

Abstract
Adenylosuccinate synthetase catalyzes the first committed step in the de novo biosynthesis of AMP, coupling L-aspartate and IMP to form adenylosuccinate. Km values of IMP and 2′-deoxy-IMP are nearly identical with each substrate supporting comparable maximal velocities. Nonetheless, the Km value for L-aspartate and the Ki value for hadacidin (a competitive inhibitor with respect to L-aspartate) are 29- to 57-fold lower in the presence of IMP than 2′-deoxy-IMP. Crystal structures of the synthetase ligated with hadacidin, GDP and either 6-phosphoryl-IMP or 2′-deoxy-6-phosphoryl-IMP are identical except for the presence of a cavity normally occupied by the 2′-hydroxyl group of IMP. In the presence of 6-phosphoryl-IMP and GDP (hadacidin absent), the L-aspartate pocket can retain its fully ligated conformation, forming hydrogen bonds between the 2′-hydroxyl group of IMP and sequence-invariant residues. In the presence of 2′-deoxy-6-phosphoryl-IMP and GDP, however, the L-aspartate pocket is poorly ordered. The absence of the 2′-hydroxyl group of the deoxy-ribonucleotide may destabilize ligand binding to the L-aspartate pocket by disrupting hydrogen bonds that maintain a favorable protein conformation and by the introduction of a cavity in the fully ligated active site. At an approximate energy cost of 2.2 kcal/mol, the unfavorable thermodynamics of cavity formation may be the major factor in destabilizing ligands at the L-aspartate pocket.
Graphical Abstract
Adenylosuccinate synthetase (IMP: L-aspartate ligase (GDP-forming); EC 6.3.4.4), converts IMP and L-aspartate to adenylosuccinate, using GTP as an energy source. The synthetase participates in de novo purine nucleotide biosynthesis, the purine nucleotide cycle, and salvage pathways for nucleotides (1–3). Evidence supports a two-step reaction mechanism for adenylosuccinate synthetases. First, the γ-phosphoryl group of GTP is transferred to atom O-6 of IMP, forming 6-phosphoryl-IMP (6-PIMP1). Second, the α-amino group of L-aspartate displaces the 6-phosphoryl group of 6-PIMP to form adenylosuccinate (1–7). The enzyme adopts a random sequential kinetic mechanism with a strong bias in favor of the association of L-aspartate after the formation of an enzyme•IMP•GTP complex (8, 9).
Bacterial systems have but one form of adenylosuccinate synthetase, but vertebrates have two forms of the enzyme: a basic isozyme, found exclusively in muscle, is a component of the purine nucleotide cycle and the de novo pathway for the biosynthesis of AMP, and an acidic isozyme, which functions exclusively in de novo AMP biosynthesis (10–15). The two isoforms in mouse are 75% identical in their amino acid sequences, and 40% identical to the enzyme from E. coli. AMP is a weak inhibitor of the mouse basic isozyme, but a potent inhibitor of the mouse acidic isozyme and the E. coli enzyme (15). On the other hand, in the presence of saturating levels of IMP, fructose 1,6-bisphosphate is a potent inhibitor of the basic (muscle) isozyme, but only a weak inhibitor of the acidic isozyme (15). Hence, in heavily exercised muscle, elevated levels of fructose 1,6-bisphosphate could inhibit the basic isozyme, but in other tissues and in bacteria, AMP is the likely regulator of the synthetase.
The de novo synthesis of purine nucleotides in general, and adenylosuccinate synthetase specifically, is the target of numerous natural products that inhibit growth. Hydantocidin, a proherbicide activated specifically in plants, binds only to the active site of adenylosuccinate synthetase (3,16–18). Hadacidin (N-formyl-N-hydroxyglycine) is an analog of L-aspartate, but inhibits only adenylosuccinate synthetase (1–3,19,20). Many cancers (30% of all T-cell acute lymphocytic leukemia, for instance) lack a salvage pathway for adenine nucleotides and rely entirely on de novo biosynthesis of purine nucleotides (21). Inhibition of adenylosuccinate synthetase in such tumors would directly decrease the pool of adenine nucleotides, whereas normal cells with intact salvage mechanisms would be impacted to a lesser extent. Allopurinol, an analogue of hypoxanthine, significantly accelerates recovery from complicated malarial infections (22). Adenylosuccinate synthetase from Plasmodia falciparum, the organism responsible for an estimated 300–500 million annual cases of malaria (23,24), assimilates allopurinol ribonucleotide into pathways that ultimately poison ribosomal translation (25,26). A clear understanding of substrate recognition in adenylosuccinate synthetase could facilitate the development of tight-binding inhibitors and substrate analogs that impair purine biosynthesis and cell growth.
In the case of inhibition by hydantocidin and hadacidin, the inhibitor molecule combines with other ligands to fill the active site of the synthetase. Hydantocidn is phosphosphorylated on its 5′-hydroxyl group to make an analogue of IMP that binds synergistically with Pi and GDP (16–18). Hadacidin coordinates the active-site Mg2+ with its N-formyl group and packs against the base moiety of IMP (5–7,27). The structural feature common to hadacidin and hydantocidin complexes of adenylosuccinate synthetase is a set of ligands that interact well with the protein and with each other. Interactions between ligands and protein and between ligands determine the stability of the complex.
The exacting fit of substrates and substrate analogs in inhibited complexes of adenylosuccinate synthetase then, was difficult to reconcile with a report claiming 2′-deoxy-IMP as an effective substrate of adenylosuccinate synthetase (28). The 2′-hydroxyl group of IMP interacts with the side chain of a sequence-conserved arginine and with the backbone carbonyl of a sequence-conserved valine. How could the loss of this hydroxyl group have little or no effect on the stability of the substrate complex?
Here we report a role for the 2′-hydroxyl group of IMP in the recognition of L-aspartate. Indeed, as indicated by the earlier study (28), recombinant synthetases from E. coli and mouse muscle have similar Km values for 2′-deoxy-IMP and IMP. Both enzymes, however, exhibit 29- to 57-fold increases in their Km values for L-aspartate (KmL-aspartate) and Ki values for hadacidin (Kihadacidin) when 2′-deoxy-IMP replaces IMP. Crystal structures of the E. coli synthetase, having bound GDP, hadacidin and either 6-PIMP or 2′-deoxy-6-PIMP, reveal no conformational differences within experimental uncertainty; the absence of the 2′-hydroxyl group introduces only a cavity. On the other hand, crystalline complexes of the mouse muscle synthetase without hadacidin, but having bound GDP and either 6-PIMP or 2′-deoxy-6-PIMP, differ significantly. Disorder in the L-aspartate pocket is correlated with the loss of hydrogen bonds normally associated with the 2′-hydroxyl group of IMP. Although disorder in the L-aspartate pocket may contribute to increases in Kihadacidin and KmL-aspartate, cavity formation in the fully ligated enzyme by itself can account for the observed increases in kinetic parameters. The packing efficiency of ligands in the active site of a multisubstrate enzyme can be a significant determinant in substrate recognition and an important consideration in screening for tight-binding inhibitors.
EXPERIMENTAL
Materials
All reagents, including GTP, IMP, L-aspartate, bovine serum albumin, and DEAE-Sepharose were from Sigma unless noted otherwise. Hadacidin was a generous gift from F. Rudolph and B. Cooper (Dept. of Biochemistry and Cell Biology, Rice University, Houston, TX). Plasmids for the expression of enzyme are those employed in previous investigations (15, 29).
Expression and Purification of Mouse Muscle and E. coli Adenylosuccinate Synthetase
The recombinant E. coli and mouse basic isozyme were prepared as described previously (29, 30). Enzyme purity was assayed by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Investigations here employed the mouse muscle isozyme with its N-terminal polyhistidyl tag in place. The presence of the polyhistidyl tag has no effect on the kinetics of the mouse basic isozyme (30); however, only the tagged form of protein has been crystallized (27, 30, 31). Recombinant E. coli adenylosuccinate synthetase used here does not have an affinity tag (29).
Enzyme Assays
Protein concentrations were determined by the method of Bradford (32), using bovine serum albumin as a standard. Enzyme activity was determined by the change in absorbance at 290 nm and at 22 °C as described previously (8). Using up to 1 μg/mL enzyme (22 nM in subunit concentration), the reaction was linear for at least 1 min. Km and Vmax values for each substrate were obtained by holding the other two substrates at saturating levels (100 μM GTP, 300 μM IMP or 2′-deoxy-IMP, and 2 mM L-aspartate with IMP or 200 mM L-aspartate with 2′-deoxy-IMP) and varying the concentration of the third substrate. In determining Km values, initial concentrations of GTP varied systematically from 5–100 μM, those of IMP (or 2′-deoxy-IMP) from 25–300 μM, and those of L-aspartate from 0.1–2 mM (for IMP) or 1–96 mM (for 2′-deoxy-IMP). In determining the Ki values of hadacidin, concentrations of GTP and IMP were maintained at saturating levels (100 and 300 μM, respectively) as initial concentrations of L-aspartate and hadacidin varied from 0.125–2 mM and 0.25–16 μM, respectively. Ki values of hadacidin in the presence of 2′-deoxy-IMP employed fixed initial concentrations of GTP and 2′-deoxy-IMP of 100 and 300 μM, respectively, as initial concentrations of L-aspartate and hadacidin varied from 4–64 mM and 10–160 μM, respectively. For the E. coli enzyme, the assay buffer contained 20 mM Hepes, pH 7.7, and 6 mM magnesium chloride, and for the mouse enzyme 20 mM Hepes, pH 7.2, and 8 mM magnesium acetate. All data were analyzed with the computer program GraFit (33). Models for competitive, noncompetitive, uncompetitive and mixed inhibition were fit to hadacidin inhibition data.
Crystallization
Crystals were grown by the method of hanging drops. The mouse muscle enzyme crystallized from equal parts of a protein solution (10 mg/ml protein in 50 mM Hepes, pH 7.5, 50 mM NaCl, 1 mM dithiothreitol (DTT), 0.5 mM EDTA, 10 mM GTP and 10 mM 2′-deoxy-IMP) and a precipitant solution (100 mM Hepes, pH 7.0, 200 mM magnesium acetate, 12–14% (w/v) polyethylene glycol 8000 or 12–17% (w/v) polyethylene glycol 3350). Crystals of the E. coli enzyme grew from equal parts of a protein solution (10 mg/ml protein in 50 mM Hepes, pH 7.0, 50 mM NaCl, 1 mM dithiothreitol (DTT), 0.5 mM EDTA, 5 mM GTP, 5 mM 2′-deoxy-IMP, 2 mM hadacidin, and 10 mM magnesium acetate) and a precipitant solution (100 mM Hepes, pH 7.0, 200 mM magnesium acetate, 12–16% (w/v) polyethylene glycol 8000). In all experiments, droplet volumes were 6 μL, and wells contained 500 μL of the precipitant solution. Equal dimensional prisms of 100 microns appeared within 3 days at 22 °C. Crystals of the E. coli and mouse muscle enzymes were transferred to a solution of 50 mM Hepes, pH 7.0, 100 mM magnesium acetate, 27% (w/v) polyethylene glycol 8000, and 21% (v/v) glycerol and then frozen in liquid nitrogen.
Data Collection, Model Building, and Refinement
Data for the mouse muscle complex was collected at Beamline 9-2 of the Stanford Synchrotron Radiation Laboratory, using a ADSC Quantum 4 CCD detector. The wavelength of radiation was 0.979 Å, and the temperature of data collection was 120 K. Data reduction employed Denzo/Scalepack (34). Data for the E. coli complex were collected at Iowa State University on a Rigaku R-AXIS IV++ image plate detector using an Osmic confocal mirror system (CuKa radiation) and a sample temperature of 100 K. Data reduction employed the software package CrystalClear (35). Intensities were converted to structure factors using the CCP4 program TRUNCATE (36). Structures were solved by molecular replacement, using the program AmoRe (37) and models for the fully ligated mouse muscle enzyme (Protein Data Bank identifier 1LON, (27)) and E. coli enzyme (Protein Data Bank identifier 1CGO, (7)). Model building and refinement employed the programs XTALVIEW (38) and CNS (39), respectively. Force constants and parameters of stereochemistry came from Engh and Huber (40). Criteria for the addition of water molecules were identical to those of previous studies (5–7). Estimates of coordinate error used the method of Luzzati (41). Evaluation of stereochemistry of the refined model employed PROCHECK (42). Superposition of structures employed software from the CCP4 package (43) or XTALVIEW (38).
Estimates of Cavitation Energies
The contribution to the total potential energy of the 2′-hydroxyl group of 6-PIMP in the fully ligated complexes of the E. coli and mouse muscle synthetases was calculated by using AutoDock 3.0 (44). Van der Waals’ interactions were determined between the 2′-O atom and atoms within a sphere of radius 10 Å. The calculation sums pair-wise interaction energies between the 2′-O atom of 6-PIMP and the approximately 200 carbon, nitrogen, and oxygen atoms within a limiting distance of 10 Å from the 2′-O atom. The interaction energies are based on a Lennard-Jones 6–12 function, using parameters in AutoDock 3.0. Parameters are not available for interactions involving Mg2+; however, as Mg2+ is approximately 9.8 Å from the 2′-hydroxyl group of 6-PIMP in synthetase structures, the contribution of a single Mg2+-O contact to the total energy is negligible.
RESULTS
Enzyme Purity and Kinetics
Proteins used in assays and crystallization were at least 95% pure on the basis of sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). 2′-Deoxy-IMP is a substrate of the synthetase (28), but prior to the present study no information was available regarding the kinetics of an adenylosuccinate synthetase reaction supported by 2′-deoxy-IMP. Km values for 2′-deoxy-IMP and IMP (Table 1) for both the E. coli and mouse muscle enzyme are equal within experimental uncertainty. All other parameters are equivalent for the two reactions except those associated with L-aspartate and hadacidin. KmL-aspartate increases 57- and 29-fold, and Kihadacidin increases 35- and 44-fold, for E. coli and mouse muscle enzymes, respectively, in reactions that use 2′-deoxy-IMP instead of IMP. Evidently, the E. coli and mouse muscle adenylosuccinate synthetases, broadly representative of microbial and vertebrate systems, have a mechanism to distinguish ribonucleotide and 2′-deoxy-ribonuceleotide substrates. That mechanism is indirect; the recognition of the nucleotide itself is equally robust, but in some way the 2′-hydroxyl group of the ribonucleotide is an essential determinant in the recognition of L-aspartate and for high-affinity binding of hadacidin.
Table 1.
Kinetic parameters for recombinant adenylosuccinate synthetases from E. coli and mouse muscle using IMP and 2′-deoxy-IMPa.
| Enzyme | E. coli | Mouse muscle |
|---|---|---|
| IMP reaction: | ||
| kcat (sec−1) | 1.00±0.05 | 5.4±0.4b |
| KmIMP (μM) | 28±1 | 45±7b |
| KmGTP (μM) | 26±2 | 12±2b |
| KmL-aspartate (μM) | 230±40 | 140±20b |
| Kihadacidin (μM) | 0.49±0.08 | 0.32±0.05 |
| 2′-Deoxy-IMP reaction: | ||
| kcat (sec−1) | 0.96±0.05 | 5.5±0.2 |
| Km2′-deoxy-IMP (μM) | 41±4 | 58±8 |
| KmGTP (μM) | 21±1 | 15±2 |
| KmL-aspartate (mM) | 13±5 | 4±0.2 |
| Kihadacidin (μM) | 17±3 | 14±3 |
Conditions of assay are in the experimental section.
From reference (23).
Existing Crystal Structures of Ribonucleotide Complexes of E. coli and Mouse Muscle Synthetases
Crystal structures of synthetases from E. coli and mouse muscle exist in ligand-free and fully ligated complexes (7, 27, 30). For the latter, GDP, 6-PIMP, hadacidin (as an analog of L-aspartate) and Mg2+ are in the active site. In addition, a structure is available for the 6-PIMP•GDP complex for the mouse muscle enzyme (27) and for the Arg303→Leu E. coli enzyme, which has low affinity for hadacidin and a high Km for L-aspartate (7, 45). In fully ligated complexes, the 2′-hydroxyl group of IMP hydrogen bonds with a backbone carbonyl group of a peptide link between sequence invariant residues (Val273 and Gly274 in the E. coli synthetase and Val305 and Gly306 in the mouse muscle enzyme). The 2′-hydroxyl group also hydrogen bonds with an arginyl side chain (Arg303 and Arg335 in the E. coli and mouse muscle enzymes, respectively). Arg303 (and probably the corresponding position in the mouse muscle isozyme Arg335) is essential for the recognition of L-aspartate, as its mutation to leucine causes a 200-fold increase in the Km for L-aspartate (45). Hadacidin makes only nonbonded contacts with 6-PIMP in fully ligated complexes.
Fully Ligated 2′-Deoxy-ribonucleotide Complex of the E. coli Synthetase (Protein Data Bank identifier 2GCQ)
Statistics of data collection and refinement for crystals of the E. coli synthetase grown in 2′-deoxy-IMP, GTP, hadacidin and Mg2+ are in Table 2. The active site contains a single Mg2+ to which are coordinated 2′-deoxy-6-PIMP, GDP, and hadacidin (Figure 1). Electron density associated with 2′-deoxy-6-PIMP is strong, but no observable density is associated with what would be the position of a 2′-hydroxyl group (Figure 2). Interactions between active site ligands and the protein are identical to those of the 6-PIMP, GDP, hadacidin and Mg2+ complex (PDB identifier 1CGO) (Figure 3). Specifically, as the focus here is on the binding of hadacidin, the oxygen atom of its N-formyl group coordinates Mg2+, its N-hydroxyl group hydrogen bonds with Asp13, and its β-carboxyl group hydrogen bonds with Thr301, Arg303, and the backbone amide groups of Thr300 and Thr301 (Figure 4). The aforementioned distances in the 6-PIMP and 2′-deoxy-6-PIMP complexes exhibit a root-mean-squared deviation of 0.22 Å, well within the estimated uncertainty in coordinates of 0.25 Å, and no distance between corresponding atoms in the two complexes exceeds 0.4 Å. In fact, the principal difference in the fully ligated E. coli structures is the void left by the absence of the 2′-hydroxyl group in the 2′-deoxy-IMP complex.
Table 2.
Statistics of data collection and refinement.
| Complex | E. coli: GDP•2′-deoxy-6-PIMP•hadacidin | Mouse muscle: GDP•2′-deoxy-6-PIMP |
|---|---|---|
| Space group | P3221 | P43212 |
| Unit cell parameters: | ||
| a (Å) | 80.59 | 70.11 |
| b (Å) | 80.59 | 70.11 |
| c (Å) | 158.5 | 199.0 |
| Resolution limits (Å) | 25–2.0 | 16–2.4 |
| No. of reflections collected | 285,758 | 244,020 |
| No. of unique reflections | 39,551 | 20,371 |
| Completeness of data (%), overall/last shella | 96.4/78.5 | 98.9/98.9 |
| Rmergeb, overall/last shella | 0.055/0.265 | 0.100/0.415 |
| No. of refls. In refinementc | 39,498 | 19,121 |
| No. of atoms | 3,384 | 3,408 |
| No. of solvent sites | 169 | 128 |
| R/Rfreed | 0.224/0.251 | 0.198/0.262 |
| Mean B values (Å2): | ||
| Protein | 38 | 35 |
| Ligands | 36 | 27 |
| Root mean square deviations: | ||
| Bond lengths(Å) | 0.006 | 0.006 |
| Bond angles(°) | 1.3 | 1.2 |
| Dihedral angles(°) | 23 | 23 |
| Improper dihedral angles(°) | 0.83 | 0.85 |
Resolution range for the last shell is 2.0–2.07 Å for the E. coli complex and 2.4–2.5 Å for mouse muscle complex.
Rmerge=ΣjΣi|Iij−<Ij>|/ΣiΣjIij, where i runs over multiple observations of the same intensity and j runs over crystallographically unique intensities.
All data for which |Fobs|>0.
R-factor=Σ||Fobs|−|Fcalc||/Σ|Fobs|, |Fobs|>0.
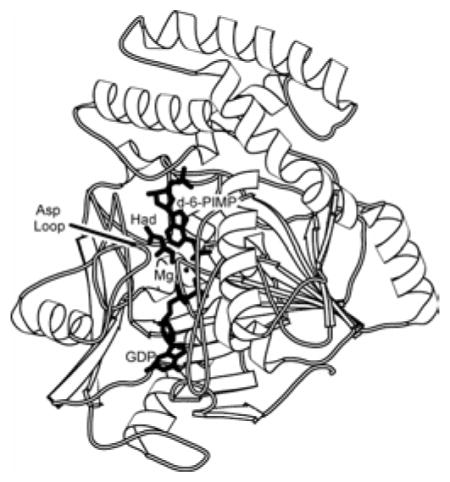
Figure 1. Overview of the 2′-deoxy-6-PIMP•GDP•hadacidin•Mg2+ complex.

Shown is a single subunit of the E. coli dimer with bound ligands defining the viewing orientation of Figures 2–6 (top). The labels Had and d-6-PIMP represent hadacidin and 2′-deoxy-6-PIMP, respectively. Structures of L-aspartate and hadacidin are compared (bottom). This figure was drawn with MOLSCRIPT (57).
Figure 2. Stereoview of 2′-deoxy-6-PIMP with electron density in the fully ligated complex of the E. coli synthetase.

The electron density is from an omit map (in which atoms for 2′-deoxy-6-PIMP have been omitted in the calculation of phase angles) with a contour level of 1.5-σ and a cutoff radius of 1.5 Å. The label d-6-PIMP represents 2′-deoxy-6-PIMP.
Figure 3. Stereoview of superpositions of fully ligated complexes of E. coli adenylosuccinate synthetase.

The complex of 6-PIMP, GDP, hadacidin and Mg2+ (white with thin lines) nearly overlays that of 2′-deoxy-6-PIMP, GDP, hadacidin and Mg2+ (black with bold lines). Labels Had and d-6-PIMP represent hadacidin and 2′-deoxy-6-PIMP, respectively. Superpositions are based on Cα alignments of Figure 2 of reference (30). This figure was drawn with MOLSCRIPT (57).
Figure 4. Schematic of the aspartyl pocket of mouse muscle adenylosuccinate synthetase.

Distances (in Å) in the 2′-deoxy-6-PIMP•GDP•hadacidin (fully ligated) complex and the 2′-deoxy-6-PIMP•GDP (partially ligated) complex are in the left and right panels, respectively. Distances from Thr300 OG1 to carbonyl 38 and amide 40 (corresponding to distances from Thr332 OG1 to carbonyl 68 and amide 70 in the mouse enzyme) provide a measure of the movement of the Asp Loop (See Figure 1 for definition) relative to the active site.
Partially Ligated 2′-Deoxy-ribonucleotide Complex of the Mouse Muscle Synthetase (Protein Data Bank identifier 2DGN)
Statistics of data collection and refinement appear in Table 2 for the mouse basic enzyme crystallized with 2′-deoxy-IMP, GTP and Mg2+. The active site contains a single Mg2+ that coordinates 2′-deoxy-6-PIMP and GDP. Electron density associated with the ligands is strong, but no density is present at what would be the position of the 2′-hydroxyl group.
Unlike the fully ligated structures of the E. coli and mouse muscle enzymes, the partially ligated complexes of the mouse basic isozyme exhibit significant conformational differences in their L-aspartate binding pockets (Figure 5). The mouse muscle enzyme with 6-PIMP, GDP and Mg2+ (Protein Database Base identifier 1LNY) has a dimer in the asymmetric unit. Backbone carbonyl groups of residues 332–334 of chain B hydrogen bond in a lattice contact that stabilizes the aspartate binding loop (residues 330–336) some 5 Å away from its position in fully ligated structures of the mouse muscle and E. coli enzymes (7, 27). Distances from Thr332 OG1 to Asn68 O and Gly70 N are 9.46 and 9.95 Å, respectively, about 6.2 Å longer than corresponding distances in the fully ligated mouse muscle enzyme. Arg335 (equivalent to Arg303 in the E. coli synthetase) and the backbone carbonyl group of Val305 (corresponding to Val273 of the E. coli enzyme) do not hydrogen bond with the 2′-hydroxyl group of the ribonucleotide in chain B. The aspartate loop of chain A participates in a different set of lattice contacts involving backbone carbonyl groups of residues 333 and 334, as well as a side chain stacking interaction involving Trp329. Nonetheless, the conformation of the aspartate loop in chain A is similar to that of the fully ligated structure. Specifically, Arg335 and the backbone carbonyl of Val305 hydrogen bond with the 2′-hydroxyl group of IMP, as observed in fully ligated systems. Distances in chain A from Thr332 OG1 to Asn68 O and Gly70 N are 6.81 and 6.77 Å, respectively, about 3.5 Å longer than those in the fully ligated mouse muscle enzyme.
Figure 5. Stereoview of superpositions of partially ligated complexes of mouse muscle adenylosuccinate synthetase.

Complexes of 6-PIMP, GDP and Mg2+ (Chain A, white with thin lines, and Chain B, gray with medium lines) are superimposed on the complex of 2′-deoxy-6-PIMP (black with bold lines). Label d-6-PIMP represents 2′-deoxy-6-PIMP. Superpositions are based on Cα alignments of Figure 2 of reference (30). This figure was drawn with MOLSCRIPT (57).
In contrast to the 6-PIMP•GDP•Mg2+ complex, the asymmetric unit of the 2′-deoxy-6-PIMP•GDP•Mg2+ mouse muscle complex has but a single subunit. The enzyme is still a dimer, however, its axis of molecular symmetry coincides with a crystallographic axis of twofold symmetry. The 6-PIMP•GDP•Mg2+ and 2′-deoxy-6-PIMP•GDP•Mg2+ complexes have nearly equivalent structures (root mean square deviation for superimposed Cα carbons of 0.4 Å) except for their aspartate loops. Interactions involving Arg335 and the backbone carbonyl of Val305 are absent as expected. Moreover, the aspartate loop is withdrawn from the active site: distances from Thr332 OG1 to Asn68 O and Gly70 N are 9.46 and 9.85 Å, respectively (Figure 4). The conformation of the aspartate loop in the 2′-deoxy-6-PIMP•GDP•Mg2+ is much like that of the aspartate loop in chain B of the 6-PIMP•GDP•Mg2+ structure, but the outwardly displaced loop cannot be attributed to a lattice contact.
Energy differences in 6-PIMP and 2′-deoxy-6-PIMP complexes
The crystal structures of the fully ligated complexes 6-PIMP and 2′-deoxy-6-PIMP of the E. coli synthetase are identical to within experimental error. Hence, the energy of interaction involving the 2′-hydroxyl group in the fully ligated 6-PIMP complex is an estimate of the energy penalty due to the absence of that group. As fully ligated 6-PIMP complexes are available for the E. coli and mouse muscle synthetases (PDB identifiers 1CG0 and 1LON, respectively), the interaction energies can be calculated for both systems. The interaction energies of the 2′-hydroxyl groups in the E. coli and mouse muscle systems from AutoDock 3.0 (Table 3) are nearly identical, in harmony with the nearly identical sequences and structures of the two complexes in the region of consideration (27).
Table 3.
Interaction energies involving the 2′-hydroxyl group of 6-PIMP in fully ligated complexes of E. coli and mouse muscle adenylosuccinate synthetase.
| Interacting atoms | Number of interactions | Energy (kcal/mol) |
|---|---|---|
| E. coli: | ||
| O2′-C | 107 | −1.487 |
| O2′-N | 45 | −0.371 |
| O2′-O | 50 | −0.481 |
| Total: | 202 | −2.339 |
| Mouse muscle: | ||
| O2′-C | 108 | −1.247 |
| O2′-N | 47 | −0.481 |
| O2′-O | 44 | −0.463 |
| Total | 199 | −2.190 |
DISCUSSION
The kinetic mechanism for the E. coli enzyme is rapid-equilibrium random (8, 9). Hence, KmL-aspartate is a reasonable approximation of the equilibrium constant governing the dissociation of L-aspartate from the enzyme•IMP•GTP•L-aspartate•Mg2+ complex. Moreover, changes in Kihadacidin are consistent with those of KmL-aspartate, further evidence of rapid equilibrium kinetics. L-Aspartate and hadacidin then dissociate more readily from the 2′-deoxy-IMP complex than from the IMP complex, and this is true to the same extent for the E. coli and mouse muscle enzymes.
A factor in the relatively weak association of L-aspartate and hadacidin in the presence of 2′-deoxy-IMP is the loss of hydrogen bonds involving the side chain of a conserved arginine (positions 303 and 335 in E. coli and mouse muscle enzymes, respectively) and the backbone carbonyl group of a conserved valine (positions 273 and 305 in E. coli and mouse muscle enzymes, respectively). Complexes of 6-PIMP, GDP and Mg2+ of the mouse muscle enzyme (Protein Data Bank identifier 1LNY, space group C2) indicate a predisposition to form hydrogen bonds involving the 2′-hydroxyl group of 6-PIMP even in the absence of hadacidin. When these hydrogen bonds are intact (as in chain A of 1LNY), the L-aspartate loop assumes a conformation much like that of the fully ligated complexes of the synthetase. Lattice contacts observed in the mouse muscle crystal structure (1LNY), however, introduce an element of uncertainty. We do not know whether hydrogen bonds involving the 2′-hydroxyl of 6-PIMP form in solution in the absence of L-aspartate. If such hydrogen bonds do form, they have no influence on the Km of IMP, and hence the free energy from such interactions must go toward stabilizing a subset of conformations favorable to the binding of L-aspartate.
A more intriguing contribution to the elevated kinetic parameters for L-aspartate and hadacidin is the cavity that replaces the 2′-hydroxyl group of the fully ligated 2′-deoxy-ribonucleotide complex. Water does not occupy the site left vacant by the absent 2′-hydroxyl group, and the protein structure does not relax to fill the void. Hence, the cost of cavity formation to a first approximation is the loss of all interactions involving the 2′-hyroxyl group in the fully ligated complex. Assuming the entropy is not changed significantly in a thermodynamic transition from a 6-PIMP to a 2′-deoxy-6-PIMP complex, the calculated difference in potential energy is a fair approximation of the free energy differences in the ribonucleotide and deoxy-ribonucleotide complexes. The energy difference ranges from 2.2 to 2.3 kcal/mol, which would increase dissociation constants for hadacidin by 41- to 49-fold. The calculated increases are close to those of observed kinetic parameters, and suggest that cavity formation is the primary cause of the observed behavior.
Curiously, the absence of the 2′-hydoxyl group only influences kinetic parameters of hadacidin and L-aspartate. One would expect an increase in the Km of 2′-deoxy-IMP relative to IMP, as the loss in hydrogen bonding potential and nonbonded interactions are due to the absence of a functional group covalently attached to the nucleotide. An explanation of this phenomenon may rest with the kinetic mechanism of the synthetase. Although that mechanism is random, isotope exchange kinetics indicate that in 90% of synthetase turnovers L-aspartate is the last substrate to bind to the enzyme (9). Evidently, when IMP binds to the active site, its base encroaches upon the L-aspartate pocket (27). Phosphorylation at the 6-position of IMP introduces numerous interactions that “bend” the purine base away from the L-aspartate pocket (31). Hence, the formation of 6-PIMP removes steric barriers to the productive binding of L-aspartate. As the cavity due to the absent 2′-hydroxyl group does not exist until L-aspartate binds (and for the vast majority of turnovers, binds last of the three substrates), the negative impact of cavity formation is felt entirely by the amino acid substrate.
The unfavorable free energy of cavity formation may be a factor in the weak binding of small amino acids (glycine, alanine and serine) to the synthetase2 and may account for the relatively weak inhibition of adenylosuccinate synthetases by succinate. Succinate has the carboxyl groups of L-aspartate, but lacks the steric encumbrance of an α-amino group, and yet KmL-aspartate is 4-fold lower than the Kisuccinate for the E. coli enzyme (46). If, however, the succinate complex of the synthetase leaves a void where an α-amino group should be, its Ki would be high relative to the Km of L-aspartate.
Cavity formation as a means of substrate discrimination is not unprecedented. In fact, the selection of the correct amino acid by aminoacyl-tRNA synthetases is due at least in part to the penalty in placing a hydrogen atom in a binding pocket designed for a larger functional group. L-Valine suffers approximately a 3.4 kcal/mol penalty relative to L-isoleucine in binding to the recognition pocket of isoleucyl-tRNA synthetase, and a penalty of similar magnitude occurs in the binding of L-α-aminobutyric acid to valyl-tRNA synthetase (47–49). Energy shortfalls for the association of L-alanine to cysteinyl-tRNA synthetase and L-phenylalanine to tyrosyl-tRNA synthetase are even greater (9.1 and 7.0 kcal/mol, respectively), and result in effective substrate discrimination without recourse to editing mechanisms (49, 50). The high energy penalties of tRNA synthetases, attributed to the loss of interactions of a missing carbon, oxygen or sulfur atom, infer a rigid pocket for the amino acid side chain, surrounded by densely packed protein atoms. By comparison, the energy penalty caused by 2′-deoxy-IMP (2.2–2.3 kcal/mol) is relatively modest, and clearly, one observes “empty space” in the vicinity 2′-hydroxy group even for the correct substrate (Figure 6).
Figure 6. Space filling models of the fully ligated complex of the E. coli synthetase.

Packing voids in the vicinity of the 2′-hydroxy group of 6-PIMP (left) and in its absence in the 2′-deoxy-6-PIMP complex (right) are indicated by dark outlines of the atoms determining the void. This figure was drawn with PyMOL (58).
Purine nucleoside phosphorylase should maintain low concentrations of 2′-deoxy-inosine, so that for normal mammalian cell-types, concentrations of 2′-deoxy-IMP should be low relative to IMP. The relatively high Km for L-aspartate in the presence of 2′-deoxy-IMP further assures that the de novo pathway would not be a source of 2′-deoxy-AMP. Certain melanomas, however, lack purine nucleoside phosphorylase, and in such cell types 2′-deoxy-inosine may become phosphorylated and enter the de novo purine nucleotide pathway (51). Adenylosuccinate synthetase discriminates against 2′-deoxy-IMP as a substrate, but IMP dehydrogenase reportedly does not distinguish between 2′-deoxy-IMP and IMP (52). Hence, at the very least, an increase in the ratio of 2′-deoxy-IMP to IMP should lead to an increase in levels of 2′-deoxy-GTP. An increase in 2′-deoxy-GTP (a substrate for adenylosuccinate synthetase) could offset reduced flux through adenylosuccinate synthetase due to the inhibitory effects of 2′-deoxy-IMP. Indeed, Musk et al. (51) report 25- and 11-fold increases in 2′-deoxy-GTP and 2′-deoxy-ATP, respectively, in a melanoma (MM96L cell line) deficient in purine nucleoside phosphorylase. Increased levels of 2′-deoxy-purine nucleotides in melanomas may be responsible for base misincorporation and the emergence of a mutator phenotype that causes the progressive transformation of the melanoma (51).
Other multisubstrate enzymes exhibit substrate-linked substrate recognition phenomena. Pantothenate synthetase from Mycobacterium tuberculosis must synthesize the pantoyl adenylate intermediate before it can bind β-alanaine (53,54). The phosphoryl group of the pantoyl adenylate intermediate hydrogen bonds with the amino group of β-alanine, positioning that group for its nucleophilic attack on the carbonyl carbon of the intermediate (54). The amino group of glycinamide ribonucleotide (GAR) may play a critical role in the binding of formate to the active site of glycinanide ribonucleotide transformylase (55), and evidently the affinity of HhaI DNA methyltransferase for DNA is enhanced some 900-fold in the presence of its cofactor S-adenosyl-L-methionine (56). The mutual recognition of substrates in the active site may be a general phenomenon in multisubstrate systems, and in such systems, an unchanged Km for a substrate alternative does not insure the proper recognition of other components of the assembled enzyme-substrate complex.
Cavity formation is a significant consideration in the design of tight-binding inhibitors. In the case of adenylosuccinate synthetase and perhaps for other multi-substrate systems, it may be possible to design or discover “complementary” inhibitors, molecules that by themselves inhibit weakly, but when combined with other ligands result in potent inhibition. In fact, hydantocidin owes its potent inhibition (Ki~20 nM) of adenylosuccinate synthetase to its tight packing with phosphate and GDP in the active site (18). A screen of adenylosuccinate synthetase in its complex with 2′-deoxy-6-PIMP and GDP or fructose 1,6-bisphosphate and GDP could lead to tight-binding alternatives to hadacidin.
Footnotes
This work was supported by National Institutes of Health Research Grant NS 10546. Coordinates and structures factors for structures reported here (2GCQ and 2DGN) have been deposited with the Research Collaboratory for Structural Bioinformatics, http://www.rcsb.org/pdb/.
The abbreviations used are: 6-PIMP, 6-phosphoryl-IMP; 2′-deoxy-6-PIMP, 2′-deoxy-6-phosphoryl-IMP.
Aaron ver Heul and Richard B. Honzatko, unpublished
References
- 1.Stayton MM, Rudolph FB, Fromm HJ. Regulation, genetics, and properties of adenylosuccinate synthetase: a review. Curr Top Cell Regul. 1983;22:103–141. doi: 10.1016/b978-0-12-152822-5.50008-7. [DOI] [PubMed] [Google Scholar]
- 2.Honzatko RB, Fromm HJ. Structure-function studies of adenylosuccinate synthetase from Escherichia coli. Arch Biochem Biophys. 1999;370:1–8. doi: 10.1006/abbi.1999.1383. [DOI] [PubMed] [Google Scholar]
- 3.Honzatko RB, Stayton MM, Fromm HJ. Adenylosuccinate synthetase: recent developments. Adv Enzymol Relat Areas Mol Biol. 1999;73:57–102. [PubMed] [Google Scholar]
- 4.Bass MB, Fromm HJ, Rudolph FB. The mechanism of the adenylosuccinate synthetase reaction as studied by positional isotope exchange. J Biol Chem. 1984;259:12330–12333. [PubMed] [Google Scholar]
- 5.Poland BW, Fromm HJ, Honzatko RB. Crystal structures of adenylosuccinate synthetase from Escherichia coli complexed with GDP, IMP hadacidin, NO3−, and Mg2+ J Mol Biol. 1996;264:1013–1027. doi: 10.1006/jmbi.1996.0693. [DOI] [PubMed] [Google Scholar]
- 6.Poland BW, Bruns C, Fromm HJ, Honzatko RB. Entrapment of 6-thiophosphoryl-IMP in the active site of crystalline adenylosuccinate synthetase from Escherichia coli. J Biol Chem. 1997;272:15200–15205. doi: 10.1074/jbc.272.24.15200. [DOI] [PubMed] [Google Scholar]
- 7.Choe JY, Poland BW, Fromm HJ, Honzatko RB. Mechanistic implications from crystalline complexes of wild-type and mutant adenylosuccinate synthetases from Escherichia coli. Biochemistry. 1999;38:6953–6961. doi: 10.1021/bi990159s. [DOI] [PubMed] [Google Scholar]
- 8.Rudolph FB, Fromm HJ. Initial rate studies of adenylosuccinate synthetase with product and competitive inhibitors. J Biol Chem. 1969;244:3832–3839. [PubMed] [Google Scholar]
- 9.Cooper BF, Fromm HJ, Rudolph FB. Isotope exchange at equilibrium studies with rat muscle adenylosuccinate synthetase. Biochemistry. 1986;25:7323–7327. doi: 10.1021/bi00371a013. [DOI] [PubMed] [Google Scholar]
- 10.Lowenstein JM. Ammonia production in muscle and other tissues: the purine nucleotide cycle. Physiol Rev. 1972;52:382–414. doi: 10.1152/physrev.1972.52.2.382. [DOI] [PubMed] [Google Scholar]
- 11.Goodman MV, Lowenstein JM. The purine nucleotide cycle. Studies of ammonia production by skeletal muscle in situ and in perfused preparations. J Biol Chem. 1977;252:5054–5060. [PubMed] [Google Scholar]
- 12.Matsuda Y, Ogawa H, Fukutome S, Shiraki H, Nakagawa H. Adenylosuccinate synthetase in rat liver: the existence of two types and their regulatory roles. Biochem Biophys Res Commun. 1977;78:766–771. doi: 10.1016/0006-291x(77)90245-5. [DOI] [PubMed] [Google Scholar]
- 13.Ogawa H, Shiraki H, Nakagawa H. Study on the regulatory role of fructose-1,6-diphosphate in the formation of AMP in rat skeletal muscle. A mechanism for synchronization of glycolysis and the purine nucleotide cycle. Biochem Biophys Res Commun. 1976;68:524–528. doi: 10.1016/0006-291x(76)91177-3. [DOI] [PubMed] [Google Scholar]
- 14.Guicherit OM, Rudolph FB, Kellems RE, Cooper BF. Molecular cloning and expression of a mouse muscle cDNA encoding adenylosuccinate synthetase. J Biol Chem. 1991;266:22582–22587. [PubMed] [Google Scholar]
- 15.Borza T, Iancu CV, Pike E, Honzatko RB, Fromm HJ. Variations in the response of mouse isozymes of adenylosuccinate synthetase to inhibitors of physiological relevance. J Biol Chem. 2003;278:6673–6679. doi: 10.1074/jbc.M210838200. [DOI] [PubMed] [Google Scholar]
- 16.Heim DR, Cseke C, Gerwick BC, Murdoch MG, Green SB. Hydantocidin: A possible proherbicide inhibiting purine biosynthesis at the site of adenylosuccinate synthetase. Pesticide Biochem Physiol. 1995;53:138–145. [Google Scholar]
- 17.Siehl DL, Subramanian MV, Walters EW, Lee S-F, Anderson RJ, Toschi AG. Adenylosuccinate synthetase: Site of action of hydantocidin, a microbial phytotoxin. Plant Physiol. 1996;110:753–758. doi: 10.1104/pp.110.3.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poland BW, Lee SF, Subramanian MV, Siehl DL, Anderson RJ, Fromm HJ, Honzatko RB. Refined crystal structure of adenylosuccinate synthetase from Escherichia coli complexed with hydantocidin 5′-phosphate, GDP, HPO42−, Mg2+, and hadacidin. Biochemistry. 1996;35:15753–15759. doi: 10.1021/bi961758r. [DOI] [PubMed] [Google Scholar]
- 19.Shigeura HT, Gordon CN. The mechanism of action of hadacidin. J Biol Chem. 1962;237:1937–1940. [PubMed] [Google Scholar]
- 20.Gale GR, Smith AB. Alanosine and hadacidin--comparison of effects on adenylosuccinate synthetase. Biochem Pharmacol. 1968;17:2495–2498. doi: 10.1016/0006-2952(68)90144-5. [DOI] [PubMed] [Google Scholar]
- 21.Batova A, Diccianni MB, Omura-Minamisawa M, Yu J, Carrera CJ, Bridgeman LJ, Kung FH, Pullen J, Amylon MD, Yu AL. Use of alanosine as a methylthioadenosine phosphorylase-selective therapy for T-cell acute lymphoblastic leukemia in vitro. Cancer Res. 1999;59:1492–1497. [PubMed] [Google Scholar]
- 22.Sarma PSA, Mandal AK, Khamis HJ. Allopurinol as an additive to quinine in the treatment of acute complicated falciparum malaria. Am J Trop Med Hyg. 1998;58:454–457. doi: 10.4269/ajtmh.1998.58.454. [DOI] [PubMed] [Google Scholar]
- 23.Greenwood B, Mutabingwa T. Malaria in 2002. Nature. 2002;415:670–672. doi: 10.1038/415670a. [DOI] [PubMed] [Google Scholar]
- 24.Sachs J, Malaney P. The economic and social burden of malaria. Nature. 2002;415:680–685. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 25.Spector T, Jones TE, Elion GB. Specificity of adenylosuccinate synthetase and adenylosuccinate lyase from Leishmania donovani. J Biol Chem. 1979;254:8422–8426. [PubMed] [Google Scholar]
- 26.Spector T, Berens RL, Marr JJ. Adenylosuccinate synthetase and adenylosuccinate lyase from Trypanosoma cruzi. Biochem Pharmacol. 1982;31:225–229. doi: 10.1016/0006-2952(82)90215-5. [DOI] [PubMed] [Google Scholar]
- 27.Iancu CV, Borza T, Fromm HJ, Honzatko RB. IMP, GTP, and 6-phosphoryl-IMP complexes of recombinant mouse muscle adenylosuccinate synthetase. J Biol Chem. 2002;277:26779–26787. doi: 10.1074/jbc.M203730200. [DOI] [PubMed] [Google Scholar]
- 28.Spector T, Miller RL. Mammalian adenylosuccinate synthetase. Nucleotide monophosphate substrates and inhibitors. Biochim Biophys Acta. 1976;445:509–517. doi: 10.1016/0005-2744(76)90104-2. [DOI] [PubMed] [Google Scholar]
- 29.Wang W, Gorrell A, Hou Z, Honzatko RB, Fromm HJ. Ambiguities in mapping the active site of a conformationally dynamic enzyme by directed mutation. Role of dynamics in structure-function correlations in Escherichia coli adenylosuccinate synthetase. J Biol Chem. 1998;273:16000–16004. doi: 10.1074/jbc.273.26.16000. [DOI] [PubMed] [Google Scholar]
- 30.Iancu CV, Borza T, Choe JY, Fromm HJ, Honzatko RB. Recombinant mouse muscle adenylosuccinate synthetase: overexpression, kinetics, and crystal structure. J Biol Chem. 2001;276:42146–42152. doi: 10.1074/jbc.M106294200. [DOI] [PubMed] [Google Scholar]
- 31.Iancu CV, Borza T, Fromm HJ, Honzatko RB. Feedback inhibition and product complexes of recombinant mouse-muscle adenylosuccinate synthetase. J Biol Chem. 2002;277:40536–40543. doi: 10.1074/jbc.M204952200. [DOI] [PubMed] [Google Scholar]
- 32.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 33.Leatherbarrow RJ. GraFit, Version 5. Erithacus Software Ltd; Horley, UK: 2001. [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray. diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Hendrixson T. CrystalClear Version 1.3.5SP2. Rigaku/MSC, Inc; The Woodlands, TX: 2002. [Google Scholar]
- 36.French GS, Wilson KS. On the treatment of negative intensity observations. Acta Crystallogr Sect A. 1978;34:517–525. [Google Scholar]
- 37.Navazza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr Sect A. 1994;50:157–163. [Google Scholar]
- 38.McRee DE. J Mol Graphics. 1992;10:44–46. [Google Scholar]
- 39.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 40.Engh RA, Huber R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr Sect A. 1991;47:392–400. [Google Scholar]
- 41.Luzzati V. Acta Crystallogr. 1956;5:802–810. [Google Scholar]
- 42.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 43.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr Sect D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 44.Goodsell DS, Olson AJ. Automated Docking of Substrates to Proteins by Simulated Annealing. Proteins: Str Func Genet. 1990;8:195–202. doi: 10.1002/prot.340080302. [DOI] [PubMed] [Google Scholar]
- 45.Wang W, Poland BW, Honzatko RB, Fromm HJ. Identification of arginine residues in the putative L-aspartate binding site of Escherichia coli adenylosuccinate synthetase. J Biol Chem. 1995;270:13160–13163. doi: 10.1074/jbc.270.22.13160. [DOI] [PubMed] [Google Scholar]
- 46.Gorrell A, Wang W, Underbakke E, Hou Z, Honzatko RB, Fromm HJ. Determinants of L-aspartate and IMP Recognition in Escherichia coli Adenylosuccinate Synthetase. J Biol Chem. 2002;277:8817–8821. doi: 10.1074/jbc.M111810200. [DOI] [PubMed] [Google Scholar]
- 47.Fersht AR. Editing mechanisms in protein synthesis. Rejection of valine by isoleucyl-tRNA synthetase. Biochemistry. 1977;16:1025–1030. doi: 10.1021/bi00624a034. [DOI] [PubMed] [Google Scholar]
- 48.Fersht AR, Dingwall C. Establishing the misacylation/deacylation of the tRNA pathway for the editing mechanism of prokaryotic and eukaryotic valyl-tRNA synthases. Biochemistry. 1977;18:1238–1245. doi: 10.1021/bi00574a019. [DOI] [PubMed] [Google Scholar]
- 49.Fersht AR, Shindler JS, Tsui W-C. Probing the limits of protein-amino acid side chain recognition with aminoacyl-tRNA synthetases. Discrimination against phenylalanine by tyrosyl-tRNA synthetases. Biochemistry. 1980;19:5520–5524. doi: 10.1021/bi00565a009. [DOI] [PubMed] [Google Scholar]
- 50.Fersht AR, Dingwall C. Cysteinyl-tRNA synthetase fro Escherichia coli does not need an editing mechanism to reject serine and alanine. High binding energy of small groups in specific molecular interactions. Biochemistry. 1979;18:1245–1249. doi: 10.1021/bi00574a020. [DOI] [PubMed] [Google Scholar]
- 51.Musk P, Clark JM, Thompson D, Dunn IS, Christopherson RI, Szabados E, Rose SE, Parsons PG. Purine deoxynucleoside metabolism in human melanoma cells with high spontaneous mutation rate. Mutation Res. 1996;350:229–238. doi: 10.1016/0027-5107(95)00111-5. [DOI] [PubMed] [Google Scholar]
- 52.Markham GD, Bock CL, Schalk-Hihi C. Acid-base catalysis in the chemical mechanism of inosine monophosphate dehydrogenase. Biochemistry. 1999;38:4433–4440. doi: 10.1021/bi9829579. [DOI] [PubMed] [Google Scholar]
- 53.Zheng R, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry. 2001;40:12904–12912. doi: 10.1021/bi011522+. [DOI] [PubMed] [Google Scholar]
- 54.Wang S, Eisenberg D. Crystal structure of the pantothenate synthetase from Mycobacterium tuberculosis, snapshots of the enzyme in action. Biochemistry. 2006;45:1554–1561. doi: 10.1021/bi051873e. [DOI] [PubMed] [Google Scholar]
- 55.Thoden JB, Firestine S, Nixon A, Benkovic SJ, Holden HM. Molecular structure of Escherichia coli purT-encoded glycinamide ribonucleotide transformylase. Biochemistry. 2000;39:8791–8802. doi: 10.1021/bi000926j. [DOI] [PubMed] [Google Scholar]
- 56.Lindstrom WN, Jr, Flynn J, Reich NO. Reconciling structure and function in HhaI DNA Cytosine-C-5 methyltransferase. J Biol Chem. 2000;275:4912–4919. doi: 10.1074/jbc.275.7.4912. [DOI] [PubMed] [Google Scholar]
- 57.Kraulis J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 58.DeLeno WL. The PyMOL molecular graphics system. DeLano Scientific; San Carlos, CA, USA: 2002. http://www.pymol.org. [Google Scholar]