydrocephalus), M‐CMTC (macrocephaly cutis marmorata telangiectasia congenita), and MCAP (macrocephaly capillary malformation; Mirzaa et al, 2004; Conway et al, 2007; Tore et al, 2009). The genetic causes of megalencephaly are only partially understood, but recent progress highlights the importance of phosphatidylinositol 3‐kinase (PI3K)‐Akt signaling, which seems to play a central role in controlling brain size (DiLiberti, 1998; Lee et al, 2012; Riviere et al, 2012; Mirzaa et al, 2013; Table 1).
Dysplasia
A very common group of malformations of cortical development related to epilepsy is focal cortical dysplasia (FCD), which classically has included patients showing a variety of histologic alterations such as cortical disorganization and architecture (abnormal layering, polymicrogyria) and cells with abnormal location or morphology (neuronal heterotopia, balloon cells, neuronal cytomegaly; Palmini et al, 2004; Barkovich et al, 2005). These alterations may appear in any part of the cortex and affect regions of different size, even multiple cortical lobes (Tassi et al, 2002), which determine the semiology of seizures. Genes in the mTOR pathway are emerging as important players on the origin of developmental cortical dysplasias (Crino et al, 2006; Barkovich et al, 2012). FCDs are distinguished in three different varieties (Blumcke et al, 2011; Barkovich et al, 2012): (i) type I (isolated), with disrupted cortical lamination that may be radial (Ia), tangential (Ib), or both (Ic); (ii) type II (isolated), presenting dysmorphic neurons with balloon cells (IIb) or without them (IIa); (iii) type III, associated with another main lesion, such as hippocampal sclerosis (IIIa), glial or glio‐neuronal tumor (IIIb), vascular malformation (IIIc), and others (trauma, ischemic injury, encephalitis) (IIId) (Blumcke et al, 2011).
Cortical folding
Alterations of cortical folding in the human brain have been classically attributed to defects of neuronal migration (Ross & Walsh, 2001). However, disruption of neuron migration and positioning also leads to other cortical defects with a mild alteration of cortical folding, such as lissencephaly type II and subcortical band heterotopia. The latter will be discussed in the next section focused on ectopias.
Lissencephaly (smooth brain)
This includes several disorders collectively characterized by the simplification of the folding pattern: agyria (complete absence of folds), pachygyria (simplified pattern of folds), and subcortical band heterotopia (gyral pattern is either normal or simplified with broad convolutions and a thickened cortex; Guerrini & Marini, 2006; Fig 4). Lissencephalies are classified into two main types: type I or classic, caused by mutations in genes related to the cytoskeleton and affecting cell migration; newborn neurons fail to migrate properly and, instead of forming the characteristic six layers, they accumulate below the preplate in only four distinguishable layers, resulting in a largely disorganized and thickened cortex (Golden & Harding, 2004). Type II, or cobblestone, is caused by alterations in the interaction between radial glia and the pial surface, which result in the disruption of the cortical surface and the overflow of neurons above the meninges (Bizzotto & Francis, 2015).
Most cases of type I lissencephaly are due to mutations in LIS1 or DCX (Pilz et al, 1998). These are proteins that interact with the tubulin cytoskeleton allowing its polymerization and stability (Sapir et al, 1997; Caspi et al, 2000). This is also the case for TUBB3 and TUBA1A, mutated in 1–4% of type I lissencephalies (Morris‐Rosendahl et al, 2008; Kumar et al, 2010) and 30% of lissencephalies with cerebellar hypoplasia (impaired growth). Interestingly, the loss and simplification of folds displayed by these patients are very similar to those associated with mutations in LIS1, suggesting a shared molecular pathway (Barkovich et al, 2012). A small number of patients with autosomal recessive type I lissencephaly with cerebellar hypoplasia have mutations in RELN, also a gene essential for radial migration and normal cortical lamination in mouse and humans (D'Arcangelo et al, 1995; Dulabon et al, 2000; Hong et al, 2000; Rice & Curran, 2001; Table 1).
Polymicrogyria (many small folds)
This includes a group of cortical malformations characterized by the formation of abnormally abundant and small cortical folds. It usually also involves the interdigitation of white matter resulting in abnormal lamination (Barkovich et al, 1999; Walsh, 2001). The defects in cortical lamination may be either simplification, with four layers similar to type I lissencephaly, or complete disruption and disorganization. Most frequently polymicrogyria (PMG) phenotypes are very complex and combined with other alterations such as microcephaly (Bilguvar et al, 2010; Yu et al, 2010). Due to this phenotypic complexity, the causative genes for human PMG have been very elusive. Genetic mutations linked to PMG include alterations in SPRX2 (Roll et al, 2006), microsomal deletions in 22q11 (Bassett et al, 2005; Robin et al, 2006), and mutations in a number of cytoskeleton‐associated genes (Table 1). Nongenetic causes of PMG have also been identified including insults during embryogenesis such as hypoxia, hypoperfusion, and congenital infections (Jacobs et al, 1999; Squier & Jansen, 2014).
Ectopia
The proper position of cortical neurons depends on a complex cellular and molecular regulation of two variables: where and when. Neurons must migrate through the entire cortical thickness and stop precisely near the cortical surface, a process determined by the time and place of their generation. Altering these events leads to misplaced neurons, a malformation generically called ectopia (out of place; Fig 4).
Subcortical band heterotopia/double cortex
This type of ectopia is characterized by the accumulation of neurons in the cortical white matter (Barkovich et al, 2001; Ross & Walsh, 2001). Typically, the cluster of ectopic neurons forms a thick band of cells below an otherwise normal cortical gray matter (Gleeson et al, 1998; Francis et al, 1999). Importantly, double cortex is accompanied by a reduction in the size of the cerebral cortex due to the loss of neurons from the normocortex. This frequently affects cortical surface area and thickness, and in some cases, it even results in microgyria (Barkovich et al, 2012). Genetic mutations causative of double cortex affect a variety of cytoskeleton‐interacting proteins (Table 1).
Cobblestone
Whereas most types of heterotopia are due to deficient neuronal migration, cobblestone (type II lissencephaly) is caused by their excessive migration. In this case, the anchoring and attachment of the radial fiber of RGCs to the pial surface is disrupted, thus altering the basement membrane (Yamamoto et al, 2004; Luo et al, 2011). Given that the cortical basement membrane and the attachment of RGCs to it are the finish line for radially migrating neurons, this disruption leads to their overmigration, which continue moving up to the meningeal space, thus resembling cobblestones on the cortical surface (van Reeuwijk et al, 2005a). Several complex syndromes cause cobblestone lissencephaly: Fukuyama congenital muscular dystrophy (FCMD), muscle–eye–brain disease (MEB), and Walker–Warburg syndrome (WWS). In spite of this wide spectrum of phenotypes, mutations linked to cobblestone are found in genes involved in the attachment of the radial glial fiber to the pial surface (Li et al, 2008; Luo et al, 2011), or associated with reduced glycosylation of alpha dystroglycan, which is fundamental to anchor the dystrophin complex to the extracellular matrix (van Reeuwijk et al, 2005a; Roscioli et al, 2012; Buysse et al, 2013). Six major genes have been identified encoding putative or confirmed glycosyltransferases (Table 1).
Periventricular heterotopia
Contrary to cobblestone, in periventricular heterotopia (PH) cortical neurons are unable to undergo radial migration. Due to defective remodeling of the actin cytoskeleton, newborn neurons cannot perform the changes in cell shape and locomotion required for their migration and completely fail to leave the germinal zones, remaining in the vicinity of the ventricular surface clustered into nodules, which eventually act as epileptic foci (Sheen et al, 2001, 2005; Sheen & Walsh, 2005; Sarkisian et al, 2006, 2008; Andrade, 2009). These periventricular nodules may appear in a variety of locations and conformations: bilateral, unilateral, laminar, sub‐ependymal, and subcortical white matter (Andrade, 2009; Ferland et al, 2009). Remarkably, most of the cortex appears completely normal and patients show no gross defects in intellectual development or performance. PH may appear alone or as part of complex syndromes, associated with other cortical malformations such as microcephaly (Parrini et al, 2006). The most frequent genetic alterations linked to periventricular nodular heterotopia affect FLNA and ARFGEF2 (Table 1; Fox et al, 1998; Sheen et al, 2001, 2004; Parrini et al, 2006; Ferland et al, 2009). Although these two proteins have very different cellular functions (FLNA acts on the actin cytoskeleton; ARFGEF2 has a role in intracellular membrane and vesicle trafficking), they may act in a common pathway and even interact directly (Sheen et al, 2004; Ferland et al, 2009). It has been proposed that the disruption of vesicle trafficking due to alterations of the cytoskeleton may impair cell adhesion and the integrity of the apical adherens junctions, thus leading to the formation of the periventricular nodules (Ferland et al, 2009).
Evolution of cortical folding
Brain size varies in several orders of magnitude between mammalian species, which is mostly the result of a disproportionate difference in size of the cerebral cortex (Finlay & Darlington, 1995). Increased cortical size is largely due to increased surface area (Rakic, 1995), and this is accompanied by cortical folding and fissuring, which in part allow its effective packing within a minimal cranial volume (Welker, 1990; Albert & Huttner, 2015). Based on our previous sections, evolution of cerebral cortex size and topology may be attributable to modifications in the abundance and behavior of cortical progenitor cells (Borrell & Reillo, 2012). Changing the duration of the cell cycle, generating IPCs, and increasing their abundance would augment exponentially the production of neurons and therefore brain size, whereas generating bRGCs would augment cortical surface area and folding (Borrell & Calegari, 2014; Lewitus et al, 2014).
Early gyrification and secondary loss
Classically, the remarkable expansion and folding of the mammalian cerebral cortex along evolution has been viewed as a unidirectional process, where the small and smooth cortex of a primitive ancestor (presumed similar to mouse) gradually evolved to be larger, more complex, and folded, and from there on, it further evolved to have an increasing number of folds, like the human cortex (Kriegstein et al, 2006; Rakic, 2009). However, this view was challenged recently with the hypothesis that gyrencephaly might be an evolutionarily ancient trait, expressed in a common ancestor to all mammals and retained during speciation (Borrell & Reillo, 2012). This hypothesis is based on two facts: a) gyrencephaly develops in species from across mammalian phylogeny, ranging from monotremes and marsupials to ungulates, carnivores, primates, and even rodents, and b) during embryonic development of the cerebral cortex, gyrencephaly differs from lissencephaly in two critical features: subdivision of SVZ into ISVZ and OSVZ and high abundance of basal progenitors (bRGCs and IPCs in ISVZ+OSVZ) that greatly outnumber apical progenitors (aRGCs in VZ; Reillo et al, 2011; Borrell & Reillo, 2012; Kelava et al, 2012; Reillo & Borrell, 2012). Based on these facts, it seems most parsimonious to propose that gyrencephaly emerged in the stem mammal ancestor upon the innovative generation of bRGCs and OSVZ, and these traits were retained along mammalian speciation. This hypothesis of early gyrification was subsequently supported by studies using a phenomic character matrix of living placental orders and fossil species, which conclude that the ancestor of placental mammals was a small gyrencephalic animal (O'Leary et al, 2013). In this scenario, the smooth cortex of lissencephalic mammals (namely rodents and lagomorphs) would have emerged by the simplification, or phenotypic reversal, of gyrencephaly. This reversal may have occurred by reducing the abundance and self‐amplificative capacity of basal neurogenic progenitors and bRGCs, as supported by recent studies (Kelava et al, 2012, 2013; Martinez‐Cerdeno et al, 2012; Borrell & Gotz, 2014; De Juan Romero & Borrell, 2015). There are various examples of phenotypic and genomic reversals documented (Teotonio & Rose, 2001), one of its attributes being the ability to acquire new evolutionary trajectories (Borowsky & Wilkens, 2002). The seeming ability of the mammalian brain to undergo significant phenotypic reversals and change in various directions during evolution may explain the remarkable adaptability of mammals along this process (Kelava et al, 2013).
Molecular evolution
As discussed previously, the biology of cortical progenitor cells is regulated by the coordinated action of multiple proteins, and the experimental manipulation of these proteins has a profound influence on cortical size and folding. However, it remains to be defined whether the development of folded versus smooth cortices is due to the differential regulation of these genes between species during their normal development, and if so how they became differently regulated during evolution. Only recently, we have begun to identify molecular changes occurred during evolution that provide answers to these questions.
Human accelerated regions (HARs)
Our understanding of the genetic basis of cerebral cortex evolution was jump‐started by the generalized interest in identifying the genetic determinants of human uniqueness (Dorus et al, 2004). Strategies to identify the genetic and molecular mechanisms underlying the distinction of humans focused on searching for variations between the genome of human and immediate relatives in phylogeny. In a seminal study, Lahn and colleagues compared the genomic sequence of humans, chimpanzee, rat, and mouse and found that the genome is moderately well conserved across these species, but the human genome showed sites of uniquely high divergence rate (Dorus et al, 2004). Subsequently, they also found evidence demonstrating that genetic evolution is still ongoing in humans, with hotspots in genes related to brain development and also relevant in pathology of human cortical development, such as ASPM and MCPH1 associated with microcephaly (Evans et al, 2004, 2005; Mekel‐Bobrov et al, 2005).
Whereas sequence changes in protein‐coding genes might seem the most straightforward mechanism to drive brain evolution (Hill & Walsh, 2005), further analyses have revealed an even more likely role for regions regulating gene expression (Prabhakar et al, 2008; Bae et al, 2014). Improved genomic comparisons between humans and chimpanzees identified hundreds of small DNA segments, the sequence of which diverged rapidly in humans (Pollard et al, 2006a,b). These segments were called “human accelerated regions” (HARs), in reference to their uniquely high rate of nucleotide substitution in the immediate human lineage. This accelerated evolution was proposed to have contributed to acquiring the unmatched size and complexity of the human brain in a relatively brief period (Pollard et al, 2006a). Importantly, instead of being part of protein‐coding genes, HARs are mainly located in introns and intergenic regions, strongly suggesting their role in gene regulation (Bejerano et al, 2006; Pollard et al, 2006b). Significantly, relevant genes nearby HARs code for transcription factors and other DNA binding proteins involved in development and morphogenesis. In fact, HAR1, the HAR with the highest level of difference, contains an RNA gene (HAR1F) expressed in Cajal–Retzius cells during development, a peculiar type of cell essential for neuronal migration and lamination of the cerebral cortex (Pollard et al, 2006b).
Novel regulatory sequences
Recent analyses of HARs focusing on their sequence, histone modification, chromatin state, and transcription factor binding sites conclude that at least 30% of HARs are developmental enhancers (Capra et al, 2013). Indeed, the activity of 29 noncoding HARs was tested by generating transgenic mice, which demonstrated that the majority of them are active enhancers in humans and chimpanzee. This confirms that HARs are good candidates as human‐specific regulatory regions and also that human‐specific brain evolution might be particularly associated with changes in spatial–temporal gene expression, instead of changes in protein sequence (King & Wilson, 1975; Mouse Genome Sequencing Consortium et al, 2002; Lunter et al, 2006; Pollard et al, 2006b; Ponting & Lunter, 2006; Prabhakar et al, 2008).
In order to investigate how the activity of enhancers influences the developing telencephalon, Visel and colleagues performed an in vivo digital atlas of transgenic mouse embryos using 145 selected enhancers to drive reporter gene expression (Visel et al, 2013). Using a similar strategy, Pattabirman and colleagues generated stable transgenic mouse lines to characterize the gene‐promoting activity of putative enhancers and demonstrated that these exhibit sharp boundaries of reporter gene expression in the E11.5 mouse pallium (Pattabiraman et al, 2014). These mouse lines were also used to determine the regional fate map of the mouse telencephalon, which demonstrated the existence of distinct progenitor protodomains defined by the activity of those enhancers at various developmental stages.
More recently, the evolution of active genomic enhancers was demonstrated with the analysis of the human GPR56 locus, mutated in malformations of cortical development including polymicrogyria (Bae et al, 2014). In this study, a collection of transgenic mice was generated driving GFP expression from a portion of the same GPR56 enhancer from various species (humans, mouse, marmoset, dolphin, and cat). The enhancer from these gyrencephalic species drove a similar GFP expression pattern, but this was different from the endogenous pattern in the lissencephalic mouse. Given that mouse and primates share a phylogenic ancestor more recent than with carnivores and cetaceans (Bininda‐Emonds et al, 2007), the similar regulation of GPR56 expression among these gyrencephalic species suggests convergent evolution.
Novel non‐protein‐coding genes
In addition to variations in enhancer sequences, an extra level of control over the spatial–temporal patterns of gene expression is provided by non‐protein‐coding RNAs. Mounting evidence demonstrates the relevance of noncoding RNAs (miRNAs, lncRNAs, etc.) on cerebral cortex development (Aprea & Calegari, 2015; Liu & Sun, 2015), and their significance on evolution is supported by their increased number at evolutionary divergence points (Heimberg et al, 2008). As for the regulation of cerebral cortex expansion and folding, hundreds of miRNAs have been found expressed in apical and basal progenitor cells of the developing cortex of macaque embryos, but not in mouse (Arcila et al, 2014). These primate miRNAs prominently target proteins regulating the cell cycle and neurogenesis, hence contributing to the differential growth, amplification, and complexification of germinal layers and cortical areas, including the OSVZ (Lukaszewicz et al, 2005; Dehay & Kennedy, 2007). Altogether, the coevolution of emergent miRNAs and their target genes suggests that novel miRNAs became integrated into ancient gene circuitry to exert additional control over cortical progenitors, ultimately leading to the acquisition of primate‐specific cortical features and, potentially, higher brain performance (Dehay et al, 2015).
Novel protein‐coding genes
As shown by Lahn and colleagues (Dorus et al, 2004), biological evolution also comes from variations in the sequence of protein‐coding genes (True & Carroll, 2002). Looking for gene innovations that might have been involved in cortical expansion and folding during evolution, the focus has recently turned specifically onto cortical progenitor cells. A transcriptomic search for genes differentially expressed in radial glial cells between species identified 56 genes expressed in human, but not mouse, RGCs (Florio et al, 2015). Among these, ARHGAP11B was uniquely intriguing because it had the highest degree of radial glia‐specific expression, and there is no mouse ortholog. In fact, ARHGAP11B arose from partial duplication of the ARHGAP11A gene on the human lineage after separation from the chimpanzees, and thus, it is a hominid‐specific gene (Antonacci et al, 2014). Most impressively, experimental expression of ARHGAP11B in aRGCs of the embryonic mouse cortex promoted the generation of self‐renewing basal progenitors (particularly bRGCs) and, in turn, increased cortical surface area and induced the formation of cortical folds in mouse (Florio et al, 2015). Therefore, the emergence of new protein‐coding genes along mammalian phylogeny, such as ARHGAP11B in the hominid lineage, may have contributed significantly to the evolutionary expansion and folding of the mammalian cerebral cortex.
Concluding remarks and open questions
The expansion and folding of the mammalian cerebral cortex during embryonic development is a rather complex process regulated by multiple factors, where the abundance, type, and lineage of cortical progenitor cells play central roles. These cellular mechanisms are subject to molecular regulation by multiple proteins and signaling pathways, the expression of which is tightly controlled by a variety of enhancer elements and non‐protein‐coding genes. The specific spatial–temporal expression patterns of some of these proteins on the embryonic cortex faithfully map the prospective pattern of folds and fissures, and their mutation frequently leads to malformations of cortical size and folding in human patients. Yet, we are still far from identifying the specific role of these genes and their spatial–temporal regulation on the normal development of the human cerebral cortex.
Studying cortical development and folding across species helps us to understand the evolution of this complex process, and in return, we hope that this helps us to understand the aspects of cortical development critical to its expansion and folding during embryogenesis. Again, more and more refined molecular and genomic analyses are shedding some light on this problem, and emerging experimental animal models in this field like the ferret are of great help, but the truth is that we remain far from having a significant level of understanding.
Given the complexity of the developmental mechanisms involved in the expansion and folding of the cerebral cortex, and thus its tremendous cost in terms of genetic, cellular, and histogenic evolution, the ecological advantages must be more than remarkable. But what are the advantages of cortical folding? Clearly, a bigger cortex contains more neurons and more neuropile, and thus in principle, it provides greater computational power. Folding brings together highly interconnected cortical areas, thus minimizing cortical wiring and favoring high speed associational communication, ultimately optimizing brain circuitry (Klyachko & Stevens, 2003). However, the human cortex is neither the largest nor the most folded among mammals, as compared with elephants or dolphins, for example, and yet it is assumed that humans have a higher intelligence over any other earthly species. Are we really more intelligent than these other species? (Roth & Dicke, 2005) Or it is only that we have the advantage of speech and manual ability to express our intellectual abilities? Is there a trade‐off between cortical size, folding, and intellectual performance? Modern humans and Neandertals co‐existed until 30,000 years ago, but we persisted and the latter perished. Was there some critical (albeit small) disadvantage in the organization or folding of the Neandertal brain? Was their extinction related to a less competitive intelligence? Were their cortical circuits or synapses less efficient? With the genomes of Neandertal and modern humans at hand, the specific differences in their DNA sequence identified (Green et al, 2010; Prufer et al, 2014), in combination with novel in vitro models of human brain development like cerebral organoids (Lancaster et al, 2013), these fascinating questions may soon be approachable. Hopefully, our understanding of the mechanisms and consequences of cortical expansion and folding will be much deeper 25 years from now.
Acknowledgements
We thank people from our laboratory for discussions and R. Toro for critical comments on the manuscript. We apologize that, because of space limitations, many primary and historical publications could not be cited. V.F. was recipient of a Formación de Personal Investigador fellowship from the Spanish Ministry of Economy and Competitivity (MINECO), and C.L. was supported by the European Union Seventh Framework Programme FP7/2007–2013 under the project DESIRE (Grant Agreement No. 602531). Work in our laboratory is also supported by grants from MINECO (BFU2012‐33473) and European Research Council (ERC StG309633) to V.B. The Instituto de Neurociencias is a “Centre of Excellence Severo Ochoa”.
Conflict of interest
The authors declare that they have no conflict of interest.
The EMBO Journal (2016) 35: 1021–1044
References
- Abdollahi MR, Morrison E, Sirey T, Molnar Z, Hayward BE, Carr IM, Springell K, Woods CG, Ahmed M, Hattingh L, Corry P, Pilz DT, Stoodley N, Crow Y, Taylor GR, Bonthron DT, Sheridan E (2009) Mutation of the variant alpha‐tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am J Hum Genet 85: 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrieu A, Kahana JA, Wood KW, Cleveland DW (2000) CENP‐E as an essential component of the mitotic checkpoint in vitro. Cell 102: 817–826 [DOI] [PubMed] [Google Scholar]
- Adachi Y, Poduri A, Kawaguch A, Yoon G, Salih MA, Yamashita F, Walsh CA, Barkovich AJ (2011) Congenital microcephaly with a simplified gyral pattern: associated findings and their significance. AJNR Am J Neuroradiol 32: 1123–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert M, Huttner WB (2015) Clever space saving‐how the cerebral cortex folds. EMBO J 34: 1845–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkuraya FS, Cai X, Emery C, Mochida GH, Al‐Dosari MS, Felie JM, Hill RS, Barry BJ, Partlow JN, Gascon GG, Kentab A, Jan M, Shaheen R, Feng Y, Walsh CA (2011) Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]. Am J Hum Genet 88: 536–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL (1997) Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278: 474–476 [DOI] [PubMed] [Google Scholar]
- Andrade DM (2009) Genetic basis in epilepsies caused by malformations of cortical development and in those with structurally normal brain. Hum Genet 126: 173–193 [DOI] [PubMed] [Google Scholar]
- Anton ES, Kreidberg JA, Rakic P (1999) Distinct functions of alpha3 and alpha(v) integrin receptors in neuronal migration and laminar organization of the cerebral cortex. Neuron 22: 277–289 [DOI] [PubMed] [Google Scholar]
- Anton ES, Marchionni MA, Lee KF, Rakic P (1997) Role of GGF/neuregulin signaling in interactions between migrating neurons and radial glia in the developing cerebral cortex. Development 124: 3501–3510 [DOI] [PubMed] [Google Scholar]
- Antonacci F, Dennis MY, Huddleston J, Sudmant PH, Steinberg KM, Rosenfeld JA, Miroballo M, Graves TA, Vives L, Malig M, Denman L, Raja A, Stuart A, Tang J, Munson B, Shaffer LG, Amemiya CT, Wilson RK, Eichler EE (2014) Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat Genet 46: 1293–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprea J, Calegari F (2015) Long non‐coding RNAs in corticogenesis: deciphering the non‐coding code of the brain. EMBO J 34: 2865–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcila ML, Betizeau M, Cambronne XA, Guzman E, Doerflinger N, Bouhallier F, Zhou H, Wu B, Rani N, Bassett DS, Borello U, Huissoud C, Goodman RH, Dehay C, Kosik KS (2014) Novel primate miRNAs coevolved with ancient target genes in germinal zone‐specific expression patterns. Neuron 81: 1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong E, Curtis M, Buxhoeveden DP, Fregoe C, Zilles K, Casanova MF, McCarthy WF (1991) Cortical gyrification in the rhesus monkey: a test of the mechanical folding hypothesis. Cereb Cortex 1: 426–432 [DOI] [PubMed] [Google Scholar]
- Attardo A, Calegari F, Haubensak W, Wilsch‐Brauninger M, Huttner WB (2008) Live imaging at the onset of cortical neurogenesis reveals differential appearance of the neuronal phenotype in apical versus basal progenitor progeny. PLoS ONE 3: e2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baala L, Briault S, Etchevers HC, Laumonnier F, Natiq A, Amiel J, Boddaert N, Picard C, Sbiti A, Asermouh A, Attie‐Bitach T, Encha‐Razavi F, Munnich A, Sefiani A, Lyonnet S (2007) Homozygous silencing of T‐box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat Genet 39: 454–456 [DOI] [PubMed] [Google Scholar]
- Bae BI, Tietjen I, Atabay KD, Evrony GD, Johnson MB, Asare E, Wang PP, Murayama AY, Im K, Lisgo SN, Overman L, Sestan N, Chang BS, Barkovich AJ, Grant PE, Topcu M, Politsky J, Okano H, Piao X, Walsh CA (2014) Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science 343: 764–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakircioglu M, Carvalho OP, Khurshid M, Cox JJ, Tuysuz B, Barak T, Yilmaz S, Caglayan O, Dincer A, Nicholas AK, Quarrell O, Springell K, Karbani G, Malik S, Gannon C, Sheridan E, Crosier M, Lisgo SN, Lindsay S, Bilguvar K, et al (2011) The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. Am J Hum Genet 88: 523–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB (2012) A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135: 1348–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkovich AJ, Hevner R, Guerrini R (1999) Syndromes of bilateral symmetrical polymicrogyria. AJNR Am J Neuroradiol 20: 1814–1821 [PMC free article] [PubMed] [Google Scholar]
- Barkovich AJ, Kuzniecky RI, Dobyns WB (2001) Radiologic classification of malformations of cortical development. Curr Opin Neurol 14: 145–149 [DOI] [PubMed] [Google Scholar]
- Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB (2005) A developmental and genetic classification for malformations of cortical development. Neurology 65: 1873–1887 [DOI] [PubMed] [Google Scholar]
- Barron DH (1950) An experimental analysis of some factors involved in the development of the fissure pattern of the cerebral cortex. J Exp Zool 113: 553–581 [Google Scholar]
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA (2005) Clinical features of 78 adults with 22q11 Deletion Syndrome. Am J Med Genet A 138: 307–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA, Altman J (1991) Neocortical Development. New York: Raven Press; [Google Scholar]
- Bayly PV, Taber LA, Kroenke CD (2014) Mechanical forces in cerebral cortical folding: a review of measurements and models. J Mech Behav Biomed Mater 29: 568–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano G, Lowe CB, Ahituv N, King B, Siepel A, Salama SR, Rubin EM, Kent WJ, Haussler D (2006) A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature 441: 87–90 [DOI] [PubMed] [Google Scholar]
- Beltran‐Valero de Bernabe D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der Zwaag B, Kayserili H, Merlini L, Chitayat D, Dobyns WB, Cormand B, Lehesjoki AE, Cruces J, Voit T, Walsh CA, van Bokhoven H, Brunner HG (2002) Mutations in the O‐mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker‐Warburg syndrome. Am J Hum Genet 71: 1033–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betizeau M, Cortay V, Patti D, Pfister S, Gautier E, Bellemin‐Menard A, Afanassieff M, Huissoud C, Douglas RJ, Kennedy H, Dehay C (2013) Precursor diversity and complexity of lineage relationships in the outer subventricular zone of the primate. Neuron 80: 442–457 [DOI] [PubMed] [Google Scholar]
- Bhat V, Girimaji SC, Mohan G, Arvinda HR, Singhmar P, Duvvari MR, Kumar A (2011) Mutations in WDR62, encoding a centrosomal and nuclear protein, in Indian primary microcephaly families with cortical malformations. Clin Genet 80: 532–540 [DOI] [PubMed] [Google Scholar]
- Bilguvar K, Ozturk AK, Louvi A, Kwan KY, Choi M, Tatli B, Yalnizoglu D, Tuysuz B, Caglayan AO, Gokben S, Kaymakcalan H, Barak T, Bakircioglu M, Yasuno K, Ho W, Sanders S, Zhu Y, Yilmaz S, Dincer A, Johnson MH et al (2010) Whole‐exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bininda‐Emonds OR, Cardillo M, Jones KE, MacPhee RD, Beck RM, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A (2007) The delayed rise of present‐day mammals. Nature 446: 507–512 [DOI] [PubMed] [Google Scholar]
- Bizzotto S, Francis F (2015) Morphological and functional aspects of progenitors perturbed in cortical malformations. Front Cell Neurosci 9: 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, Jacques TS, Avanzini G, Barkovich AJ, Battaglia G, Becker A, Cepeda C, Cendes F, Colombo N, Crino P, Cross JH, Delalande O, Dubeau F, Duncan J, Guerrini R et al (2011) The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 52: 158–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland E, Clayton‐Smith J, Woo VG, McKee S, Manson FD, Medne L, Zackai E, Swanson EA, Fitzpatrick D, Millen KJ, Sherr EH, Dobyns WB, Black GC (2007) Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet 81: 292–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF, Walsh CA, Woods CG (2002) ASPM is a major determinant of cerebral cortical size. Nat Genet 32: 316–320 [DOI] [PubMed] [Google Scholar]
- Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, Costa SM, Baralle D, Raponi M, Karbani G, Rashid Y, Jafri H, Bennett C, Corry P, Walsh CA, Woods CG (2005) A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 37: 353–355 [DOI] [PubMed] [Google Scholar]
- Borowsky R, Wilkens H (2002) Mapping a cave fish genome: polygenic systems and regressive evolution. J Hered 93: 19–21 [DOI] [PubMed] [Google Scholar]
- Borrell V, Calegari F (2014) Mechanisms of brain evolution: regulation of neural progenitor cell diversity and cell cycle length. Neurosci Res 86: 14–24 [DOI] [PubMed] [Google Scholar]
- Borrell V, Cardenas A, Ciceri G, Galceran J, Flames N, Pla R, Nobrega‐Pereira S, Garcia‐Frigola C, Peregrin S, Zhao Z, Ma L, Tessier‐Lavigne M, Marin O (2012) Slit/Robo signaling modulates the proliferation of central nervous system progenitors. Neuron 76: 338–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell V, Gotz M (2014) Role of radial glial cells in cerebral cortex folding. Curr Opin Neurobiol 27C: 39–46 [DOI] [PubMed] [Google Scholar]
- Borrell V, Reillo I (2012) Emerging roles of neural stem cells in cerebral cortex development and evolution. Dev Neurobiol 72: 955–971 [DOI] [PubMed] [Google Scholar]
- Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, Ponting CP, Estournet B, Romero NB, Mercuri E, Voit T, Sewry CA, Guicheney P, Muntoni F (2001) Mutations in the fukutin‐related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha‐dystroglycan. Am J Hum Genet 69: 1198–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultje RS, Castaneda‐Castellanos DR, Jan LY, Jan YN, Kriegstein AR, Shi SH (2009) Mammalian Par3 regulates progenitor cell asymmetric division via notch signaling in the developing neocortex. Neuron 63: 189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buysse K, Riemersma M, Powell G, van Reeuwijk J, Chitayat D, Roscioli T, Kamsteeg EJ, van den Elzen C, van Beusekom E, Blaser S, Babul‐Hirji R, Halliday W, Wright GJ, Stemple DL, Lin YY, Lefeber DJ, van Bokhoven H (2013) Missense mutations in beta‐1,3‐N‐acetylglucosaminyltransferase 1 (B3GNT1) cause Walker‐Warburg syndrome. Hum Mol Genet 22: 1746–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch‐Brauninger M, Lewitus E, Sykes A, Hevers W, Lancaster M, Knoblich JA, Lachmann R, Paabo S, Huttner WB, Treutlein B (2015) Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci USA 112: 15672–15677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantagrel V, Lossi AM, Lisgo S, Missirian C, Borges A, Philip N, Fernandez C, Cardoso C, Figarella‐Branger D, Moncla A, Lindsay S, Dobyns WB, Villard L (2007) Truncation of NHEJ1 in a patient with polymicrogyria. Hum Mutat 28: 356–364 [DOI] [PubMed] [Google Scholar]
- Cappello S, Gray MJ, Badouel C, Lange S, Einsiedler M, Srour M, Chitayat D, Hamdan FF, Jenkins ZA, Morgan T, Preitner N, Uster T, Thomas J, Shannon P, Morrison V, Di Donato N, Van Maldergem L, Neuhann T, Newbury‐Ecob R, Swinkells M et al (2013) Mutations in genes encoding the cadherin receptor‐ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat Genet 45: 1300–1308 [DOI] [PubMed] [Google Scholar]
- Capra JA, Erwin GD, McKinsey G, Rubenstein JL, Pollard KS (2013) Many human accelerated regions are developmental enhancers. Philos Trans R Soc Lond B Biol Sci 368: 20130025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi M, Atlas R, Kantor A, Sapir T, Reiner O (2000) Interaction between LIS1 and doublecortin, two lissencephaly gene products. Hum Mol Genet 9: 2205–2213 [DOI] [PubMed] [Google Scholar]
- Castro DS, Skowronska‐Krawczyk D, Armant O, Donaldson IJ, Parras C, Hunt C, Critchley JA, Nguyen L, Gossler A, Gottgens B, Matter JM, Guillemot F (2006) Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev Cell 11: 831–844 [DOI] [PubMed] [Google Scholar]
- Chada S, Lamoureux P, Buxbaum RE, Heidemann SR (1997) Cytomechanics of neurite outgrowth from chick brain neurons. J Cell Sci 110(Pt 10): 1179–1186 [DOI] [PubMed] [Google Scholar]
- Chenn A, McConnell SK (1995) Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell 82: 631–641 [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA (2002) Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297: 365–369 [DOI] [PubMed] [Google Scholar]
- Conti V, Carabalona A, Pallesi‐Pocachard E, Parrini E, Leventer RJ, Buhler E, McGillivray G, Michel FJ, Striano P, Mei D, Watrin F, Lise S, Pagnamenta AT, Taylor JC, Kini U, Clayton‐Smith J, Novara F, Zuffardi O, Dobyns WB, Scheffer IE et al (2013) Periventricular heterotopia in 6q terminal deletion syndrome: role of the C6orf70 gene. Brain 136: 3378–3394 [DOI] [PubMed] [Google Scholar]
- Conway RL, Pressman BD, Dobyns WB, Danielpour M, Lee J, Sanchez‐Lara PA, Butler MG, Zackai E, Campbell L, Saitta SC, Clericuzio CL, Milunsky JM, Hoyme HE, Shieh J, Moeschler JB, Crandall B, Lauzon JL, Viskochil DH, Harding B, Graham JM Jr (2007) Neuroimaging findings in macrocephaly‐capillary malformation: a longitudinal study of 17 patients. Am J Med Genet A 143A: 2981–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coogan TA, Van Essen DC (1996) Development of connections within and between areas V1 and V2 of macaque monkeys. J Comp Neurol 372: 327–342 [DOI] [PubMed] [Google Scholar]
- Costa MR, Wen G, Lepier A, Schroeder T, Gotz M (2008) Par‐complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development 135: 11–22 [DOI] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Henske EP (2006) The tuberous sclerosis complex. N Engl J Med 355: 1345–1356 [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T (1995) A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374: 719–723 [DOI] [PubMed] [Google Scholar]
- De Carlos JA, O'Leary DD (1992) Growth and targeting of subplate axons and establishment of major cortical pathways. J Neurosci 12: 1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Juan Romero C, Borrell V (2015) Coevolution of radial glial cells and the cerebral cortex. Glia 63: 1303–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lussanet MH (2016) Comments on “Cortical folding scales universally with surface area and thickness, not number of neurons”. Science 351: 2 [DOI] [PubMed] [Google Scholar]
- Dehay C, Kennedy H (2007) Cell‐cycle control and cortical development. Nat Rev Neurosci 8: 438–450 [DOI] [PubMed] [Google Scholar]
- Dehay C, Kennedy H, Kosik KS (2015) The outer subventricular zone and primate‐specific cortical complexification. Neuron 85: 683–694 [DOI] [PubMed] [Google Scholar]
- Dennerll TJ, Joshi HC, Steel VL, Buxbaum RE, Heidemann SR (1988) Tension and compression in the cytoskeleton of PC‐12 neurites. II: quantitative measurements. J Cell Biol 107: 665–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desir J, Cassart M, David P, Van Bogaert P, Abramowicz M (2008) Primary microcephaly with ASPM mutation shows simplified cortical gyration with antero‐posterior gradient pre‐ and post‐natally. Am J Med Genet A 146A: 1439–1443 [DOI] [PubMed] [Google Scholar]
- DiLiberti JH (1998) Inherited macrocephaly‐hamartoma syndromes. Am J Med Genet 79: 284–290 [DOI] [PubMed] [Google Scholar]
- Dorus S, Vallender EJ, Evans PD, Anderson JR, Gilbert SL, Mahowald M, Wyckoff GJ, Malcom CM, Lahn BT (2004) Accelerated evolution of nervous system genes in the origin of Homo sapiens. Cell 119: 1027–1040 [DOI] [PubMed] [Google Scholar]
- Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES (2000) Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron 27: 33–44 [DOI] [PubMed] [Google Scholar]
- Elias LA, Wang DD, Kriegstein AR (2007) Gap junction adhesion is necessary for radial migration in the neocortex. Nature 448: 901–907 [DOI] [PubMed] [Google Scholar]
- Elsen GE, Hodge RD, Bedogni F, Daza RA, Nelson BR, Shiba N, Reiner SL, Hevner RF (2013) The protomap is propagated to cortical plate neurons through an Eomes‐dependent intermediate map. Proc Natl Acad Sci USA 110: 4081–4086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF (2005) Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25: 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PD, Anderson JR, Vallender EJ, Gilbert SL, Malcom CM, Dorus S, Lahn BT (2004) Adaptive evolution of ASPM, a major determinant of cerebral cortical size in humans. Hum Mol Genet 13: 489–494 [DOI] [PubMed] [Google Scholar]
- Evans PD, Gilbert SL, Mekel‐Bobrov N, Vallender EJ, Anderson JR, Vaez‐Azizi LM, Tishkoff SA, Hudson RR, Lahn BT (2005) Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans. Science 309: 1717–1720 [DOI] [PubMed] [Google Scholar]
- Fallet‐Bianco C, Loeuillet L, Poirier K, Loget P, Chapon F, Pasquier L, Saillour Y, Beldjord C, Chelly J, Francis F (2008) Neuropathological phenotype of a distinct form of lissencephaly associated with mutations in TUBA1A. Brain 131: 2304–2320 [DOI] [PubMed] [Google Scholar]
- Feng Y, Walsh CA (2004) The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol 6: 1034–1038 [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Batiz LF, Neal J, Lian G, Bundock E, Lu J, Hsiao YC, Diamond R, Mei D, Banham AH, Brown PJ, Vanderburg CR, Joseph J, Hecht JL, Folkerth R, Guerrini R, Walsh CA, Rodriguez EM, Sheen VL (2009) Disruption of neural progenitors along the ventricular and subventricular zones in periventricular heterotopia. Hum Mol Genet 18: 497–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fietz SA, Huttner WB (2011) Cortical progenitor expansion, self‐renewal and neurogenesis‐a polarized perspective. Curr Opin Neurobiol 21: 23–35 [DOI] [PubMed] [Google Scholar]
- Fietz SA, Kelava I, Vogt J, Wilsch‐Brauninger M, Stenzel D, Fish JL, Corbeil D, Riehn A, Distler W, Nitsch R, Huttner WB (2010) OSVZ progenitors of human and ferret neocortex are epithelial‐like and expand by integrin signaling. Nat Neurosci 13: 690–699 [DOI] [PubMed] [Google Scholar]
- Fietz SA, Lachmann R, Brandl H, Kircher M, Samusik N, Schroder R, Lakshmanaperumal N, Henry I, Vogt J, Riehn A, Distler W, Nitsch R, Enard W, Paabo S, Huttner WB (2012) Transcriptomes of germinal zones of human and mouse fetal neocortex suggest a role of extracellular matrix in progenitor self‐renewal. Proc Natl Acad Sci USA 109: 11836–11841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay BL, Darlington RB (1995) Linked regularities in the development and evolution of mammalian brains. Science 268: 1578–1584 [DOI] [PubMed] [Google Scholar]
- Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB (2006) Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci USA 103: 10438–10443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, Albert M, Taverna E, Namba T, Brandl H, Lewitus E, Haffner C, Sykes A, Wong FK, Peters J, Guhr E, Klemroth S, Prufer K, Kelso J, Naumann R, Nusslein I, Dahl A, Lachmann R, Paabo S, Huttner WB (2015) Human‐specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 347: 1465–1470 [DOI] [PubMed] [Google Scholar]
- Florio M, Huttner WB (2014) Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141: 2182–2194 [DOI] [PubMed] [Google Scholar]
- Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA, Scheffer IE, Dobyns WB, Hirsch BA, Radtke RA, Berkovic SF, Huttenlocher PR, Walsh CA (1998) Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21: 1315–1325 [DOI] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald‐Netter Y, Denoulet P, Chelly J (1999) Doublecortin is a developmentally regulated, microtubule‐associated protein expressed in migrating and differentiating neurons. Neuron 23: 247–256 [DOI] [PubMed] [Google Scholar]
- Gaiano N, Nye JS, Fishell G (2000) Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron 26: 395–404 [DOI] [PubMed] [Google Scholar]
- Gao P, Postiglione MP, Krieger TG, Hernandez L, Wang C, Han Z, Streicher C, Papusheva E, Insolera R, Chugh K, Kodish O, Huang K, Simons BD, Luo L, Hippenmeyer S, Shi SH (2014) Deterministic progenitor behavior and unitary production of neurons in the neocortex. Cell 159: 775–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz CC, Kriegstein AR (2015) Neuronal migration dynamics in the developing Ferret cortex. J Neurosci 35: 14307–14315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh L, Jessberger S (2013) Supersize me‐new insights into cortical expansion and gyration of the mammalian brain. EMBO J 32: 1793–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore EC, Walsh CA (2013) Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip Rev Dev Biol 2: 461–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA (1998) Doublecortin, a brain‐specific gene mutated in human X‐linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92: 63–72 [DOI] [PubMed] [Google Scholar]
- Golden JA, Harding BN (2004) Pathology and genetics In Developmental Neuropathology, Golden JA, Harding BN. (eds) p 386 Basel: Neuropath Press; [Google Scholar]
- Gotz M, Huttner WB (2005) The cell biology of neurogenesis. Nature Rev Mol Cell Biol 6: 777–788 [DOI] [PubMed] [Google Scholar]
- Graser S, Stierhof YD, Nigg EA (2007) Cep68 and Cep215 (Cdk5rap2) are required for centrosome cohesion. J Cell Sci 120: 4321–4331 [DOI] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, Hansen NF, Durand EY, Malaspinas AS, Jensen JD, Marques‐Bonet T, Alkan C, Prufer K, Meyer M, Burbano HA, Good JM et al (2010) A draft sequence of the Neandertal genome. Science 328: 710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith E, Walker S, Martin CA, Vagnarelli P, Stiff T, Vernay B, Al Sanna N, Saggar A, Hamel B, Earnshaw WC, Jeggo PA, Jackson AP, O'Driscoll M (2008) Mutations in pericentrin cause Seckel syndrome with defective ATR‐dependent DNA damage signaling. Nat Genet 40: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross RE, Mehler MF, Mabie PC, Zang Z, Santschi L, Kessler JA (1996) Bone morphogenetic proteins promote astroglial lineage commitment by mammalian subventricular zone progenitor cells. Neuron 17: 595–606 [DOI] [PubMed] [Google Scholar]
- Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart PO, Wang ZQ (2011) MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1‐Cdc25 pathway. Nat Cell Biol 13: 1325–1334 [DOI] [PubMed] [Google Scholar]
- Guerrini R, Dobyns WB, Barkovich AJ (2008) Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci 31: 154–162 [DOI] [PubMed] [Google Scholar]
- Guerrini R, Marini C (2006) Genetic malformations of cortical development. Exp Brain Res 173: 322–333 [DOI] [PubMed] [Google Scholar]
- Gul A, Hassan MJ, Mahmood S, Chen W, Rahmani S, Naseer MI, Dellefave L, Muhammad N, Rafiq MA, Ansar M, Chishti MS, Ali G, Siddique T, Ahmad W (2006) Genetic studies of autosomal recessive primary microcephaly in 33 Pakistani families: novel sequence variants in ASPM gene. Neurogenetics 7: 105–110 [DOI] [PubMed] [Google Scholar]
- Gupta A, Sanada K, Miyamoto DT, Rovelstad S, Nadarajah B, Pearlman AL, Brunstrom J, Tsai LH (2003) Layering defect in p35 deficiency is linked to improper neuronal‐glial interaction in radial migration. Nat Neurosci 6: 1284–1291 [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PR, Kriegstein AR (2010) Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464: 554–561 [DOI] [PubMed] [Google Scholar]
- Harrison‐Uy SJ, Siegenthaler JA, Faedo A, Rubenstein JL, Pleasure SJ (2013) CoupTFI interacts with retinoic acid signaling during cortical development. PLoS ONE 8: e58219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama J, Bessho Y, Katoh K, Ookawara S, Fujioka M, Guillemot F, Kageyama R (2004) Hes genes regulate size, shape and histogenesis of the nervous system by control of the timing of neural stem cell differentiation. Development 131: 5539–5550 [DOI] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, Huttner WB (2004) Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci USA 101: 3196–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimberg AM, Sempere LF, Moy VN, Donoghue PC, Peterson KJ (2008) MicroRNAs and the advent of vertebrate morphological complexity. Proc Natl Acad Sci USA 105: 2946–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgetag CC, Barbas H (2006) Role of mechanical factors in the morphology of the primate cerebral cortex. PLoS Comput Biol 2: e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RS, Walsh CA (2005) Molecular insights into human brain evolution. Nature 437: 64–67 [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N, Gotoh Y (2004) The Wnt/beta‐catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 131: 2791–2801 [DOI] [PubMed] [Google Scholar]
- Hobert O (2011) Regulation of terminal differentiation programs in the nervous system. Annu Rev Cell Dev Biol 27: 681–696 [DOI] [PubMed] [Google Scholar]
- Hofman MA (1985) Size and shape of the cerebral cortex in mammals. I. The Cortical Surface. Brain Behav Evol 27: 28–40 [DOI] [PubMed] [Google Scholar]
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA (2000) Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet 26: 93–96 [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Yamaguchi M, Mori K, Kageyama R (2010) Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J Neurosci 30: 3489–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, Carr IM, Roberts E, Hampshire DJ, Crow YJ, Mighell AJ, Karbani G, Jafri H, Rashid Y, Mueller RF, Markham AF, Woods CG (2002) Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet 71: 136–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KM, Hwang BJ, Prince DA (1999) Focal epileptogenesis in a rat model of polymicrogyria. J Neurophysiol 81: 159–173 [DOI] [PubMed] [Google Scholar]
- Jaglin XH, Chelly J (2009) Tubulin‐related cortical dysgeneses: microtubule dysfunction underlying neuronal migration defects. Trends Genet 25: 555–566 [DOI] [PubMed] [Google Scholar]
- Jansen AC, Oostra A, Desprechins B, De Vlaeminck Y, Verhelst H, Regal L, Verloo P, Bockaert N, Keymolen K, Seneca S, De Meirleir L, Lissens W (2011) TUBA1A mutations: from isolated lissencephaly to familial polymicrogyria. Neurology 76: 988–992 [DOI] [PubMed] [Google Scholar]
- Johnson MB, Wang PP, Atabay KD, Murphy EA, Doan RN, Hecht JL, Walsh CA (2015) Single‐cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat Neurosci 18: 637–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Juan Romero C, Bruder C, Tomasello U, Sanz‐Anquela JM, Borrell V (2015) Discrete domains of gene expression in germinal layers distinguish the development of gyrencephaly. EMBO J 34: 1859–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, Wong LC, Shi SH, Hebert JM (2009) The transition from radial glial to intermediate progenitor cell is inhibited by FGF signaling during corticogenesis. J Neurosci 29: 14571–14580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariminejad A, Radmanesh F, Rezayi AR, Tonekaboni SH, Gleeson JG (2013) Megalencephaly‐polymicrogyria‐polydactyly‐hydrocephalus syndrome: a case report. J Child Neurol 28: 651–657 [DOI] [PubMed] [Google Scholar]
- Keays DA (2007) Neuronal migration: unraveling the molecular pathway with humans, mice, and a fungus. Mamm Genome 18: 425–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelava I, Lewitus E, Huttner WB (2013) The secondary loss of gyrencephaly as an example of evolutionary phenotypical reversal. Front Neuroanat 7: 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelava I, Reillo I, Murayama AY, Kalinka AT, Stenzel D, Tomancak P, Matsuzaki F, Lebrand C, Sasaki E, Schwamborn JC, Okano H, Huttner WB, Borrell V (2012) Abundant Occurrence of Basal Radial Glia in the Subventricular Zone of Embryonic Neocortex of a Lissencephalic Primate, the Common Marmoset Callithrix jacchus. Cereb Cortex 22: 469–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehl TR, Chow EW, Mikulis DJ, George SR, Bassett AS (2009) Neuropathologic features in adults with 22q11.2 deletion syndrome. Cereb Cortex 19: 153–164 [DOI] [PubMed] [Google Scholar]
- Kielar M, Tuy FP, Bizzotto S, Lebrand C, de Juan Romero C, Poirier K, Oegema R, Mancini GM, Bahi‐Buisson N, Olaso R, Le Moing AG, Boutourlinsky K, Boucher D, Carpentier W, Berquin P, Deleuze JF, Belvindrah R, Borrell V, Welker E, Chelly J et al (2014) Mutations in Eml1 lead to ectopic progenitors and neuronal heterotopia in mouse and human. Nat Neurosci 17: 923–933 [DOI] [PubMed] [Google Scholar]
- King MC, Wilson AC (1975) Evolution at two levels in humans and chimpanzees. Science 188: 107–116 [DOI] [PubMed] [Google Scholar]
- Kingsbury MA, Rehen SK, Contos JJ, Higgins CM, Chun J (2003) Non‐proliferative effects of lysophosphatidic acid enhance cortical growth and folding. Nat Neurosci 6: 1292–1299 [DOI] [PubMed] [Google Scholar]
- Klyachko VA, Stevens CF (2003) Connectivity optimization and the positioning of cortical areas. Proc Natl Acad Sci USA 100: 7937–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornack DR, Rakic P (1998) Changes in cell‐cycle kinetics during the development and evolution of primate neocortex. Proc Natl Acad Sci USA 95: 1242–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk T, Pontious A, Englund C, Daza RA, Bedogni F, Hodge R, Attardo A, Bell C, Huttner WB, Hevner RF (2009) Intermediate neuronal progenitors (basal progenitors) produce pyramidal‐projection neurons for all layers of cerebral cortex. Cereb Cortex 19: 2439–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez‐Buylla A (2009) The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32: 149–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Noctor S, Martinez‐Cerdeno V (2006) Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat Rev Neurosci 7: 883–890 [DOI] [PubMed] [Google Scholar]
- Kumar A, Blanton SH, Babu M, Markandaya M, Girimaji SC (2004) Genetic analysis of primary microcephaly in Indian families: novel ASPM mutations. Clin Genet 66: 341–348 [DOI] [PubMed] [Google Scholar]
- Kumar RA, Marshall CR, Badner JA, Babatz TD, Mukamel Z, Aldinger KA, Sudi J, Brune CW, Goh G, Karamohamed S, Sutcliffe JS, Cook EH, Geschwind DH, Dobyns WB, Scherer SW, Christian SL (2009) Association and mutation analyses of 16p11.2 autism candidate genes. PLoS ONE 4: e4582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, Pilz DT, Babatz TD, Cushion TD, Harvey K, Topf M, Yates L, Robb S, Uyanik G, Mancini GM, Rees MI, Harvey RJ, Dobyns WB (2010) TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet 19: 2817–2827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMonica BE, Lui JH, Hansen DV, Kriegstein AR (2013) Mitotic spindle orientation predicts outer radial glial cell generation in human neocortex. Nat Commun 4: 1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA (2013) Cerebral organoids model human brain development and microcephaly. Nature 501: 373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Huttner WB, Calegari F (2009) Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 5: 320–331 [DOI] [PubMed] [Google Scholar]
- Le Gros Clark ME (1945) Deformation patterns on the cerebral cortex In Essays on Growth and Form, Le Gros Clark WE, Medawar PB. (eds), pp 1–22. London: Clarendon; [Google Scholar]
- Lee JH, Huynh M, Silhavy JL, Kim S, Dixon‐Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, Gleeson JG (2012) De novo somatic mutations in components of the PI3K‐AKT3‐mTOR pathway cause hemimegalencephaly. Nat Genet 44: 941–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Walsh CA (2011) Neurogenesis at the brain‐cerebrospinal fluid interface. Annu Rev Cell Dev Biol 27: 653–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, D'Ercole AJ, Wong ET, LaMantia AS, Walsh CA (2011) The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron 69: 893–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewitus E, Kelava I, Kalinka AT, Tomancak P, Huttner WB (2014) An adaptive threshold in Mammalian neocortical evolution. PLoS Biol 12: e1002000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin Z, Koirala S, Bu L, Xu L, Hynes RO, Walsh CA, Corfas G, Piao X (2008) GPR56 regulates pial basement membrane integrity and cortical lamination. J Neurosci 28: 5817–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cogswell CA, LoTurco JJ (1998) Neuronal differentiation of precursors in the neocortical ventricular zone is triggered by BMP. J Neurosci 18: 8853–8862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sun T (2015) microRNAs and molecular pathogenesis of microcephaly. Curr Mol Pharmacol doi:10.2174/1874467208666150928153949 [DOI] [PubMed] [Google Scholar]
- Lohmann G, von Cramon DY, Colchester AC (2008) Deep sulcal landmarks provide an organizing framework for human cortical folding. Cereb Cortex 18: 1415–1420 [DOI] [PubMed] [Google Scholar]
- Lohmann G, von Cramon DY, Steinmetz H (1999) Sulcal variability of twins. Cereb Cortex 9: 754–763 [DOI] [PubMed] [Google Scholar]
- Longman C, Brockington M, Torelli S, Jimenez‐Mallebrera C, Kennedy C, Khalil N, Feng L, Saran RK, Voit T, Merlini L, Sewry CA, Brown SC, Muntoni F (2003) Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha‐dystroglycan. Hum Mol Genet 12: 2853–2861 [DOI] [PubMed] [Google Scholar]
- Lui JH, Hansen DV, Kriegstein AR (2011) Development and evolution of the human neocortex. Cell 146: 18–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewicz A, Savatier P, Cortay V, Giroud P, Huissoud C, Berland M, Kennedy H, Dehay C (2005) G1 phase regulation, area‐specific cell cycle control, and cytoarchitectonics in the primate cortex. Neuron 47: 353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewicz A, Savatier P, Cortay V, Kennedy H, Dehay C (2002) Contrasting effects of basic fibroblast growth factor and neurotrophin 3 on cell cycle kinetics of mouse cortical stem cells. J Neurosci 22: 6610–6622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, Ponting CP, Hein J (2006) Genome‐wide identification of human functional DNA using a neutral indel model. PLoS Comput Biol 2: e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Jeong SJ, Jin Z, Strokes N, Li S, Piao X (2011) G protein‐coupled receptor 56 and collagen III, a receptor‐ligand pair, regulates cortical development and lamination. Proc Natl Acad Sci USA 108: 12925–12930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabie PC, Mehler MF, Kessler JA (1999) Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J Neurosci 19: 7077–7088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machon O, van den Bout CJ, Backman M, Kemler R, Krauss S (2003) Role of beta‐catenin in the developing cortical and hippocampal neuroepithelium. Neuroscience 122: 129–143 [DOI] [PubMed] [Google Scholar]
- Mairet‐Coello G, Tury A, DiCicco‐Bloom E (2009) Insulin‐like growth factor‐1 promotes G(1)/S cell cycle progression through bidirectional regulation of cyclins and cyclin‐dependent kinase inhibitors via the phosphatidylinositol 3‐kinase/Akt pathway in developing rat cerebral cortex. J Neurosci 29: 775–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatesta P, Hartfuss E, Gotz M (2000) Isolation of radial glial cells by fluorescent‐activated cell sorting reveals a neuronal lineage. Development 127: 5253–5263 [DOI] [PubMed] [Google Scholar]
- Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul‐Gilloteaux P, Basto R (2013) Centrosome amplification causes microcephaly. Nat Cell Biol 15: 731–740 [DOI] [PubMed] [Google Scholar]
- Martinez‐Cerdeno V, Cunningham CL, Camacho J, Antczak JL, Prakash AN, Cziep ME, Walker AI, Noctor SC (2012) Comparative analysis of the subventricular zone in rat, ferret and macaque: evidence for an outer subventricular zone in rodents. PLoS ONE 7: e30178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B, Drechsel D, Guillemot F (2012) Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb Perspect Biol 4: a008359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda K, Toda T, Shinmyo Y, Ebisu H, Hoshiba Y, Wakimoto M, Ichikawa Y, Kawasaki H (2015) Pathophysiological analyses of cortical malformation using gyrencephalic mammals. Sci Rep 5: 15370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan LD, Alaama RA, Freise AC, Huang JC, Charvet CJ, Striedter GF (2012) Expansion, folding, and abnormal lamination of the chick optic tectum after intraventricular injections of FGF2. Proc Natl Acad Sci USA 109 (Suppl 1): 10640–10646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekel‐Bobrov N, Gilbert SL, Evans PD, Vallender EJ, Anderson JR, Hudson RR, Tishkoff SA, Lahn BT (2005) Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science 309: 1720–1722 [DOI] [PubMed] [Google Scholar]
- Mineyko A, Doja A, Hurteau J, Dobyns WB, Das S, Boycott KM (2010) A novel missense mutation in LIS1 in a child with subcortical band heterotopia and pachygyria inherited from his mildly affected mother with somatic mosaicism. J Child Neurol 25: 738–741 [DOI] [PubMed] [Google Scholar]
- Mirzaa G, Dodge NN, Glass I, Day C, Gripp K, Nicholson L, Straub V, Voit T, Dobyns WB (2004) Megalencephaly and perisylvian polymicrogyria with postaxial polydactyly and hydrocephalus: a rare brain malformation syndrome associated with mental retardation and seizures. Neuropediatrics 35: 353–359 [DOI] [PubMed] [Google Scholar]
- Mirzaa GM, Enyedi L, Parsons G, Collins S, Medne L, Adams C, Ward T, Davitt B, Bicknese A, Zackai E, Toriello H, Dobyns WB, Christian S (2014) Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: five novel mutations and review of the literature. Am J Med Genet A 164A: 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa GM, Riviere JB, Dobyns WB (2013) Megalencephaly syndromes and activating mutations in the PI3K‐AKT pathway: MPPH and MCAP. Am J Med Genet C Semin Med Genet 163C: 122–130 [DOI] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M (1984) Dynamic instability of microtubule growth. Nature 312: 237–242 [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Saito K, Kawano M, Muto T, Ogawa M (2004) Asymmetric production of surface‐dividing and non‐surface‐dividing cortical progenitor cells. Development 131: 3133–3145 [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi D, Kawaguchi A, Gotoh Y (2010) Mechanisms that regulate the number of neurons during mouse neocortical development. Curr Opin Neurobiol 20: 22–28 [DOI] [PubMed] [Google Scholar]
- Mizutani K, Yoon K, Dang L, Tokunaga A, Gaiano N (2007) Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature 449: 351–355 [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD (2007) Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci 8: 427–437 [DOI] [PubMed] [Google Scholar]
- Morris‐Rosendahl DJ, Najm J, Lachmeijer AM, Sztriha L, Martins M, Kuechler A, Haug V, Zeschnigk C, Martin P, Santos M, Vasconcelos C, Omran H, Kraus U, Van der Knaap MS, Schuierer G, Kutsche K, Uyanik G (2008) Refining the phenotype of alpha‐1a Tubulin (TUBA1A) mutation in patients with classical lissencephaly. Clin Genet 74: 425–433 [DOI] [PubMed] [Google Scholar]
- Mota B, Herculano‐Houzel S (2015) BRAIN STRUCTURE. Cortical folding scales universally with surface area and thickness, not number of neurons. Science 349: 74–77 [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium, Waterston RH, Lindblad‐Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T et al (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Muckli L, Naumer MJ, Singer W (2009) Bilateral visual field maps in a patient with only one hemisphere. Proc Natl Acad Sci USA 106: 13034–13039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munji RN, Choe Y, Li G, Siegenthaler JA, Pleasure SJ (2011) Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci 31: 1676–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas AK, Khurshid M, Desir J, Carvalho OP, Cox JJ, Thornton G, Kausar R, Ansar M, Ahmad W, Verloes A, Passemard S, Misson JP, Lindsay S, Gergely F, Dobyns WB, Roberts E, Abramowicz M, Woods CG (2010) WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet 42: 1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR (2001) Neurons derived from radial glial cells establish radial units in neocortex. Nature 409: 714–720 [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez‐Cerdeno V, Ivic L, Kriegstein AR (2004) Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7: 136–144 [DOI] [PubMed] [Google Scholar]
- Nonaka‐Kinoshita M, Reillo I, Artegiani B, Martinez‐Martinez MA, Nelson M, Borrell V, Calegari F (2013) Regulation of cerebral cortex size and folding by expansion of basal progenitors. EMBO J 32: 1817–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, Plajzer‐Frick I, Shoukry M, Afzal V, Rubenstein JL, Rubin EM, Pennacchio LA, Visel A (2013) Rapid and pervasive changes in genome‐wide enhancer usage during mammalian development. Cell 155: 1521–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata S, Aoki R, Kinoshita S, Yamaguchi M, Tsuruoka‐Kinoshita S, Tanaka H, Wada H, Watabe S, Tsuboi T, Masai I, Okamoto H (2011) Dual roles of Notch in regulation of apically restricted mitosis and apicobasal polarity of neuroepithelial cells. Neuron 69: 215–230 [DOI] [PubMed] [Google Scholar]
- O'Leary DD, Borngasser D (2006) Cortical ventricular zone progenitors and their progeny maintain spatial relationships and radial patterning during preplate development indicating an early protomap. Cereb Cortex 16(Suppl 1): i46–i56 [DOI] [PubMed] [Google Scholar]
- O'Leary MA, Bloch JI, Flynn JJ, Gaudin TJ, Giallombardo A, Giannini NP, Goldberg SL, Kraatz BP, Luo ZX, Meng J, Ni X, Novacek MJ, Perini FA, Randall ZS, Rougier GW, Sargis EJ, Silcox MT, Simmons NB, Spaulding M, Velazco PM et al (2013) The placental mammal ancestor and the post‐K‐Pg radiation of placentals. Science 339: 662–667 [DOI] [PubMed] [Google Scholar]
- Olson EC, Walsh CA (2002) Smooth, rough and upside‐down neocortical development. Curr Opin Genet Dev 12: 320–327 [DOI] [PubMed] [Google Scholar]
- Palmini A, Najm I, Avanzini G, Babb T, Guerrini R, Foldvary‐Schaefer N, Jackson G, Luders HO, Prayson R, Spreafico R, Vinters HV (2004) Terminology and classification of the cortical dysplasias. Neurology 62: S2–S8 [DOI] [PubMed] [Google Scholar]
- Parrini E, Ramazzotti A, Dobyns WB, Mei D, Moro F, Veggiotti P, Marini C, Brilstra EH, Dalla Bernardina B, Goodwin L, Bodell A, Jones MC, Nangeroni M, Palmeri S, Said E, Sander JW, Striano P, Takahashi Y, Van Maldergem L, Leonardi G et al (2006) Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain 129: 1892–1906 [DOI] [PubMed] [Google Scholar]
- Passemard S, Titomanlio L, Elmaleh M, Afenjar A, Alessandri JL, Andria G, de Villemeur TB, Boespflug‐Tanguy O, Burglen L, Del Giudice E, Guimiot F, Hyon C, Isidor B, Megarbane A, Moog U, Odent S, Hernandez K, Pouvreau N, Scala I, Schaer M et al (2009) Expanding the clinical and neuroradiologic phenotype of primary microcephaly due to ASPM mutations. Neurology 73: 962–969 [DOI] [PubMed] [Google Scholar]
- Pattabiraman K, Golonzhka O, Lindtner S, Nord AS, Taher L, Hoch R, Silberberg SN, Zhang D, Chen B, Zeng H, Pennacchio LA, Puelles L, Visel A, Rubenstein JL (2014) Transcriptional regulation of enhancers active in protodomains of the developing cerebral cortex. Neuron 82: 989–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoenix TN, Temple S (2010) Spred1, a negative regulator of Ras‐MAPK‐ERK, is enriched in CNS germinal zones, dampens NSC proliferation, and maintains ventricular zone structure. Genes Dev 24: 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao X, Basel‐Vanagaite L, Straussberg R, Grant PE, Pugh EW, Doheny K, Doan B, Hong SE, Shugart YY, Walsh CA (2002) An autosomal recessive form of bilateral frontoparietal polymicrogyria maps to chromosome 16q12.2‐21. Am J Hum Genet 70: 1028–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao X, Chang BS, Bodell A, Woods K, Benzeev B, Topcu M, Guerrini R, Goldberg‐Stern H, Sztriha L, Dobyns WB, Barkovich AJ, Walsh CA (2005) Genotype‐phenotype analysis of human frontoparietal polymicrogyria syndromes. Ann Neurol 58: 680–687 [DOI] [PubMed] [Google Scholar]
- Piao X, Hill RS, Bodell A, Chang BS, Basel‐Vanagaite L, Straussberg R, Dobyns WB, Qasrawi B, Winter RM, Innes AM, Voit T, Ross ME, Michaud JL, Descarie JC, Barkovich AJ, Walsh CA (2004) G protein‐coupled receptor‐dependent development of human frontal cortex. Science 303: 2033–2036 [DOI] [PubMed] [Google Scholar]
- Pillay P, Manger PR (2007) Order‐specific quantitative patterns of cortical gyrification. Eur J Neurosci 25: 2705–2712 [DOI] [PubMed] [Google Scholar]
- Pilz DT, Matsumoto N, Minnerath S, Mills P, Gleeson JG, Allen KM, Walsh CA, Barkovich AJ, Dobyns WB, Ledbetter DH, Ross ME (1998) LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum Mol Genet 7: 2029–2037 [DOI] [PubMed] [Google Scholar]
- Pilz GA, Shitamukai A, Reillo I, Pacary E, Schwausch J, Stahl R, Ninkovic J, Snippert HJ, Clevers H, Godinho L, Guillemot F, Borrell V, Matsuzaki F, Gotz M (2013) Amplification of progenitors in the mammalian telencephalon includes a new radial glial cell type. Nat Commun 4: 2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L, Mader MT, Irmler M, Gentilini M, Santoni F, Drechsel D, Blum R, Stahl R, Bulfone A, Malatesta P, Beckers J, Gotz M (2008) Prospective isolation of functionally distinct radial glial subtypes–lineage and transcriptome analysis. Mol Cell Neurosci 38: 15–42 [DOI] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Walsh CA (2013) Somatic mutation, genomic variation, and neurological disease. Science 341: 1237758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, Parrini E, Valence S, Pierre BS, Oger M, Lacombe D, Genevieve D, Fontana E, Darra F, Cances C, Barth M, Bonneau D, Bernadina BD, N'Guyen S, Gitiaux C et al (2013) Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet 45: 639–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, Rosenbloom KR, Kent J, Haussler D (2006a) Forces shaping the fastest evolving regions in the human genome. PLoS Genet 2: e168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, Kern AD, Dehay C, Igel H, Ares M Jr, Vanderhaeghen P, Haussler D (2006b) An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443: 167–172 [DOI] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval‐Espinosa C, Nicholas CR, Shuga J, Liu SJ, Oldham MC, Diaz A, Lim DA, Leyrat AA, West JA, Kriegstein AR (2015) Molecular Identity of Human Outer Radial Glia during Cortical Development. Cell 163: 55–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Shuga J, Wang X, Leyrat AA, Lui JH, Li N, Szpankowski L, Fowler B, Chen P, Ramalingam N, Sun G, Thu M, Norris M, Lebofsky R, Toppani D, Kemp DW 2nd, Wong M, Clerkson B, Jones BN et al (2014) Low‐coverage single‐cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol 32: 1053–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poluch S, Juliano SL (2015) Fine‐tuning of neurogenesis is essential for the evolutionary expansion of the cerebral cortex. Cereb Cortex 25: 346–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, Lunter G (2006) Evolutionary biology: human brain gene wins genome race. Nature 443: 149–150 [DOI] [PubMed] [Google Scholar]
- Postiglione MP, Juschke C, Xie Y, Haas GA, Charalambous C, Knoblich JA (2011) Mouse inscuteable induces apical‐basal spindle orientation to facilitate intermediate progenitor generation in the developing neocortex. Neuron 72: 269–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer‐Frick I, Morrison H, Fitzpatrick DR, Afzal V, Pennacchio LA, Rubin EM, Noonan JP (2008) Human‐specific gain of function in a developmental enhancer. Science 321: 1346–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prufer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, Li H, Mallick S, Dannemann M, Fu Q, Kircher M, Kuhlwilm M, Lachmann M, Meyer M, Ongyerth M, Siebauer M et al (2014) The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D (1988) Body and Brain: A Trophic Theory of Neural Connections. Cambridge, MA: Harvard University Press; [DOI] [PubMed] [Google Scholar]
- Raballo R, Rhee J, Lyn‐Cook R, Leckman JF, Schwartz ML, Vaccarino FM (2000) Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci 20: 5012–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P (1972) Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol 145: 61–83 [DOI] [PubMed] [Google Scholar]
- Rakic P (1978) Neuronal migration and contact guidance in the primate telencephalon. Postgrad Med J 54(Suppl 1): 25–40 [PubMed] [Google Scholar]
- Rakic P (1995) A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci 18: 383–388 [DOI] [PubMed] [Google Scholar]
- Rakic P (2009) Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci 10: 724–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P, Stensas LJ, Sayre E, Sidman RL (1974) Computer‐aided three‐dimensional reconstruction and quantitative analysis of cells from serial electron microscopic montages of foetal monkey brain. Nature 250: 31–34 [DOI] [PubMed] [Google Scholar]
- Rash BG, Lim HD, Breunig JJ, Vaccarino FM (2011) FGF signaling expands embryonic cortical surface area by regulating Notch‐dependent neurogenesis. J Neurosci 31: 15604–15617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash BG, Tomasi S, Lim HD, Suh CY, Vaccarino FM (2013) Cortical gyrification induced by fibroblast growth factor 2 in the mouse brain. J Neurosci 33: 10802–10814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Reeuwijk J, Brunner HG, van Bokhoven H (2005a) Glyc‐O‐genetics of Walker‐Warburg syndrome. Clin Genet 67: 281–289 [DOI] [PubMed] [Google Scholar]
- van Reeuwijk J, Janssen M, van den Elzen C, Beltran‐Valero de Bernabe D, Sabatelli P, Merlini L, Boon M, Scheffer H, Brockington M, Muntoni F, Huynen MA, Verrips A, Walsh CA, Barth PG, Brunner HG, van Bokhoven H (2005b) POMT2 mutations cause alpha‐dystroglycan hypoglycosylation and Walker‐Warburg syndrome. J Med Genet 42: 907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reillo I, Borrell V (2012) Germinal zones in the developing cerebral cortex of ferret: ontogeny, cell cycle kinetics, and diversity of progenitors. Cereb Cortex 22: 2039–2054 [DOI] [PubMed] [Google Scholar]
- Reillo I, de Juan Romero C, Garcia‐Cabezas MA, Borrell V (2011) A role for intermediate radial glia in the tangential expansion of the Mammalian cerebral cortex. Cereb Cortex 21: 1674–1694 [DOI] [PubMed] [Google Scholar]
- Ribes V, Stutzmann F, Bianchetti L, Guillemot F, Dolle P, Le Roux I (2008) Combinatorial signalling controls Neurogenin2 expression at the onset of spinal neurogenesis. Dev Biol 321: 470–481 [DOI] [PubMed] [Google Scholar]
- Rice DS, Curran T (2001) Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci 24: 1005–1039 [DOI] [PubMed] [Google Scholar]
- Richman DP, Stewart RM, Hutchinson JW, Caviness VS Jr (1975) Mechanical model of brain convolutional development. Science 189: 18–21 [DOI] [PubMed] [Google Scholar]
- Riviere JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, St‐Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA, Albrecht B, Armour CM, Armstrong L, Caluseriu O, Cytrynbaum C et al (2012) De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet 44: 934–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin NH, Taylor CJ, McDonald‐McGinn DM, Zackai EH, Bingham P, Collins KJ, Earl D, Gill D, Granata T, Guerrini R, Katz N, Kimonis V, Lin JP, Lynch DR, Mohammed SN, Massey RF, McDonald M, Rogers RC, Splitt M, Stevens CA et al (2006) Polymicrogyria and deletion 22q11.2 syndrome: window to the etiology of a common cortical malformation. Am J Med Genet A 140: 2416–2425 [DOI] [PubMed] [Google Scholar]
- Roll P, Rudolf G, Pereira S, Royer B, Scheffer IE, Massacrier A, Valenti MP, Roeckel‐Trevisiol N, Jamali S, Beclin C, Seegmuller C, Metz‐Lutz MN, Lemainque A, Delepine M, Caloustian C, de Saint Martin A, Bruneau N, Depetris D, Mattei MG, Flori E et al (2006) SRPX2 mutations in disorders of language cortex and cognition. Hum Mol Genet 15: 1195–1207 [DOI] [PubMed] [Google Scholar]
- Ronan L, Fletcher PC (2015) From genes to folds: a review of cortical gyrification theory. Brain Struct Funct 220: 2475–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan L, Voets N, Rua C, Alexander‐Bloch A, Hough M, Mackay C, Crow TJ, James A, Giedd JN, Fletcher PC (2014) Differential tangential expansion as a mechanism for cortical gyrification. Cereb Cortex 24: 2219–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscioli T, Kamsteeg EJ, Buysse K, Maystadt I, van Reeuwijk J, van den Elzen C, van Beusekom E, Riemersma M, Pfundt R, Vissers LE, Schraders M, Altunoglu U, Buckley MF, Brunner HG, Grisart B, Zhou H, Veltman JA, Gilissen C, Mancini GM, Delree P et al (2012) Mutations in ISPD cause Walker‐Warburg syndrome and defective glycosylation of alpha‐dystroglycan. Nat Genet 44: 581–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross ME, Walsh CA (2001) Human brain malformations and their lessons for neuronal migration. Annu Rev Neurosci 24: 1041–1070 [DOI] [PubMed] [Google Scholar]
- Roth G, Dicke U (2005) Evolution of the brain and intelligence. Trends Cogn Sci 9: 250–257 [DOI] [PubMed] [Google Scholar]
- Sahara S, O'Leary DD (2009) Fgf10 regulates transition period of cortical stem cell differentiation to radial glia controlling generation of neurons and basal progenitors. Neuron 63: 48–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai D, Dixon J, Dixon MJ, Trainor PA (2012) Mammalian neurogenesis requires Treacle‐Plk1 for precise control of spindle orientation, mitotic progression, and maintenance of neural progenitor cells. PLoS Genet 8: e1002566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom SN, Livesey FJ (2009) Gradients in the brain: the control of the development of form and function in the cerebral cortex. Cold Spring Harb Perspect Biol 1: a002519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapir T, Elbaum M, Reiner O (1997) Reduction of microtubule catastrophe events by LIS1, platelet‐activating factor acetylhydrolase subunit. EMBO J 16: 6977–6984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisian MR, Bartley CM, Chi H, Nakamura F, Hashimoto‐Torii K, Torii M, Flavell RA, Rakic P (2006) MEKK4 signaling regulates filamin expression and neuronal migration. Neuron 52: 789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisian MR, Bartley CM, Rakic P (2008) Trouble making the first move: interpreting arrested neuronal migration in the cerebral cortex. Trends Neurosci 31: 54–61 [DOI] [PubMed] [Google Scholar]
- Sauer F (1935) The interkinetic migration of embryonic epithelial nuclei. J Morphol 60: 1–11 [Google Scholar]
- Sheen VL, Dixon PH, Fox JW, Hong SE, Kinton L, Sisodiya SM, Duncan JS, Dubeau F, Scheffer IE, Schachter SC, Wilner A, Henchy R, Crino P, Kamuro K, DiMario F, Berg M, Kuzniecky R, Cole AJ, Bromfield E, Biber M et al (2001) Mutations in the X‐linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet 10: 1775–1783 [DOI] [PubMed] [Google Scholar]
- Sheen VL, Ganesh VS, Topcu M, Sebire G, Bodell A, Hill RS, Grant PE, Shugart YY, Imitola J, Khoury SJ, Guerrini R, Walsh CA (2004) Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet 36: 69–76 [DOI] [PubMed] [Google Scholar]
- Sheen VL, Jansen A, Chen MH, Parrini E, Morgan T, Ravenscroft R, Ganesh V, Underwood T, Wiley J, Leventer R, Vaid RR, Ruiz DE, Hutchins GM, Menasha J, Willner J, Geng Y, Gripp KW, Nicholson L, Berry‐Kravis E, Bodell A et al (2005) Filamin A mutations cause periventricular heterotopia with Ehlers‐Danlos syndrome. Neurology 64: 254–262 [DOI] [PubMed] [Google Scholar]
- Sheen VL, Torres AR, Du X, Barry B, Walsh CA, Kimonis VE (2010) Mutation in PQBP1 is associated with periventricular heterotopia. Am J Med Genet A 152A: 2888–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen VL, Walsh CA (2005) Periventricular heterotopia: new insights into Ehlers‐Danlos syndrome. Clin Med Res 3: 229–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Eyaid W, Mochida GH, Al‐Moayyad F, Bodell A, Woods CG, Walsh CA (2005) ASPM mutations identified in patients with primary microcephaly and seizures. J Med Genet 42: 725–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitamukai A, Konno D, Matsuzaki F (2011) Oblique radial glial divisions in the developing mouse neocortex induce self‐renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J Neurosci 31: 3683–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicca F, Kelemen A, Genton P, Das S, Mei D, Moro F, Dobyns WB, Guerrini R (2003) Mosaic mutations of the LIS1 gene cause subcortical band heterotopia. Neurology 61: 1042–1046 [DOI] [PubMed] [Google Scholar]
- Sidman RL, Rakic P (1973) Neuronal migration, with special reference to developing human brain: a review. Brain Res 62: 1–35 [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA, Folias AE, Choe Y, May SR, Kume T, Napoli JL, Peterson AS, Pleasure SJ (2009) Retinoic acid from the meninges regulates cortical neuron generation. Cell 139: 597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sir JH, Barr AR, Nicholas AK, Carvalho OP, Khurshid M, Sossick A, Reichelt S, D'Santos C, Woods CG, Gergely F (2011) A primary microcephaly protein complex forms a ring around parental centrioles. Nat Genet 43: 1147–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart IH, Dehay C, Giroud P, Berland M, Kennedy H (2002) Unique morphological features of the proliferative zones and postmitotic compartments of the neural epithelium giving rise to striate and extrastriate cortex in the monkey. Cereb Cortex 12: 37–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart IH, McSherry GM (1986) Gyrus formation in the cerebral cortex of the ferret. II. Description of the internal histological changes. J Anat 147: 27–43 [PMC free article] [PubMed] [Google Scholar]
- Soriano E, Dumesnil N, Auladell C, Cohen‐Tannoudji M, Sotelo C (1995) Molecular heterogeneity of progenitors and radial migration in the developing cerebral cortex revealed by transgene expression. Proc Natl Acad Sci USA 92: 11676–11680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squier W, Jansen A (2014) Polymicrogyria: pathology, fetal origins and mechanisms. Acta Neuropathol Commun 2: 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl R, Walcher T, De Juan Romero C, Pilz GA, Cappello S, Irmler M, Sanz‐Aquela JM, Beckers J, Blum R, Borrell V, Gotz M (2013) Trnp1 regulates expansion and folding of the mammalian cerebral cortex by control of radial glial fate. Cell 153: 535–549 [DOI] [PubMed] [Google Scholar]
- Stancik EK, Navarro‐Quiroga I, Sellke R, Haydar TF (2010) Heterogeneity in ventricular zone neural precursors contributes to neuronal fate diversity in the postnatal neocortex. J Neurosci 30: 7028–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenzel D, Wilsch‐Brauninger M, Wong FK, Heuer H, Huttner WB (2014) Integrin alphavbeta3 and thyroid hormones promote expansion of progenitors in embryonic neocortex. Development 141: 795–806 [DOI] [PubMed] [Google Scholar]
- Striedter GF, Srinivasan S, Monuki ES (2015) Cortical folding: when, where, how, and why? Annu Rev Neurosci 38: 291–307 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS Jr (1993) Cell cycle parameters and patterns of nuclear movement in the neocortical proliferative zone of the fetal mouse. J Neurosci 13: 820–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallinen T, Chung JY, Biggins JS, Mahadevan L (2014) Gyrification from constrained cortical expansion. Proc Natl Acad Sci USA 111: 12667–12672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassi L, Colombo N, Garbelli R, Francione S, Lo Russo G, Mai R, Cardinale F, Cossu M, Ferrario A, Galli C, Bramerio M, Citterio A, Spreafico R (2002) Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain 125: 1719–1732 [DOI] [PubMed] [Google Scholar]
- Taverna E, Gotz M, Huttner WB (2014) The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol 30: 465–502 [DOI] [PubMed] [Google Scholar]
- Taverna E, Huttner WB (2010) Neural progenitor nuclei IN motion. Neuron 67: 906–914 [DOI] [PubMed] [Google Scholar]
- Teotonio H, Rose MR (2001) Perspective: reverse evolution. Evolution 55: 653–660 [DOI] [PubMed] [Google Scholar]
- Thompson D (1917) On Growth and Form. Cambridge: University Cambridge Press; [Google Scholar]
- Thornton GK, Woods CG (2009) Primary microcephaly: do all roads lead to Rome? Trends Genet 25: 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischfield MA, Cederquist GY, Gupta ML Jr, Engle EC (2011) Phenotypic spectrum of the tubulin‐related disorders and functional implications of disease‐causing mutations. Curr Opin Genet Dev 21: 286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tore HG, McKinney AM, Nagar VA, Lohman B, Truwit CL, Raybaud C (2009) Syndrome of megalencephaly, polydactyly, and polymicrogyria lacking frank hydrocephalus, with associated MR imaging findings. AJNR Am J Neuroradiol 30: 1620–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro R, Burnod Y (2005) A morphogenetic model for the development of cortical convolutions. Cereb Cortex 15: 1900–1913 [DOI] [PubMed] [Google Scholar]
- True JR, Carroll SB (2002) Gene co‐option in physiological and morphological evolution. Annu Rev Cell Dev Biol 18: 53–80 [DOI] [PubMed] [Google Scholar]
- Tyler WA, Medalla M, Guillamon‐Vivancos T, Luebke JI, Haydar TF (2015) Neural precursor lineages specify distinct neocortical pyramidal neuron types. J Neurosci 35: 6142–6152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valence S, Poirier K, Lebrun N, Saillour Y, Sonigo P, Bessieres B, Attie‐Bitach T, Benachi A, Masson C, Encha‐Razavi F, Chelly J, Bahi‐Buisson N (2013) Homozygous truncating mutation of the KBP gene, encoding a KIF1B‐binding protein, in a familial case of fetal polymicrogyria. Neurogenetics 14: 215–224 [DOI] [PubMed] [Google Scholar]
- Van Essen DC (1997) A tension‐based theory of morphogenesis and compact wiring in the central nervous system. Nature 385: 313–318 [DOI] [PubMed] [Google Scholar]
- Visel A, Taher L, Girgis H, May D, Golonzhka O, Hoch RV, McKinsey GL, Pattabiraman K, Silberberg SN, Blow MJ, Hansen DV, Nord AS, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, Plajzer‐Frick I, Shoukry M, Afzal V, Kaplan T et al (2013) A high‐resolution enhancer atlas of the developing telencephalon. Cell 152: 895–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viti J, Gulacsi A, Lillien L (2003) Wnt regulation of progenitor maturation in the cortex depends on Shh or fibroblast growth factor 2. J Neurosci 23: 5919–5927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CA (1999) Genetic malformations of the human cerebral cortex. Neuron 23: 19–29 [DOI] [PubMed] [Google Scholar]
- Walsh CA (2001) Neuroscience in the post‐genome era: an overview. Trends Neurosci 24: 363–364 [DOI] [PubMed] [Google Scholar]
- Wang X, Tsai JW, Lamonica B, Kriegstein AR (2011) A new subtype of progenitor cell in the mouse embryonic neocortex. Nat Neurosci 14: 555–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker W (1990) Why does cerebral cortex fissure and fold? A review of determinants of gyri and sulci In Cerebral Cortex, Peters A, Jones EG. (eds), pp 3–136. New York: Plenum Press; [Google Scholar]
- Willer T, Lee H, Lommel M, Yoshida‐Moriguchi T, de Bernabe DB, Venzke D, Cirak S, Schachter H, Vajsar J, Voit T, Muntoni F, Loder AS, Dobyns WB, Winder TL, Strahl S, Mathews KD, Nelson SF, Moore SA, Campbell KP (2012) ISPD loss‐of‐function mutations disrupt dystroglycan O‐mannosylation and cause Walker‐Warburg syndrome. Nat Genet 44: 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong FK, Fei JF, Mora‐Bermudez F, Taverna E, Haffner C, Fu J, Anastassiadis K, Stewart AF, Huttner WB (2015) Sustained Pax6 Expression Generates Primate‐like Basal Radial Glia in Developing Mouse Neocortex. PLoS Biol 13: e1002217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead GJ, Mutch CA, Olson EC, Chenn A (2006) Cell‐autonomous beta‐catenin signaling regulates cortical precursor proliferation. J Neurosci 26: 12620–12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods CG, Bond J, Enard W (2005) Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am J Hum Genet 76: 717–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Juschke C, Esk C, Hirotsune S, Knoblich JA (2013) The phosphatase PP4c controls spindle orientation to maintain proliferative symmetric divisions in the developing neocortex. Neuron 79: 254–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Knutsen AK, Dikranian K, Kroenke CD, Bayly PV, Taber LA (2010) Axons pull on the brain, but tension does not drive cortical folding. J Biomech Eng 132: 071013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Kato Y, Karita M, Kawaguchi M, Shibata N, Kobayashi M (2004) Expression of genes related to muscular dystrophy with lissencephaly. Pediatr Neurol 31: 183–190 [DOI] [PubMed] [Google Scholar]
- Yao X, Abrieu A, Zheng Y, Sullivan KF, Cleveland DW (2000) CENP‐E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat Cell Biol 2: 484–491 [DOI] [PubMed] [Google Scholar]
- Yingling J, Youn YH, Darling D, Toyo‐Oka K, Pramparo T, Hirotsune S, Wynshaw‐Boris A (2008) Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell 132: 474–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, Herrmann R, Straub V, Talim B, Voit T, Topaloglu H, Toda T, Endo T (2001) Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 1: 717–724 [DOI] [PubMed] [Google Scholar]
- Yu TW, Mochida GH, Tischfield DJ, Sgaier SK, Flores‐Sarnat L, Sergi CM, Topcu M, McDonald MT, Barry BJ, Felie JM, Sunu C, Dobyns WB, Folkerth RD, Barkovich AJ, Walsh CA (2010) Mutations in WDR62, encoding a centrosome‐associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat Genet 42: 1015–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CJ, Borello U, Rubenstein JL, Pleasure SJ (2006) Neuronal production and precursor proliferation defects in the neocortex of mice with loss of function in the canonical Wnt signaling pathway. Neuroscience 142: 1119–1131 [DOI] [PubMed] [Google Scholar]