

Abstract
Background
Self-incompatibility (SI) is under genetic control and prevents inbreeding depression in angiosperms. SI mechanisms are quite complicated and still poorly understood in many plants. Tea (Camellia sinensis L.) belonging to the family of Theaceae, exhibits high levels of SI and high heterozygosity. Uncovering the molecular basis of SI of the tea plant may enhance breeding and simplify genomics research for the whole family.
Results
The growth of pollen tubes following selfing and crossing was observed using fluorescence microscopy. Self-pollen tubes grew slower than cross treatments from 24 h to 72 h after pollination. RNA-seq was employed to explore the molecular mechanisms of SI and to identify SI-related genes in C. sinensis. Self and cross-pollinated styles were collected at 24 h, 48 h and 72 h after pollination. Six RNA-seq libraries (SP24, SP48, SP72, CP24 CP48 and CP72; SP = self-pollinated, CP = cross-pollinated) were constructed and separately sequenced. In total, 299.327 million raw reads were generated. Following assembly, 63,762 unigenes were identified, and 27,264 (42.76 %) unigenes were annotated in five public databases: NR, KOG, KEGG, Swiss-Port and GO. To identify SI-related genes, the fragments per kb per million mapped reads (FPKM) values of each unigene were evaluated. Comparisons of CP24 vs. SP24, CP48 vs. SP48 and CP72 vs. SP72 revealed differential expression of 3,182, 3,575 and 3,709 genes, respectively. Consequently, several ubiquitin-mediated proteolysis, Ca2+ signaling, apoptosis and defense-associated genes were obtained. The temporal expression pattern of genes following CP and SP was analyzed; 6 peroxidase, 1 polyphenol oxidase and 7 salicylic acid biosynthetic process-related genes were identified. The RNA-seq data were validated by qRT-PCR of 15 unigenes. Finally, a unigene (CL25983Contig1) with strong homology to the S-RNase was analyzed. It was mainly expressed in styles, with dramatically higher expression in self-pollinated versus cross-pollinated tissues at 24 h post-pollination.
Conclusions
The present study reports the transcriptome of styles after cross- and self-pollination in tea and offers novel insights into the molecular mechanism behind SI in C. sinensis. We believe that this RNA-seq dataset will be useful for improvement in C. sinensis as well as other plants in the Theaceae family.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2703-5) contains supplementary material, which is available to authorized users.
Keywords: Camellia sinensis, Theaceae, Tea, Transcriptome, Self-incompatibility, S-RNase, Gametophytic
Background
Self-incompatibility (SI) is genetically controlled, and promotes outcrossing while preventing inbreeding depression in flowering plants [1]. Although SI has emerged as a model for cell-cell communication and signal transduction [2, 3], it is also an agriculturally important trait [4, 5]. Although SI has been widely demonstrated in angiosperms, its mechanism has been elucidated in only a few plants [6]. Sporophytic SI (SSI) in Brassicaceae is controlled by the stigma-specific S-receptor kinase (SRK) and pollen-specific S-locus cysteine-rich protein (SCR), which inhibits germination and growth of self-pollen on the stigma [7]. While during gametophytic SI (GSI) in Rosaceae, Solanaceae and Plantaginaceae, the interaction between the female determinant S-RNase and the male component S-locus F-box protein (SLF/SFB) mediate the rejection response of self-pollen tubes in the pistil [8–10]. Another GSI mechanism exists in Papaveraceae in which a Ca2+ signaling cascade leads to programmed cell death (PCD) [11]. Additionally, many species demonstrate SI from pollen inhibition in the ovary, which is called either ovarian SI (OSI) or late-acting SI (LSI). This form of SI was defined according to the location of the pollen inhibition instead of the genetic mechanism of SI as with GSI and SSI [12]. LSI is more widespread among the basal groups of Angiosperms, which indicates a conserved ancestral mechanism of SI; nevertheless, its molecular basis still remains unclear [6, 13]. Some types of LSI may be controlled by either the gametophyte or sporophyte independently, whereas other types may be jointly controlled [13].
Overall, SI is regulated by multiple genes and associated with different metabolic pathways. Next Generation Sequencing (NGS) and transcriptome analysis has revealed candidate genes that contribute to SI interaction in Citrus clementine [14], Citrus limon [4] and Leymus chinensis [3].
The Theaceae family consists of 20 genera and over 600 species [15]. Several of the species in these subclasses have significant economic value, such as Tea (C. sinensis L.), Tea-oil tree (C. oleifera Abel.) and the ornamental Sasanqua (C. sasanqua Thunb.). Due to their self-incompatibility, Theaceae species are highly heterogeneous, making crop improvement via classical breeding and genetics difficult [16], it and has hindered genetic map construction and contig assembly from whole-genome sequencing [16–18]. Therefore, uncovering the molecular basis of SI in Theaceae may simplify and enhance breeding and genomics in this family.
Tea is one of the most prominent beverages in the world [16] and is widespread in tropical and subtropical regions [15]. It is also a model for studying SI [19, 20]. Tea has exhibited GSI because of a self-pollen tube growth inhibiting at the base of the style [21, 22]. However, more recent evidence suggests LSI because the pollen tubes from self-fertilized flowers were inhibited in the ovary [23, 24]. Similarly, the edible oil plant of C. oleifera also manifests LSI behavior [5]. In these studies, although pollen tubes from self-pollinations penetrated the ovary, pollen tube growth was inhibited to some extent in the style. In contrast, pollen tubes from cross-pollination reached the ovary with no interference [5, 22–24]. Therefore, the differential growth of the pollen tube from self and cross-pollinations was a critical trait in understanding SI in C. sinensis.
Attempts have been made to understand the molecular mechanisms underlying tea SI [16, 21, 25]. A pistil-specific pathogenesis-related-1 (PR-1) protein was identified in tea, but the relationship between PR-1 and SI was not clearly demonstrated [21]. cDNA-AFLP method was also used to analyze differentially expressed genes in self-incompatible lines of tea, and 34 genes were isolated which were involved in energy metabolism, signal transduction and defense [26]. A C. sinensis floral transcriptome analysis revealed the presence of several pollen/pistil specific expression unigenes [16]. The objective of this study was to gain deeper insights into the molecular basis of SI and to provide a global view of candidate SI-related genes in tea using RNA-seq. We believe that the identification of candidate SI-related genes will contribute to a more complete understanding of the SI mechanism in C. sinensis, which will improve breeding and genomics research in the Theaceae.
Results and discussion
Observation of pollen tube growth
Pollens from both the cross- and self-pollination germinated at 6 h post-pollination, and no visible differences were observed in pollen tube growth from 6 h to 12 h. At 24 h after pollination, a small number of tubes from the cross-pollination reached the style base (Fig. 1). However, pollen tubes from self-pollination grew more slowly, reaching just the middle of the style. At 48 h after pollination, most tubes from cross-pollination reached the style base, but the pollen tubes from the self-pollination took 72 h to reach the same place. Self-pollen tubes grew slower than cross-pollen tubes from 24 h to 72 h after pollination. This is consistent with previous studies that also showed reduced pollen tube growth from a SI cross [5, 21–23]. The delay in pollen tube growth may be critical for the SI reaction because, by the time that the self-pollen tubes penetrate the ovary, the pistil may have already been primed for abscission [6].
Fig. 1.
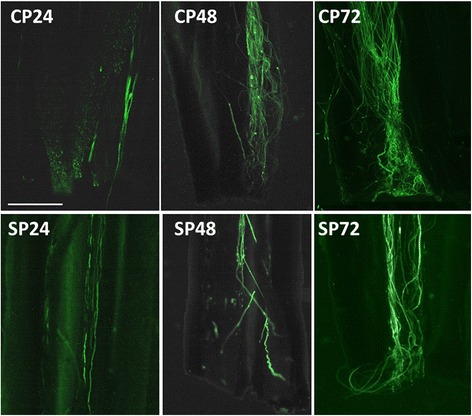
Pollen tubes growth after CP and SP in C. sinensis. CP24: a few pollen tubes reached the style base; CP48 and CP72: a larger number of pollen tubes arrived the style base; SP24: pollen tubes in the middle of the style; SP48: a few pollen tubes reached the style base; SP72: a large number of pollen tubes arrived the style base. Bar = 500 μm
RNA-seq and transcriptome assembly
To uncover the mechanism behind SI reaction in C. sinensis, six RNA-seq libraries (CP24, CP48, CP72, SP24, SP48 and SP72) were separately constructed and sequenced. In total, 299.327 million raw reads with a Q30 over 83 % were generated. After removing the low quality sequences (length < 35 bp, Q < 20), a total of 294.144 million clean reads were retained (Table 1). After de novo assembly using Trinity software, 63,762 unigenes were identified. The lengths of unigenes ranged from 301 bp to 14,580 bp, with an average of 1,018.26 bp and a N50 length of 1,354 bp (Table 2).
Table 1.
Summary for the RNA-seq outcomes of six separately pooled samples
| Sample | CP24a | CP48 | CP72 | SP24 | SP48 | SP72 | Total |
|---|---|---|---|---|---|---|---|
| Raw reads (million) | 43.798 | 55.519 | 54.843 | 48.094 | 49.118 | 47.956 | 299.327 |
| Clean reads (million) | 43.075 | 54.536 | 53.905 | 47.337 | 48.283 | 47.008 | 294.144 |
| Total base (Gb) | 5.007 | 6.339 | 6.266 | 5.502 | 5.612 | 5.464 | 34.189 |
| Q30 | 83.3 % | 83.4 % | 83.5 % | 83.3 % | 83.1 % | 83.1 % | — |
aCP = cross pollination, SP = self pollination, numbers after CP and SP indicate hours after pollination
Table 2.
Summary for the de novo assembly
| 300–499 (bp) | 500–999 (bp) | ≥1000 (bp) | N50 (bp) | Total Length (Mbp) | Max Length (bp) | Min Length (bp) | Average Length (bp) | |
|---|---|---|---|---|---|---|---|---|
| Unigene | 19228 | 24178 | 20356 | 1354 | 61.918 | 14580 | 301 | 1018.26 |
Functional annotation
To predict potential functions of the assembled unigenes, all of them were blastx (E-value ≤ 10−5) against 5 public databases, including the National Center for Biotechnology Information (NCBI) Non-redundant Protein (NR), Clusters of Orthologous Groups for Eukaryotic Complete Genomes (KOG), Kyoto Encyclopedia of Genes and Genomes (KEGG), Swiss-Prot Protein Database (Swiss-Port) and Gene Ontology (GO) (Fig. 2). Most unigenes (26,984, 42.32 %) were annotated to the NR database, and 16,210 (60.08 %) sequences had significant homology (blastx E-value < 10−45). Among the annotated unigenes, 37 % (9975) and 9.76 % (2631) had strong homology to Vitis vinifera and Theobroma cacao, respectively (Additional file 1).
Fig. 2.
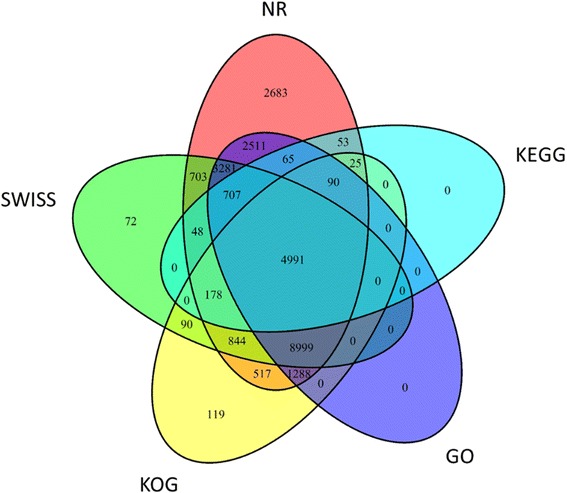
Blastx results of the transcriptome to five databases
The unigenes were further annotated and classified using the KOG database (Fig. 3). A total of 27,618 unigenes were assigned KOG classifications. Among the 25 KOG categories, the “general function prediction only” (6391, 23.15 %) was the most abundant, followed by “posttranslational modification, protein turnover, chaperones” (3054, 11.06 %), “signal transduction mechanisms” (2,803, 10.15 %) and “translation, ribosomal structure and biogenesis” (1394, 5.05 %).
Fig. 3.
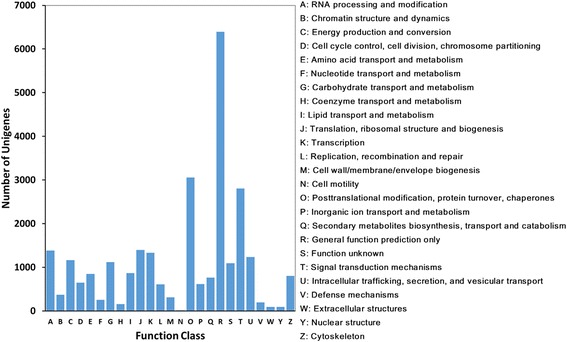
KOG functional classification of the C. sinensis transcriptome
The unigenes were then matched to the GO terms (Additional file 2). A total of 21,932 unigenes were classified into 58 sub-categories belonging to 3 categories (biological process, cellular component and molecular function). Among the biological process category, “metabolic process” and “cellular process” were the main functional groups, which were followed by “single organism process” and “response to stimulus”. In terms of cellular component, “cell part” and “cell” were the most highly represented subcategories. For the molecular function category, “binding” and “catalytic activity” were the two main groups.
To predict putative functions of the unigenes, KEGG annotation was also performed. Altogether, 6158 unigenes were assigned to 330 pathways, including “plant hormone signal transduction”, “calcium signaling pathway”, “plant-pathogen interaction”, “flavonoid biosynthesis” and “ubiquitin mediated proteolysis”. Plant hormones play an important role in incompatibility response; for example, in Theobroma cacao, auxin exhibited a strong increase after compatibility pollination, while ethylene exhibited a strong increase after incompatibility pollination [27], and higher 1-naphthaleneacetic acid (NAA) concentrations improved flower retention after incompatibility pollination [28]. Plant hormone signal transduction-related pathways were enriched in tomatoes when comparing the unpollinated and pollinated styles of self-compatible and self-incompatible species [29]. Calcium is essential to pollen tube germination and growth, and its temporal and spatial changes in cytosol generate Ca2+ signals [30, 31]. Ca2+ signaling cascade leads to PCD, which mediates SI reaction in Papaveraceae [11]. RNA-seq has also revealed that the calcium-signaling pathway is associated with SI in Citrus [14, 32] and Leymus chinensis [3]. SI systems presumably have evolved from pathogen defense mechanisms because similar biochemical and molecular processes occur in epidermal cells when fungal invasion occurs and during incompatible pollen tube penetration [13]. A large number of defense-related genes were speculated to be involved in incompatible pollen tube rejection [3, 32]. Ubiquitin mediated proteolysis pathway has a pivotal role in S-RNase-based GSI system. The Skp-1-Cullin1-F-box-Rbx1 complex (SCF) causes ubiquitin-mediated degradation of non-self S-RNase [33].
Differentially expressed genes involve in SI
To identify differentially expressed genes associated with SI in tea, the expression levels of unigenes were measured based on the fragments per kb per million of the mapped reads (FPKM) value. Totals of 3182, 3575 and 3709 differentially expressed genes were obtained between groups of CP24 vs. SP24, CP48 vs. SP48 and CP72 vs. SP72, respectively (Fig. 4, Additional file 3). To determine the putative functions of these genes, GO and KEGG analyses were implemented. Between the group comparing CP24 vs. SP24, 1826 genes were up-regulated and 1356 genes were down-regulated. Among these differentially expressed genes, 1008 were associated with GO categories, and 272 were mapped to 232 KEGG pathways. Similarly, between the group of CP48 vs. SP48, 1946 genes were up-regulated and 1629 genes were down-regulated; 1501 of them were associated with GO categories, and 566 were mapped to 251 KEGG pathways. In addition, between the group of CP72 vs. SP72, 1659 genes were up-regulated and 2050 genes were down-regulated; 1465 of them were associated with GO categories, and 485 were mapped to 266 KEGG pathways. The high number of differentially expressed unigenes provided an abundant list of candidate SI-related genes. In contrast, cDNA-AFLP analysis of self-incompatible lines of tea previously yielded only 100 polymorphic bands [26]. Therefore, RNA-seq seems to be more productive than cDNA-AFLP for identifying potential SI related genes.
Fig. 4.
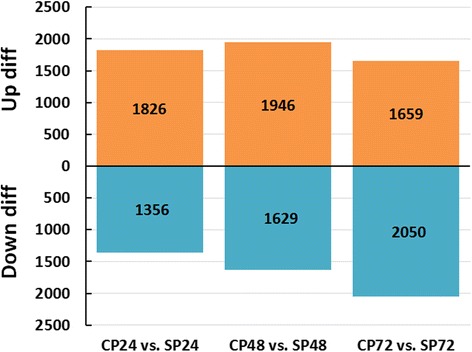
Number of differentially expressed unigenes between each two sample combination
In the above comparisons, some pathways occurred in all three comparisons, such as “phenylpropanoid biosynthesis”, “phenylalanine metabolism” and “plant-pathogen interaction”. In addition, “ubiquitin mediated proteolysis” [34–36], “apoptosis” [37, 38], “calcium signaling pathway” [30] and several defense-related pathways [3, 39] were also identified as up-regulated pathways that are associated with the SI reaction in plants. Therefore, the unigenes associated with the above pathways may also be associated with SI interaction in tea.
For “ubiquitin mediated proteolysis”, a SCF complex plays a key role in the S-RNase-based GSI system [1, 40]. S-phase kinase-associated protein 1 (Skp-1), F-box, RING-box protein 1 (Rbx1) and Cullin-1 (CUL1) also function as components of the SCF complex [33]. The SCF complex uniquely mediates the ubiquitination of non-self S-RNase, which is then degraded by the 26 s proteasome [33]. In this study, 7 differentially expressed unigenes annotated as Skp-1 (CL1Contig5289 and CL18002Contig1), F-box (CL1Contig1120 and CL1Contig2140), Rbx1 (CL1Contig679) and CUL1 (CL1Contig1054 and comp136094_c0_seq6) were identified. These unigenes might be directly related to SI in tea, which warrant further exploration.
Calcium (Ca2+) is a second messenger, which plays a key role in pollen germination and tube growth and serves an important function in the prevention of self-fertilization [30]. Several Ca2+ signaling-related genes were identified that may function during SI. Calcium-dependent protein kinase (CDPK) is a Ca2+ response element, which localized at the tip of the pollen tube. In Petunia, overexpression the Pi CDPK1 and the Pi CDPK2 will disrupt growth polarity or inhibit extension of the pollen tube, respectively [41]. Five predicted CDPKs (CL1Contig1101, CL1Contig1911, comp91595_c0_seq1, CL41260Contig1 and comp128850_c0_seq1) were detected; the latter three have higher expression levels in SP samples than CP, which might affect the growth polarity or inhibit the extension of the pollen tubes from self-pollination. The protein families of calmodulin (CaM), calmodulin-like (CML), and calcineurin B-like (CBL) act as sensors during Ca2+ signal transduction. Several members of them are expressed pollen-specifically and highly expressed during pollen tube germination and elongation [42]. Here, three CaMs (CL20031Contig1, comp152366_c1_seq4 and CL2413Contig1), four CBLs (CL1Contig702, CL23964Contig1, CL26666Contig1 and comp88134_c0_seq1), and three CMLs (CL27939Contig1, CL33074Contig1, and CL39651Contig1) were identified to be differentially expressed. Therefore, the unigenes annotated as CDPKs, CaMs and CMLs merit special attention.
Gene temporal expression patterns of CP and SP
To identify the temporal expression patterns of genes following self-pollination and cross-pollination treatments (Additional file 4), the short time-series expression miner software (STEM) was employed. The unigenes in CP and SP groups were clustered into 8 profiles (Fig. 5). Each profile represents a group of genes that exhibited similar expression trend. The highest number of genes in group CP was found in profile 7 (1,001, 31.51 %), followed by profile 2 (586, 18.45 %), profile 6 (535, 16.84 %) and finally profile 5 (319, 10.04 %). While in group SP, profile 7 (813, 25.70 %) was also the largest category, followed by profile 6 (636, 20.11 %), profile 2 (452, 14.29 %) and profile 5 (359, 11.35 %).
Fig. 5.

Different gene expression patterns between CP and SP. Each square represents a pattern. Top number indicates the profile ID number, bottom number indicates the number of time series genes. The patterns were ordered based on the number of genes. The colored squares were significant modules. The horizontal axis represents different times after pollination
Following GO-term analysis to identify the putative functions of genes in profile 7 of both treatments, genes were classified into 3 categories including biological process, cellular component and molecular function (Fig. 6). For the biological process category, “oxidation-reduction process” was the most abundant subcategory in group CP, while 31 genes were classified into “defense response to fungus” (16), “recognition of pollen” (8) and “salicylic acid biosynthetic process” (7) categories. In group SP, “carbohydrate metabolic process” and “response to ethylene” were the dominant groups, while 12 genes were involved in “ethylene-activated signaling pathway” (6) and “cell wall macromolecule catabolic process” (6) subcategories. These results show that cross- and self-pollination result in different gene expression responses in the style.
Fig. 6.

GO functional classification of the genes in profile 7. The letters C, F and P represent “Cellular component”, “Molecular function”, and “Biological process”, respectively
A total of 83 unigenes were clustered into the “oxidation-reduction process” group, and a heat map was constructed based on the FPKM values of six samples (Fig. 7). Expression of these unigenes steadily increased from 24 h to 72 h in cross-pollinated styles. A similar trend was observed in self-pollinated styles but with a delay. Previous studies have revealed that reactive oxygen species (ROS) regulate stress resistance and pollen tube growth in plants [43, 44]. ROS concentrations increase in incompatible pollen tubes, leading to deleterious effects and SI-mediated PCD [45]. Various antioxidant systems take part in biological processes to maintain the proper concentration of ROS and prevent the damage from ROS [46]. Here, 6 peroxidases (CL39261Contig1, CL36955Contig1, CL799Contig1, CL31720Contig1, CL1Contig803 and CL13267Contig1), 1 polyphenol oxidase (CL28614Contig1) and several other redox-related genes were greatly increased in cross-pollinated styles, which is consistent with a previous study showing higher activity of peroxidase and polyphenol oxidase in cross-pollinated styles than self-pollinated ones of C. sinensis [47]. Among the 6 putative peroxidase genes, 3 unigenes (CL799Contig1, CL39261Contig1 and CL31720Contig1) contained intact ORFs and were homologues to AT5G05340. A multiple sequence alignment of AT5G05340 and the deduced amino acid sequences of CL799Contig1, CL39261Contig1 and CL31720Contig1 were constructed, which share approximately 70 %, 45 % and 41 % identity, respectively (Fig. 8). The gene AT5G05340 encodes a peroxidase (AtPrx 53) enzyme, which is closely related to the cell wall synthesis [48]. Therefore, the higher expression levels of these genes might promote pollen tube elongation during compatible styles.
Fig. 7.

A heat map of 83 oxidation-reduction process-related unigenes generated using a MeV software. Along the horizontal axis (from left to right): CP24, CP48, CP72, SP24, SP48, SP72. The vertical axis represent the unigenes
Fig. 8.

A multiple alignment of amino acid sequences of AT5G05340, CL799Contig1, CL39261Contig1 and CL31720Contig1 constructed by DNAMAN
Furthermore, 7 unigenes (CL1Contig2132, CL1Contig5305, CL20835Contig1, CL25392Contig1, CL29231Contig1, CL35779Contig1 and CL38632Contig1) associated with “salicylic acid biosynthetic process” were also consistently up-regulated in 3 samples of CP. Salicylic acid (SA) may contribute to regulating ROS accumulation [49], and then induce the defense responses and impact the self/non-self recognition as well as the pollen tube growth [50, 51].
Differentially expressed gene validation by qRT-PCR
Fifteen differentially expressed genes were selected to test the reliability of RNA-seq data (Table 3). These genes were associated with “plant-pathogen interaction” (CL1Contig2502, CL25362Contig1, CL25650Contig1, CL21816Contig1 and CL2609Contig1), “flavonoid biosynthesis” (CL32645Contig1, CL28991Contig1 and CL26013Contig1), “ubiquitin mediated proteolysis” (CL1Contig5289 and CL1Contig1054), “apoptosis” (CL12551Contig1), “calcium signaling pathway” (CL23964Contig1), “plant hormone response” (CL25362Contig1 and CL31783Contig1) and “negative regulation of programmed cell death” (CL20835Contig1). The expression patterns for 11 genes (73.3 %) were highly consistent with the sequencing data, while 4 genes (26.7 %, CL1Contig2502, CL25362Contig1, CL25362Contig1 and CL1Contig5289) were partially consistent (Fig. 9). The qRT-PCR results confirmed that the RNA-seq results were relatively accurate.
Table 3.
Primers used for qRT-PCR validation
| Unigenes | KEEG Annotation | Gene | Forward 5'–3' | Reverse 5'–3' |
|---|---|---|---|---|
| CL12551Contig1 | Apoptosis | Serine/threonine-protein kinase | TATTCCCTTTTCCGATCTTCGCTAT | CAAAGATGAGTTGCTTTTCGTGTTG |
| CL23964Contig1 | Calcium signaling pathway | calcineurin B-like protein | AAATTTGCATATCTGCCTGTGTCAA | ATGTAAAACATCCAAAACCCCAGTC |
| CL32645Contig1 | Flavonoid biosynthesis | ANR | AGTATGGCTTTTGTGCTTTGATTGA | TAGAGGTGTCAAGGTTTCAGTTTCA |
| CL28991Contig1 | Flavonoid biosynthesis | CYP75A(F3'5'H) | TTGTAAGTATGGGAGTTTGGGTAGG | CGACACATCATCAAGCGTAATAGAG |
| CL26013Contig1 | Flavonoid biosynthesis | LDOX | AGTTGGCTAATAATGCTAGTGGTCA | TTTTCTAGTCGGCCTTCTTCTAGTC |
| CL20835Contig1 | negative regulation of programmed cell death | Probable WRKY 40 | ATTGTACTTGTCGCAAATGTCTGTT | CAATGTTAACCCACTTCCACTACAC |
| CL1Contig2502 | Plant-pathogen interaction | caffeoyl-CoA O-methyltransferase | TCACAAGAACCAGACACAAACATTC | TTAATGGGCACAAGGGTTATGTTTC |
| CL41260Contig1 | Plant-pathogen interaction | CPK | GATTCCTAAACATTTCCAAGCCACA | CTTTTAATGGTTGGACGGTGAGAAA |
| CL25650Contig1 | Plant-pathogen interaction | PR1 | ACCGTTATTTGTACACTGAACCCTA | TATGCTAATTCGAGGATGGGTGATT |
| CL21816Contig1 | Plant-pathogen interaction | RPS2 | CCAATGGGACCTCTGAACTATTTTG | TGCTTAAATTGCCTTTCAACGAAGA |
| CL2609Contig1 | Plant-pathogen interaction | CALM | TGAAATCTATCATTCCATCCCCGTT | GAAGGGAGCTTTTGATGTGTTTGAT |
| CL1Contig5289 | ubiquitin mediated proteolysis | Skp1 | TGGGTTCCAATGGTATTGTTTGAAG | CGAGTGGATTATCGTTCAGTTGATG |
| CL1Contig1054 | ubiquitin mediated proteolysis | Cullin-1 | ATATGTGTGGTAACCCTATTGGTCC | AATTCCCCGTAAACCAGTTCATAGA |
| CL25362Contig1 | Response to auxin | Protein TIFY 9 | ATTGACGATTTTCTACAACGGAACC | TCTCCAGAAGTTGATGTTGATCTGT |
| CL31783Contig1 | Response to salicylic acid | 12-oxophytodienoate reductase 11 | TCATAACAAGGGTGGGATCTTCTTT | CACTGTATCTTTCATGAACTGGTCG |
| CL25983Contig1 | - | Ribonuclease, T2 family | CTGCGATAGCCGCAACTCTT | GGAAGTAGCTGTGCTGGTCAA |
| GE651107 | Reference gene | GAPDH | GGAAGCATCATGAACTCAAAGTGAA | ATCCTTCTCATTGACACCCATAACA |
Fig. 9.

Validation of RNA-seq results by qRT-PCR, 15 genes were selected for qRT-PCR validation. The GADPH gene was chosen as the reference gene
Analysis of a putative S-RNase gene
S-RNase is associated with GSI in plants of Rosaceae, Solanaceae and Plantaginaceae [8, 10]. Pistil-expressed S-RNase acts as a type of cytotoxin for self-pollen rejection [52]. It was first cloned from Nicotiana alata [51], then transgenic analyses in Petunia inflate prove that the S-RNase is the pistil determinant during the pollen rejection interaction [53]. Recently, S-RNase has been identified in various species, such as Solanaceae [51], Prunus [54], Malus [55], Pyrus [56], Antirrhinum [57], and Citrus [4].
A putative S-RNase gene (CL25983Contig1), which had strong homology to ribonuclease T2 gene, was obtained. A phylogenetic tree, based on the amino acid sequence of CL25983Contig1 and S-RNases from other species (Fig. 10), revealed 4 main groups. S-RNases from Malus and Pyrus were clustered in group I. The sequence from Coffea canephora was in group II. CL25983Contig1 and other S-RNases from Petunia, Solanum, Nicotiana, Medicago, Antirrhinum and Citrus were clustered in group III. All of the sequences from Prunus were clustered together in group IV. CL25983Contig1 was most closely related to S-RNases from Citrus reticulata, Antirrhinum hispanicum and Medicago truncatulata.
Fig. 10.

Phylogenetic analysis of the putative S-RNase (CL25983Contig1) from C. sinensis and S-RNases from other species using Neighbor-joining method. Blue highlighting indicates the position of CL25983Contig1. Yellow highlighting indicates the position of 4 genes, which have the highest similarity to CL25983Contig1
qRT-PCR of CL25983Contig1 in different tissues of tea (Fig. 11) confirmed that this gene was expressed at much higher levels in styles than other tissues such as filaments, leaves, petals and pollens. Moreover, the expression level of CL25983Contig1 increased at 24 h after self-pollination and was 7.45 times higher than that of cross-pollination (Fig. 12). Interestingly, the pollen tube growth in SP was hindered at 24 h. This suggests that CL25983Contig1 plays an important role in the SI reaction.
Fig. 11.

The expression pattern of the putative S-RNase gene (CL25983Contig1) in different tissues in C. sinensis
Fig. 12.
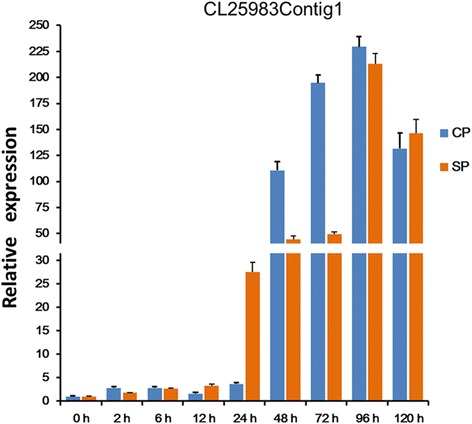
Expression pattern of the putative S-RNase gene (CL25983Contig1) after CP and SP
Recently, LSI has been demonstrated in many different species; however, the molecular mechanism for LSI is not well understood [6, 13]. LSI is defined based on the location of self-pollen inhibition rather than the molecular mechanism [12]. LSI can be regulated in different ways, through gametophytic or sporophytic control, either independently or acting together [13, 58]. For the pollen tube growth from a self-pollination that was arrested in the ovary, self-incompatibility in C. sinensis was assumed to occur via LSI [6, 23, 24]. Because growth of pollen tubes from self-pollination was halted in the style to a certain extent, and several SCF components-encoding genes and a putative S-RNase were identified, LSI in tea is likely under gametophytic control.
Conclusions
Transcriptome analysis of styles after cross- and self-pollination in tea has identified a set of candidate genes involved in self-incompatibility. High levels of expression of several SCF components-encoding genes and a putative S-RNase gene suggest that LSI in C. sinensis might be under gametophytic control. Our study offers novel insights into the molecular mechanism behind SI in C. sinensis. We believe that this RNA-seq data will be useful for breeding and genomics research in C. sinensis as well as other plants in the Theaceae family.
Methods
Plant material
Two cultivars of ‘Fuding Dabaicha’ and ‘Zhongcha 108’, grown in the China National Germplasm Hangzhou Tea Repository, were used in this study. ‘Fuding Dabaicha’ is used in many tea-breeding programs in China and more than 18 % of national elite cultivars are derived from it [51]. While ‘Zhongcha 108’ is a radiation-induced cultivar selected by the Tea Research Institute Chinese Academy of Agricultural Sciences. The reproductive organs and seed-setting rate of these two cultivars are normal. The following tissues were collected from ‘Fuding Dabaicha’: buds, leaves, filaments, petals, pollens, sepals, unpollinated styles and ovaries with 3 biological replicates. The samples were frozen immediately in liquid nitrogen and stored at −80 °C.
Pollination treatment
Pollens of ‘Fuding Dabaicha’ and ‘Zhongcha 108’ were periodically collected from “balloon” stage flowers by placing them in a desiccator at room temperature for 8 h. Pollination was performed from 9:00-11:00 am and 1:00-3:00 pm on sunny days in November. Pollination combinations included self-pollination (SP) of ‘Fuding Dabaicha’ × ‘Fuding Dabaicha’ and cross-pollination (CP) of ‘Fuding Dabaicha’ × ‘Zhongcha 108’. Flowers in the “balloon” stage were bagged after emasculation and self- or cross-pollinated. The styles were harvested at different intervals (0 h, 2 h, 6 h, 12 h, 24 h, 48 h, 72 h, 96 h and 120 h) after pollination. Therefore, the number of self-pollinated samples ranged from SP0 (0 h after pollination) to SP120 (120 h after pollination). Similarly, the number of cross-pollinated samples ranged from CP0 to CP120. After harvesting, half of the styles of each sample were fixed in FAA (formalin:acetic acid:alcohol 5:5:90 v/v) for 3 h. The remaining styles were divided into two parts, one for RNA-seq and another for qRT-PCR with three biological replicates. Finally, the materials were frozen in liquid nitrogen and stored at −80 °C.
Observation of pollen tube growth
After removing the styles from FAA, the styles were macerated with 3 M NaOH for 3 h. Then, the styles were split along the vertical axis into 3 sections, and stained by 1 % aniline blue for 3 h. Finally, the pollen tubes were observed using a Nikon Eclipse 80i fluorescence microscope (Nikon Instech Co. Ltd., Kawasaki, Japan).
RNA extraction, library construction and sequencing
Total RNAs were extracted from self- and cross-pollinated styles using an RNAprep pure Plant Kit (Tiangen, Beijing, China). The quality of the total RNA was confirmed by electrophoresis through a 1 % agarose gel followed by viewing, and using a NanoDrop 2000 (Thermo Scientific, DE, USA). RNAs from 6 samples (SP24, SP48, SP72, CP24, CP48, and CP72) were used for RNA-seq. The mRNA was isolated from 6 μg total RNA using the Truseq RNASample Prep Kit (Illumina, San Diego, CA, USA). Next, the mRNA was mixed with fragmentation buffer for mRNA fragmentation. After the short fragment, mRNAs were used as template for the first-strand cDNA synthesis using random hexamer-primers, the second-strand cDNA was synthesized and then purified using a SuperScript Double-Stranded cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA) and PCR extraction kit (Takara Bio, Otsu, Japan), respectively. The cDNA library was then created following end-repair, polyA addition, adaptor ligation and PCR amplification. After that, cDNA libraries were evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, Calif.) and sequenced on an Illumina HiSeq™ 2000.
De novo assembly and annotation
Raw RNA-seq data were preprocessed by first removing adaptor and low quality sequences (Q < 20 or less than 35 bp) and the qualified reads were then assembled into non-redundant transcripts by Trinity [59] and TGICL [60]. All non-redundant transcripts were annotated to the NR, SWISSPROT, KOG and KEGG databases by blastx using a threshold of 1e−5. GO term distribution for each transcript was carried out using Blast2GO software [61].
Differential gene expression analysis
The expression of each gene was estimated by FPKM, which was calculated based on the universal reads from 6 separate libraries [62]. Seven groups including CP24 vs. CP48, CP48 vs. CP72, CP24 vs. SP24, CP48 vs. SP48, CP72 vs. SP72, SP24 vs. SP48 and SP48 vs. SP72 were compared. To identify differentially expressed genes, differential expression analysis of the comparisons was performed using a DESeq program (http://bioconductor.org/packages/release/bioc/html/DESeq.html). P-values related to the analysis of differentially expressed genes were adjusted for multiple testing by the Benjamini-Hochberg false discovery rate (FDR) method [63]. For each comparison, we extracted those unigenes with a criteria of FDR-adjusted P-value ≤ 0.05 and |log2Ratio| > 1. Furthermore, the differentially expressed unigenes were annotated to the KEGG database using blastx. GO term distribution was performed by a Blast2go software using an FDR-adjusted P-value ≤ 0.05 as the cutoff. The genes, which expressed differentially in the comparison of CP24 vs. SP24, CP48 vs. SP48 and CP72 vs. SP72, were used to screen candidate genes for SI.
Comparison of differential gene expression patterns between CP and SP
A STEM software (Carnegie Mellon University, USA) was employed to analyze different patterns of gene expression between CP and SP. Differentially expressed genes in the comparisons of CP24 vs. CP48 and CP48 vs. CP72 were used to cluster changing patterns of CP treatments (Additional file 3). Similarly, differentially expressed genes in the comparison of SP24 vs. SP48 and SP48 vs. SP72 were used to cluster changing patterns of SP treatments (Additional file 3). GO-term analysis was utilized to determine the potential functions of times series genes. GO term with a FDR-adjusted P-values ≤ 0.05 was considered to be significantly enriched. The heat map was generated using MeV software (www.tm4.org/mev.html). The multiple alignments of amino acid sequences were constructed using DNAMAN software (www.lynnon.com).
Validation by quantitative real-time PCR (qRT-PCR)
Fifteen differentially expressed transcripts were selected for validation of sequencing data using qRT-PCR. Specific primers were designed by Primer 5 software (Premier Biosoft International, Palo Alto, CA). Total RNA was extracted using an RNAprep pure Plant Kit (Tiangen, Beijing, China) from the styles. One microgram of total RNA was used to synthesize cDNA using a PrimeScript® 1st Strand cDNA Synthesis Kit (Takara, Dalian, China). Then, qRT-PCR was performed on the ABI 7500 Real-Time PCR System (Applied Biosystems) using a PrimeScript™ RT reagent qPCR Kit (Takara, Dalian, China). A GADPH (GE651107) gene was utilized as the reference gene. The real time PCR program was as follows: 95 °C for 15 s, 40 cycles at 95 °C for 5 s, 60 °C for 34 s. Each reaction was repeated three times for biological and technical replicates, respectively. The relative quantitation of the gene expression was calculated using the 2−ΔΔCt method [64].
Expression analysis of a putative S-RNase gene
The amino acid sequence of CL25983Contig1 was predicted using a DNAstar program Editseq option (DNASTAR, Inc., Madison, Wis.). A phylogenetic tree was constructed using the amino acid sequence deduced from CL25983Contig1 and 93 S-RNases from other species by a Neighbor-joining method by using MEGA 5.2 (www.megasoftware.net). Total RNA was extracted from 8 different tissues. The expression pattern of CL25983Contig1 was examined using qRT-PCR with a reference gene GAPDH. Furthermore, the expression profiling of CL25983Contig1 was detected in both two pollination treatments from 0 h to 120 h after pollination. Each reaction was performed with three biological replicates and three technical replicates.
Availability of supporting data
Sequence data of 6 samples have been deposited in the NCBI SRA database (SRR3290042, SRR3290055, SRR3290062, SRR3290078, SRR3290084 and SRR3290124).
Acknowledgments
The study was supported by the Modern Agro-industry Technology Research System (nycytx-23) and The Major Science and Technology Special Project of variety breeding of Zhejiang Province (2012C2905-8).
Abbreviations
- SP
self-pollination
- CP
cross-pollination
- FPKM
fragments per kb per million mapped reads
- SSI
sporophytic self-incompatibility
- SRK
S-receptor kinase
- SCR
S-locus cysteine-rich protein
- GSI
gametophytic self-incompatibility
- SLF/SFB
S-locus F-box protein
- PCD
programmed cell death
- OSI
ovarian self-incompatibility
- LSI
late-acting self-incompatibility
- NGS
next generation sequencing
- C. sinensis
Camellia sinensis
- PR-1
pathogenesis-related-1 protein
- NAA
1-naphthaleneacetic acid
- SCF
Skp-1-Cullin1-F-box-Rbx1 complex
- Skp-1
S-phase kinase-associated protein 1
- Rbx1
RING-box protein 1
- CUL1
Cullin-1
- CDPK
calcium-dependent protein kinase
- CaM
calmodulin protein
- CML
calmodulin-like protein
- CBL
calcineurin B-like protein
- STEM
short time-series expression miner software
- ROS
reactive oxygen species
- SA
salicylic acid
- qRT-PCR
Quantitative real-time PCR
- KOG
clusters of orthologous groups for eukaryotic complete genomes
- NR
NCBI non-redundant protein
- KOG
clusters of orthologous groups for eukaryotic complete genomes
- KEGG
Kyoto encyclopedia of genes and genomes
- Swiss-Port
Swiss-Prot protein database
- GO
gene ontology
- NCBI
National Center for Biotechnology Information
- RNA-Seq
RNA sequencing
- FDR
false-discovery rate
Additional files
The species distribution of all of the unigenes. (TIF 3926 kb)
Gene Ontology terms of all of the unigenes. (TIF 9114 kb)
Differentially expressed genes between cross- and self-pollination samples. (XLSX 1339 kb)
Temporal expression of genes in cross- and self-pollination samples. (XLSX 1737 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HC and CCZ conceived and designed the study. CCZ and LY Wang drafted the manuscript. HC and DJN critically reviewed the manuscript. CCZ, LY Wu, HLL and FZ collected and prepared the samples for pollen tube growth observation and RNA sequencing. LY Wang, KW and CCZ analyzed the data. All authors read and approved the final manuscript.
Contributor Information
Hao Cheng, Email: chenghao@tricaas.com.
De-Jiang Ni, Email: nidj@mail.hzau.edu.cn.
References
- 1.Wheeler MJ, de Graaf BHJ, Hadjiosif N, Poulter NS, Osman K, Perry RM, et al. Identification of the pollen self-incompatibility determinant in Papaver rhoeas. Nature. 2009;459:992–995. doi: 10.1038/nature08027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franklin-Tong VE, Ride JP, Franklin FCH. Recombinant stigmatic self-incompatibility (S-) protein elicits a Ca2+ transient in pollen of Papaver rhoeas. Plant J. 1995;8:299–307. doi: 10.1046/j.1365-313X.1995.08020299.x. [DOI] [Google Scholar]
- 3.Zhou Q, Jia J, Huang X, Yan X, Cheng L, Chen S, et al. The large-scale investigation of gene expression in Leymus chinensis stigmas provides a valuable resource for understanding the mechanisms of poaceae self-incompatibility. BMC Genomics. 2014;15:399. doi: 10.1186/1471-2164-15-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang S, Ding F, He X, Luo C, Huang G, Hu Y. Characterization of the ‘Xiangshui’ lemon transcriptome by de novo assembly to discover genes associated with self-incompatibility. Mol Genet Genomics. 2015;290:365–375. doi: 10.1007/s00438-014-0920-7. [DOI] [PubMed] [Google Scholar]
- 5.Liao T, Yuan DY, Zou F, Gao C, Yang Y, Zhang L, et al. Self-Sterility in Camellia oleifera May Be Due to the Prezygotic Late-Acting Self-Incompatibility. PloS One. 2014;9:e99639. doi: 10.1371/journal.pone.0099639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gibbs PE. Late-acting self-incompatibility - the pariah breeding system in flowering plants. New Phytol. 2014;203:717–734. doi: 10.1111/nph.12874. [DOI] [PubMed] [Google Scholar]
- 7.Wang L, Peng H, Ge T, Liu T, Hou X, Li Y. Identification of differentially accumulating pistil proteins associated with self-incompatibility of non-heading Chinese cabbage. Plant Biol. 2014;16:49–57. doi: 10.1111/plb.12016. [DOI] [PubMed] [Google Scholar]
- 8.Murfett J, Atherton TL, Mou B, Gassert CS, McClure BA. S-RNase expressed in transgenic Nicotiana causes S-allele-specific pollen rejection. Nature. 1994;367:563–566. doi: 10.1038/367563a0. [DOI] [PubMed] [Google Scholar]
- 9.Sijacic P, Wang X, Skirpan AL, et al. Identification of the pollen determinant of S-RNase-mediated self-incompatibility. Nature. 2004;429:302–305. doi: 10.1038/nature02523. [DOI] [PubMed] [Google Scholar]
- 10.McClure B. New views of S-RNase-based self-incompatibility. Curr Opin Plant Biol. 2006;9:639–646. doi: 10.1016/j.pbi.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 11.Thomas SG, Franklin-Tong VE. Self-incompatibility triggers programmed cell death in Papaver pollen. Nature. 2004;429:305–309. doi: 10.1038/nature02540. [DOI] [PubMed] [Google Scholar]
- 12.LaDoux T, Friar EA. Late-Acting Self-Incompatibility in Ipomopsis tenuifolia (Gray) V. Grant (Polemoniaceae) Int J Plant Sci. 2006;167:463–471. doi: 10.1086/500985. [DOI] [Google Scholar]
- 13.Allen AM, Hiscock SJ. Evolution and phylogeny of self-incompatibility systems in angiosperms. In: Franklin-Tong VE, editor. Self-incompatibility in flowering plants-evolution, diversity and mechanisms. Berlin: Springer; 2008. pp. 73–100. [Google Scholar]
- 14.Caruso M, Merelo P, Distefano G, La Malfa S, Piero ARL, Tadeo F, et al. Comparative transcriptome analysis of stylar canal cells identifies novel candidate genes implicated in the self-incompatibility response of Citrus clementina. BMC Plant Biol. 2012;12:20. doi: 10.1186/1471-2229-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luna I, Ochoterena H. Phylogenetic relationships of the genera of Theaceae based on morphology. Cladistics. 2004;20:223–270. doi: 10.1111/j.1096-0031.2004.00024.x. [DOI] [PubMed] [Google Scholar]
- 16.Tan LQ, Wang LY, Wei K, Zhang CC, Wu LY, Qi QN, et al. Floral transcriptome sequencing for SSR marker development and linkage map construction in the tea plant (Camellia sinensis) Plos One. 2013;8(11):e81611. doi: 10.1371/journal.pone.0081611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma JQ, Zhou YH, Ma CL, Yao MZ, Jin JQ, Wang XC, et al. Identification and characterization of 74 novel polymorphic EST-SSR markers in the tea plant, Camellia sinensis (Theaceae) Am J Bot. 2010;97:e153–e156. doi: 10.3732/ajb.1000376. [DOI] [PubMed] [Google Scholar]
- 18.Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 19.Wang XC, Zhao QY, Ma CL, Zhang ZH, Cao HL, Kong YM, et al. Global transcriptome profiles of Camellia sinensis during cold acclimation. BMC Genomics. 2013;14:415. doi: 10.1186/1471-2164-14-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, Jiang CJ, et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genomics. 2011;12:131. doi: 10.1186/1471-2164-12-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomimoto Y, Ikehashi H, Kakeda K, Kowyama Y. A pistil-specific PR-1 like protein of Camellia, its expression, sequence and genealogical position. Breeding Sci. 1999;49:97–104. doi: 10.1270/jsbbs.49.97. [DOI] [Google Scholar]
- 22.Wang Y, Jiang CJ, Zhang HY. Observation on the Self-incompatibility of Pollen Tubes in Self-pollination of Tea Plant in Style in vivo. Tea Science. 2008;28:429–435. [Google Scholar]
- 23.Wachira FN, Kamunya SK. Pseudo-self-incompatibility in some tea clones (Camellia sinensis (L) O Kuntze) J Hortic Sci Biotech. 2005;80:716–720. doi: 10.1080/14620316.2005.11512004. [DOI] [Google Scholar]
- 24.Chen X, Hao S, Wang L, Fang W, Wang Y, Li X. Late-acting self-incompatibility in tea plant (Camellia sinensis) Biologia. 2012;67:347–351. [Google Scholar]
- 25.Wang YH, Li XC, Zhu-Ge Q, Jiang X, Wang WD, Fang WP, et al. Nitric oxide participates in cold-inhibited Camellia sinensis pollen germination and tube growth partly via cGMP in vitro. PLoS One. 2012;7:e52436. [DOI] [PMC free article] [PubMed]
- 26.Chen X, Liu T, Hao S, Fang W, Wang Y, Li X. Differential gene expression analysis of self-incompatible lines in tea by cDNA-AFLP. Afr J Biotechnol. 2011;10:10906–10913. [Google Scholar]
- 27.Baker RP, Hasenstein KH, Zavada MS. Hormonal changes after compatible and incompatible pollination in Theobroma cacao L. HortScience. 1997;32:1231–1234. [Google Scholar]
- 28.Hasenstein KH, Zavada MS. Auxin modification of the incompatibility response in Theobroma cacao. Physiol Plantarum. 2001;112:113–118. doi: 10.1034/j.1399-3054.2001.1120115.x. [DOI] [PubMed] [Google Scholar]
- 29.Zhao P, Zhang L, Zhao L. Dissection of the style’s response to pollination using transcriptome profiling in self-compatible (Solanum pimpinellifolium) and self-incompatible (Solanum chilense) tomato species. BMC Plant Biol. 2015;15:119. doi: 10.1186/s12870-015-0492-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinhorst L, Kudla J. Calcium-a central regulator of pollen germination and tube growth. BBA-Mol Cell Res. 1833;2013:1573–1581. doi: 10.1016/j.bbamcr.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Brewbaker JL, Kwack BH. The essential role of calcium ion in pollen germination and pollen tube growth. Am J Bot. 1963;50:859–865. doi: 10.2307/2439772. [DOI] [Google Scholar]
- 32.Miao HX, Ye ZX, Qin YH, Hua GB. Identification of differentially expressed genes in 72 h styles from self-incompatible Citrus reticulate. Scientia Horticulturae. 2013;161:278–285. doi: 10.1016/j.scienta.2013.07.013. [DOI] [Google Scholar]
- 33.Hua ZH, Kao TH. Identification of major lysine residues of S3-RNase of Petunia inflata involved in ubiquitin-26S proteasome-mediated degradation in vitro. Plant J. 2008;54:1094–1104. doi: 10.1111/j.1365-313X.2008.03487.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Zhao Z, Xue Y. Roles of proteolysis in plant self-incompatibility. Annual Rev Plant Biol. 2009;60:21–42. doi: 10.1146/annurev.arplant.043008.092108. [DOI] [PubMed] [Google Scholar]
- 35.Zhao L, Huang J, Zhao Z, Li Q, Sims TL, Xue Y. The Skp1-like protein SSK1 is required for cross-pollen compatibility in S-RNase-based self-incompatibility. Plant J. 2010;62:52–63. doi: 10.1111/j.1365-313X.2010.04123.x. [DOI] [PubMed] [Google Scholar]
- 36.Matsumoto D, Yamane H, Abe K, Tao R. Identification of a Skp1-like protein interacting with SFB, the pollen S determinant of the gametophytic self-incompatibility in Prunus. Plant Physiol. 2012;159:1252–1262. doi: 10.1104/pp.112.197343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li S, Šamaj J, Franklin-Tong VE. A mitogen-activated protein kinase signals to programmed cell death induced by self-incompatibility in Papaver pollen. Plant Physiol. 2007;145:236–245. doi: 10.1104/pp.107.101741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hua Z, Kao TH. Identification and characterization of components of a putative Petunia S-locus F-box-containing E3 ligase complex involved in S-RNase-based self-incompatibility. Plant Cell. 2006;18:2531–2553. doi: 10.1105/tpc.106.041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hiscock SJ, Allen AM. Diverse cell signalling pathways regulate pollen-stigma interactions: the search for consensus. New Phytol. 2008;179:286–317. doi: 10.1111/j.1469-8137.2008.02457.x. [DOI] [PubMed] [Google Scholar]
- 40.Kao T, Tsukamoto T. The molecular and genetic bases of S-RNase-based self-incompatibility. Plant Cell. 2004;16(suppl 1):72–83. doi: 10.1105/tpc.016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoon GM, Dowd PE, Gilroy S, McCubbin AG. Calcium-dependent protein kinase isoforms in Petunia have distinct functions in pollen tube growth, including regulating polarity. Plant Cell. 2006;18:867–878. doi: 10.1105/tpc.105.037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou L, Fu Y, Yang Z. A genome-wide functional characterization of Arabidopsis regulatory calcium sensors in pollen tubes. J Integr Plant Biol. 2009;51:751–761. doi: 10.1111/j.1744-7909.2009.00847.x. [DOI] [PubMed] [Google Scholar]
- 43.Potocky´ M, Jones MA, Bezvoda R, Smirnoff N, Za´rsky´ V. Reactive oxygen species produced by NADPH oxidase are involved in pollen tube growth. New Phytol. 2007;174:742–751. doi: 10.1111/j.1469-8137.2007.02042.x. [DOI] [PubMed] [Google Scholar]
- 44.Wang CL, Wu J, Xu GH, Gao YB, Chen G, Wu JY, et al. S-RNase disrupts tip-localized reactive oxygen species and induces nuclear DNA degradation in incompatible pollen tubes of Pyrus pyrifolia. J Cell Sci. 2010;123:4301–4309. doi: 10.1242/jcs.075077. [DOI] [PubMed] [Google Scholar]
- 45.Wilkins KA, Bancroft J, Bosch M, Ings J, Smirnoff N, Franklin-Tong VE. Reactive oxygen species and nitric oxide mediate actin reorganization and programmed cell death in the self-incompatibility response of papaver. Plant Physiol. 2011;156:404–416. doi: 10.1104/pp.110.167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neog B, Yadav RNS, Singh ID. Peroxidase, polyphenol oxidase and acid phosphatase activities in the stigma-style tissue of Camellia sinensis (L) O. Kuntze following compatible and incompatible pollination. J Indian Inst Sci. 2004;84:47–52. [Google Scholar]
- 48.Fernandez-Perez F, Pomar F, Pedreño MA, Novo-Uzal E. The suppression of AtPrx52 affects fibers but not xylem lignification in Arabidopsis by altering the proportion of syringyl units. Physiologia Plantarum. 2015;154:395–406. doi: 10.1111/ppl.12310. [DOI] [PubMed] [Google Scholar]
- 49.Torres MA, Jones JDG, Dangl JL. Reactive oxygen species signaling in response to pathogens. Plant Physiol. 2006;141:373–378. doi: 10.1104/pp.106.079467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Brien M, Kapfer C, Major G, Laurin M, Bertrand C, Kondo K, et al. Molecular analysis of the stylar-expressed Solanum chacoense small asparagine-rich protein family related to the HT modifier of gametophytic self-incompatibility in Nicotiana. Plant J. 2002;32:985–996. doi: 10.1046/j.1365-313X.2002.01486.x. [DOI] [PubMed] [Google Scholar]
- 51.Anderson MA, Cornish EC, Mau S-L, Williams EG, Hoggart R, Atkinson A, et al. Cloning of cDNA for a stylar glycoprotein associated with expression of self-incompatibility in Nicotiana alata. Nature. 1986;321:38–44. doi: 10.1038/321038a0. [DOI] [Google Scholar]
- 52.Yu J, Huang H, Yao M, Yang YJ. Analysis of Genetic Diversity and Relationship of Half-sib Tea Cultivars Related to Fuding Dabai and Yunnan Daye Using EST-SSR Markers. J Tea Sci. 2010;30:184–190. [Google Scholar]
- 53.Lee HS, Huang S, Kao TH. S proteins control rejection of incompatible pollen in Petunia inflata. Nature. 1994;367:560–563. doi: 10.1038/367560a0. [DOI] [PubMed] [Google Scholar]
- 54.Ushijima K, Sassa H, Tao R, Yamane H, Dandekar AM, Gradziel TM, et al. Cloning and characterization of cDNAs encoding S-RNases from almond (Prunus dulcis): primary structural features and sequence diversity of the S-RNases in Rosaceae. Mol Gen Genet. 1998;260:261–268. doi: 10.1007/s004380050894. [DOI] [PubMed] [Google Scholar]
- 55.Broothaerts W, Janssens GA, Proost P, Broekaert WF. cDNA cloning and molecular analysis of two self-incompatibility alleles from apple. Plant Mol Biol. 1995;27:499–511. doi: 10.1007/BF00019317. [DOI] [PubMed] [Google Scholar]
- 56.Castillo C, Takasaki T, Saito T, Norioka S, Nakanishi T. Cloning of the S8-RNase (S8-allele) of Japanese Pear (Pyrus pyrifolia Nakai) Plant Biotechnol J. 2002;19:1–6. doi: 10.5511/plantbiotechnology.19.1. [DOI] [Google Scholar]
- 57.Xue Y, Carpenter R, Dickinson HG, Coen ES. Origin of allelic diversity in Antirrhinum S locus RNases. Plant Cell. 1996;8:805–814. doi: 10.1105/tpc.8.5.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de MF Suassuna T, Bruckner C, de Carvalho C, Borém A. Self-incompatibility in passionfruit: evidence of gametophytic-sporophytic control. Theor Appl Genet. 2003;106:298–302. doi: 10.1007/s00122-002-1103-1. [DOI] [PubMed] [Google Scholar]
- 59.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nature biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19:651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 61.Conesa A, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 62.Mortazavi A, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 63.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995;57:289–300. [Google Scholar]
- 64.Livaka KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]