

Abstract
Age-associated cognitive decline is affected by factors produced inside and outside the brain. We found in aged mice and humans, that the choroid plexus (CP), an epithelial interface between the brain and the circulation, shows a type I interferon (IFN-I)-dependent expression profile, often associated with anti-viral responses. This signature was induced by brain-derived signals present in the cerebrospinal fluid of aged mice. Blocking IFN-I signaling within the brain of cognitively-impaired aged mice, using IFN-I receptor neutralizing antibody, led to partial restoration of cognitive function and hippocampal neurogenesis, and reestablished IFN-II-dependent CP activity, lost in aging. Our data identify an aging-induced IFN-I signature at the CP, and demonstrate its negative influence on brain function, thereby suggesting a potential target for therapeutic intervention for age-related cognitive decline.
One of the mysteries of brain senescence is to what extent it is influenced by aging of other body tissues (1, 2). Recent studies have suggested that age-related changes in both circulating soluble factors (3–5) and peripheral immunity (6) are involved in this process. Nevertheless, given the fact that the mammalian central nervous system (CNS) is secluded behind barriers from directly interacting with the blood circulation (7), the underlying mechanism remains unclear.
The choroid plexus (CP), an epithelial monolayer that forms the blood-cerebrospinal fluid barrier (B-CSF-B) and produces the CSF, serves as a neuro-immunological interface in shaping brain function in health and pathology by integrating signals from the brain with signals coming from the circulation (6, 8–11). We envisioned that understanding how CP activity is altered in aging might lead to identification of strategies to attenuate age-associated decline in brain function.
To determine whether aging of the CP reflects aging of the brain and other body tissues, we systematically characterized mRNA expression profile of young (3-month old) and aged (22-month old) mice by RNA-sequencing. Whereas all examined organs from aged mice showed increased expression of mRNA transcripts associated with age-related processes, such as “immune response” (p-value=2.66E-05; cluster XIX; fig. S1A, B), the aged CP exhibited an expression profile corresponding to “type I interferon (IFN-I) response” (p-value=4.25E-07; cluster IX; Fig. 1A and fig. S1A–C), classically associated with anti-viral activity (12). To exclude a non-age-related effect, such as viral infection, we confirmed the increased expression of IFN-I-dependent genes (e.g. interferon regulatory factor 7 (irf7), interferon-beta 1 (ifnβ), and interferon-induced protein with tetratricopeptide repeats 1 (ifit1)) by real-time quantitative PCR (qPCR) in aged mice received from different animal centers (Fig. 1B). Closer examination by qPCR and immunohistochemical staining showed that whereas IFN-I-associated gene expression was increased in the aged CP, expression of type II IFN (IFN-γ)-dependent genes (10) (e.g. intercellular adhesion molecule 1 (icam1), interferon gamma-induced protein 10 (cxcl10), and chemokine (C-C motif) ligand 17 (ccl17)) was decreased (Fig. 1C, 1D). Immunohistochemical staining of brain sections of young and aged mice, as well as postmortem brain sections from non-CNS-diseased young and aged humans, showed an age-associated increase of IFN-β and IRF-7 at the CP (Fig. 1E), and indicated that this effect is conserved among species.
Fig. 1. Type I interferon expression program in the aging CP.

(A) Heat map and hierarchical clustering of gene expression in bone marrow (BM), cervical lymph nodes (CLN), choroid plexus (CP), colon (CL), hippocampus (HC), inguinal lymph nodes (ILN), liver (LV), lung (LU), mesenteric lymph nodes (MLN), spleen (SP) and thymus (TH) of aged mice, analyzed by high-throughput RNA-sequencing. Results are displayed as log of differential expression relative to young controls (n=2–3). (B–C) mRNA abundance of type I and type II IFN-dependent genes in the CP of aged animals, presented as fold change relative to expression in young mice CPs (n=10 mice per group; bars represent mean ± SEM; *, P < 0.05; **, P < 0.01;***, P < 0.001; Student’s t test for each pair; data are representative of at least three independent experiments performed). (D) Representative images of brain sections of young and aged mice, immunostained for Claudin-1 (marking epithelial tight junctions; in green), and either CXCL10 or ICAM1 (in red) (Scale bar, 25μm). (E) Representative micrographs of immunohistochemical staining for IRF7 in young and aged mice, and for IRF7 and IFN-β in young and aged human postmortem CPs (Scale bar, 50μm).
Because the CP epithelium is exposed from its apical side to brain-derived signals, via the CSF, and from its basal side to peripheral signals, via the circulation (13), we investigated which of these compartments induces the transcriptional signature of the aged CP. To differentially examine these effects, we made use of parabiosis settings, in which young and aged mice were surgically connected to share vasculature. Under these settings, young heterochronic parabionts display reduced levels of hippocampal neurogenesis, whereas aged heterochronic parabionts exhibit increased hippocampal neurogenesis and improved cognitive functions (3, 4). Transcriptome and qPCR analyses revealed that whereas the circulation of aged mice failed to induce expression of IFN-I-dependent genes in the CP of young heterochronic partners, it did affect IFN-II-dependent gene expression (Fig. 2A, B, fig. S2A, B (clusters III and VIII)), including increased expression of the chemokine CCL11 (Fig. 2C), the levels of which increase at the CP in brain aging, and reflect local reduction in IFN-γ (3, 6). Aged heterochronic parabionts, exposed to the young circulation, showed increased expression of IFN-γ-regulated genes at the CP (10), when compared to their isochronic age-matched counterparts, with no changes in the expression program of IFN-I-dependent genes, or in CCL11 ((Fig. 2A–C, fig. S2A, B). These results indicated that while IFN-II expression program at the CP could be partially modulated by the circulation, IFN-I in this compartment might be induced by factors emitted from the aged brain. To test whether aged mice CSF can affect IFN-I response at the CP, we exposed primary cultures of CP epithelial cells from young mice to CSF aspirated from either aged or young mice and found that the CSF of aged, but not of young animals, could induce IFN-I-dependent gene expression (Fig. 2D). This response was recapitulated when epithelial cells of the CP were treated with a mixture of pro-inflammatory cytokines that accumulate in the brain during aging and neurodegeneration (14) (fig. S3). These results suggested that in the aged CP, the induced IFN-I response, and the reduced IFN-II response, are controlled by signals present in the aged CSF, and in the circulation, respectively.
Fig. 2. Effect of signals from the cerebrospinal fluid, but not from the blood circulation, on type I IFN-dependent genes in the CP.

(A) Heat map and hierarchical clustering of gene expression in the CPs of aged (‘A’) and young (‘Y’), iso- (‘A-A’, ‘Y-Y’) and hetero- (‘A–Y’) chronic parabionts, analyzed by RNA-sequencing, displayed as log of differential expression relative to an average expression in young-isochronic CP (n=2–3). (B–C) mRNA expression of type I IFN-dependent genes, ifit1 and irf7 (B) and the type II IFN-dependent gene, ccl11 (C) in the CP of parabiotic mice (n=6–8 mice per group). (D) mRNA expression of ifit1 and irf7 in primary cultures of CP cells treated with PBS or CSF aspirated from young (3 months old) or aged (22 months old) mice (n=4–5 per group). Throughout the figure, bars represent mean ± SEM; *, P < 0.05; **, P < 0.01; ***, P < 0.001; one-way ANOVA with Newmann-Kleus post-hoc test.
IFN-II has an essential role in regulating CP function (10), and its diminished levels in this compartment were associated with brain aging (6). Follow up of transgenic mice deficient in IFN-II signaling, including IFN-γ receptor knockout (IFN-γR−/−) mice (which lack the ability to directly respond to IFN-γ and Tbx21−/− mice (which lack Tbx21, a transcription factor critical for the control of IFN-γ production in CD4+ T cells (15)), revealed that these mice developed spatial learning and memory deficits (fig. S4A to H), and reduced hippocampal neurogenesis during adulthood (fig. S4I to K). Brain function decline in IFN-γR−/− mice was accompanied by impaired CP activity in supporting leukocyte trafficking to the CSF (fig. S4L to M). These results indicated that impaired ability to respond to IFN-γ, or reduced IFN-γ production by T cells, could lead to premature cognitive decline and restrict hippocampal neurogenesis in adulthood (fig. S5). The apparent inverse relation between type I and type II IFN expression programs, observed here in the aged CP (Fig. 1, 2), together with the fact that chronically activated IFN-I signaling was shown to prevent IFN-II-dependent resolution of inflammation under various lingering inflammatory conditions (16–18), prompted us to examine whether chronic IFN-I in the aged CP could affect brain function, and if so, whether it involves interfering with local IFN-II responses.
To test whether aging-induced IFN-I response in the CP might influence brain function, we examined both the direct effect of the major IFN-I cytokine, IFN-β, on CP epithelial cells of young mice, and that of the neutralization of IFN-I responses in the brain of aged mice. Exposure of CP epithelial cells to IFN-β induced the classical “anti-viral” response (Fig. 3A), and resulted in reduced expression of insulin-like growth factor (igf-1) and brain derived neurotrophic factor (bdnf) (Fig. 3B), two CP-secreted molecules that support neuronal growth, differentiation and survival (11, 19). Intracerebroventricular (i.c.v.) administration of IFN-I receptor-neutralizing antibody (α-IFNAR) to the CSF of aged mice blocked IFN-I response program at the CP (Fig. 3C), and led to restoration of igf1 and bdnf expression (Fig. 3D, fig. S6), and of ccl11 and ccl17 expression (Fig. 3E), to levels similar to those found in the CP of young mice. These results showed that neutralizing IFN-I response within the aged CSF affected CP function, and suggested that IFN-II response in the aged CP is suppressed by chronically elevated IFN-I.
Fig. 3. Restored CP function after neutralization of the age-induced type I IFN response in the brain.
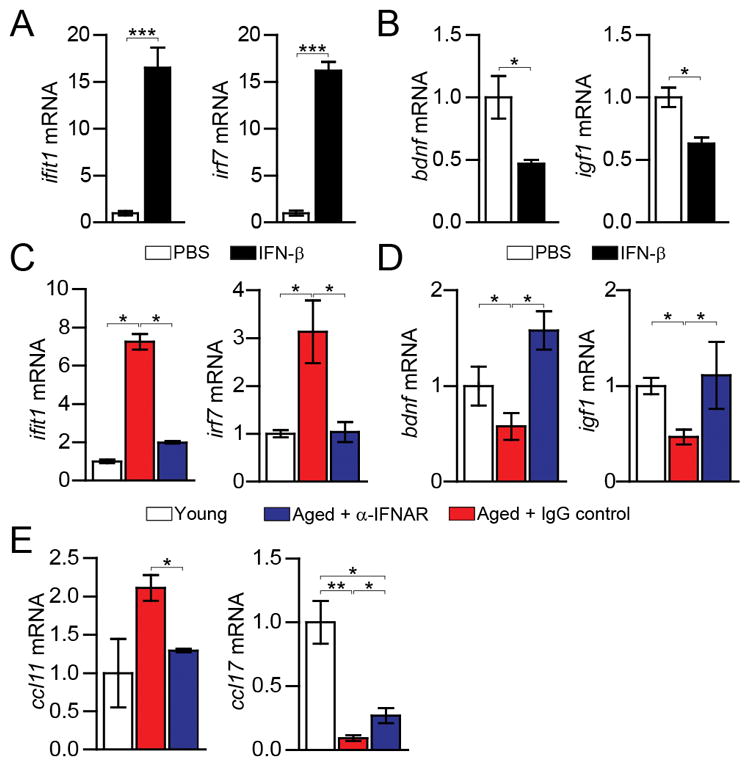
(A–B) ifit1, irf7 (A), and bdnf and igf1 (B) mRNA expression in CP epithelial cells cultured for 24h with murine IFN-β (n=4 per group; bars represent mean ± SEM; *, P < 0.05; ***, P < 0.001, Student’s t test). (C) ifit1 and irf7 mRNA expression in the CP 3 days after α-IFNAR or IgG control i.c.v. administration. (D–E) bdnf and igf1 (D) and ccl11 and ccl17 (E) mRNA expression in the CP 7 days after α-IFNAR or IgG control i.c.v. administration (n=6–7 per group). Throughout the figure, bars represent mean ± SEM; *, P < 0.05; **, P < 0.01; one-way ANOVA with Newmann-Kleus post-hoc test.
To further test whether in vivo neutralization of IFN-I in aging could reverse brain function loss, we assessed spatial learning and memory abilities and hippocampal neurogenesis, two independent measurements of brain function and plasticity (20) (Fig. 4A). Because age-associated cognitive decline varies across individuals, we first scored aged mice for their learning and memory, using the novel location recognition (NLR) test, in which mice are freely exploring two objects within an arena, and the time of exploration of each object in measured. After 24 hours, one of the objects is relocated. Under these conditions, cognitively-unimpaired mice spent more time exploring the displaced object (21). Cognitively-impaired aged mice were i.c.v. injected with either α-IFNAR or control IgG, and tested again a week after for their cognitive performance (Fig. 4B). Cognitively-impaired aged mice treated with α-IFNAR spent more time exploring the relocated object, whereas exploration preference of control IgG-injected mice was not changed (Fig. 4C). Subsequently, mice of all groups were administered with 5-bromo-2′-deoxyuridine (BrdU) for detection of newly-formed cells in the hippocampus (22). The number of newly-born neurons, stained for BrdU and Doublecortin (DCX), was increased in the α-IFNAR-treated group (Fig. 4D); enhanced hippocampal neurogenesis to a similar extent in aged mice was associated with improved cognitive function (3, 4). We also examined the hippocampal parenchyma for the effect on neuroinflammation, which detrimentally affect adult neurogenesis (23), and found increased mRNA expression of the anti-inflammatory cytokine, IL-10 (Fig. 4E), and a marked decrease in astrogliosis and microgliosis (Fig. 4F, G) in the α-IFNAR-treated group. Thus, blocking IFN-I in the CNS of aged mice attenuated age-related neuroinflammation and partially reversed decline in brain function.
Fig. 4. Restored brain function after neutralization of the age-induced type I IFN response in the brain.

(A) Scheme illustrating the experimental design. (B) Performance of “cognitively unimpaired” and “cognitively impaired” aged mice in the NLR task (n=5–18 per group). (C) Performance of α-IFNAR and control IgG injected cognitively-impaired aged mice in the NLR task (n=5–6 per group). (D–E) Analysis of the hippocampal dentate gyrus (DG) of α-IFNAR or control IgG i.c.v.-injected “cognitively impaired” aged mice, and of young and aged “cognitively unimpaired” mice, 14 days after treatment (n=4–6 per group). Quantification of hippocampal DCX and BrdU labeled newly-born neurons (notably, in this experiment, young mice showed significantly higher numbers of DCX and BrdU labeled cells, 16832±873 and 4403±51, respectively) (D); il-10 mRNA expression in the hippocampus (E). (F–G) Quantification (F), and representative images (G), of GFAP and IBA-1 staining intensity in hippocampal brain sections immunostained for GFAP (in red), IBA-1 (in green), and Hoechst nuclear staining (in blue) (scale bar, 50μm). Throughout the figure, bars represent mean ± SEM; *, P < 0.05; **, P < 0.01; one-way ANOVA with Newmann-Kleus post-hoc test.
Though originally identified by their anti-viral properties, type I IFNs have important roles in attenuating inflammation, both outside and inside the CNS (24–26). Accordingly, it is possible that IFN-I response program at the aged CP may represent a physiological mechanism evoked to mitigate age-related neuroinflammation. Our results suggest, however, that IFN-I response of the CP during normal aging negatively regulates adult neurogenesis and spatial learning and memory, suggesting that persistent expression of IFN-I by the CP becomes counterproductive (fig. S7). Such a detrimental influence of IFN-I in CNS function was also found in patients suffering from diseases in which cerebral levels of type I IFNs are increased (25), and in chronic inflammatory diseases in which IFN-I are therapeutically administrated, often accompanied by neurological and neuropsychiatric complications (27). Thus, neutralizing the response to type I IFNs within the CNS might provide an approach to reverse or slow down age-related cognitive decline.
Supplementary Material
References
- 1.Stranahan AM, Mattson MP. Nat Rev Neurosci. 2012;13:209–216. doi: 10.1038/nrn3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yeoman M, Scutt G, Faragher R. Nat Rev Neurosci. 2012;13:435–445. doi: 10.1038/nrn3230. [DOI] [PubMed] [Google Scholar]
- 3.Villeda SA, et al. Nature. 2011;477:90–94. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villeda SA, et al. Nat Med. 2014;20:659–663. doi: 10.1038/nm.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katsimpardi L, et al. Science. 2014;344:630–634. doi: 10.1126/science.1251141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baruch K, et al. Proc Natl Acad Sci U S A. 2013;110:2264–2269. doi: 10.1073/pnas.1211270110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ransohoff RM, Engelhardt B. Nat Rev Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 8.Baruch K, Schwartz M. Brain Behav Immun. 2013;34:11–16. doi: 10.1016/j.bbi.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz M, Baruch K. EMBO J. 2013 [Google Scholar]
- 10.Kunis G, et al. Brain. 2013;136:3427–3440. doi: 10.1093/brain/awt259. [DOI] [PubMed] [Google Scholar]
- 11.Falcao AM, et al. Front Cell Neurosci. 2012;6:34. doi: 10.3389/fncel.2012.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez-Navajas JM, Lee J, David M, Raz E. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johanson C, et al. Toxicol Pathol. 2011;39:186–212. doi: 10.1177/0192623310394214. [DOI] [PubMed] [Google Scholar]
- 14.Lucin KM, Wyss-Coray T. Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabo SJ, et al. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 16.Wilson EB, et al. Science. 2013;340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teles RM, et al. Science. 2013;339:1448–1453. doi: 10.1126/science.1233665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teijaro JR, et al. Science. 2013;340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emerich DF, et al. Bioessays. 2005;27:262–274. doi: 10.1002/bies.20193. [DOI] [PubMed] [Google Scholar]
- 20.Deng W, Aimone JB, Gage FH. Nat Rev Neurosci. 2010;11:339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oliveira AM, Hemstedt TJ, Bading H. Nat Neurosci. 2012;15:1111–1113. doi: 10.1038/nn.3151. [DOI] [PubMed] [Google Scholar]
- 22.van Praag H, et al. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ekdahl CT, et al. Proc Natl Acad Sci U S A. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Axtell RC, Steinman L. Immunity. 2008;28:600–602. doi: 10.1016/j.immuni.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Hofer MJ, Campbell IL. Cytokine Growth Factor Rev. 2013;24:257–267. doi: 10.1016/j.cytogfr.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Prinz M, et al. Immunity. 2008;28:675–686. doi: 10.1016/j.immuni.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Wichers M, Maes M. Int J Neuropsychopharmacol. 2002;5:375–388. doi: 10.1017/S1461145702003103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.