

Abstract
The aim of this study was to evaluate four different platinated bioconjugates containing a cisplatin (cis-diamminedichloroplatinum [cis-DDP]) fragment and epidermal growth factor receptor (EGFR)-targeting moieties as potential therapeutic agents for the treatment of brain tumors using a human EGFR-expressing transfectant of the F98 rat glioma (F98EGFR) to assess their efficacy. The first two bioconjugates employed the monoclonal antibody cetuximab (C225 or Erbitux®) as the targeting moiety, and the second two used genetically engineered EGF peptides. C225-G5-Pt was produced by reacting cis-DDP with a fifth-generation polyamidoamine dendrimer (G5) and then linking it to C225 by means of two heterobifunctional reagents. The second bioconjugate (C225-PG-Pt) employed the same methodology except that polyglutamic acid was used as the carrier. The third and fourth bioconjugates used two different EGF peptides, PEP382 and PEP455, with direct coordination to the Pt center of the cis-DDP fragment. In vivo studies with C225-G5-Pt failed to demonstrate therapeutic activity following intracerebral (ic) convection-enhanced delivery (CED) to F98EGFR glioma-bearing rats. The second bioconjugate, C225-PG-Pt, failed to show in vitro cytotoxicity. Furthermore, because of its high molecular weight, we decided that lower molecular weight peptides might provide better targeting and microdistribution within the tumor. Both PEP382-Pt and PEP455-Pt bioconjugates were cytotoxic in vitro and, based on this, a pilot study was initiated using PEP455-Pt. The end point for this study was tumor size at 6 weeks following tumor cell implantation and 4 weeks following ic CED of PEP455-Pt to F98 glioma-bearing rats. Neuropathologic examination revealed that five of seven rats were either tumor-free or only had microscopic tumors at 42 days following tumor implantation compared to a mean survival time of 20.5 and 26.3 days for untreated controls. In conclusion, we have succeeded in reformatting the toxicity profile of cis-DDP and demonstrated the therapeutic efficacy of the PEP455-Pt bioconjugate in F98 glioma-bearing rats.
Keywords: cisplatin, F98EGFR rat glioma, molecular targets, peptides, monoclonal antibodies
Introduction
Malignant gliomas are fast growing, highly invasive brain tumors that usually lead to death within 12–18 months following diagnosis.1 These tumors are resistant to all current forms of therapy, including surgery, radiotherapy, and chemotherapy.2 Although the 2-year survival has increased to almost 25%, the 5-year survival of patients has remained very low2 despite intensive efforts to develop more effective therapies.3 Significant advances have been made in understanding the molecular genetic events leading to the development and progression of gliomas.4 However, this still has not resulted in therapeutic agents that could specifically target gliomas and alter their highly invasive pattern of growth within the brain.5,6 One significant problem associated with drug delivery to the brain relates to the failure of most systemically administered therapeutic agents to cross the blood–brain barrier (BBB) and attain therapeutically effective concentrations within the tumor.7 Over the past 10 years, various studies at the European Synchrotron Research Facility8–10 and at The Ohio State University11,12 have been carried out on the use of either cisplatin (cis-dichlorodiammineplatinum [cis-DDP]) or carboplatin, administered intracerebrally (ic) by means of convection-enhanced delivery (CED).13 This technique can deliver therapeutic agents directly to the site of the brain tumor, thereby completely bypassing the BBB and resulting in carboplatin concentrations up to 1,000× greater than that achieved by systemic administration.11
The epidermal growth factor receptor (EGFR) and its mutant isoform EGFRvIII are frequently overexpressed in brain tumors, which have made them attractive molecular targets for the treatment of gliomas.14–16 A number of drugs and monoclonal antibodies (MoAbs) have been developed to target EGFR and its signaling pathways.15,16 Among these is the MoAb cetuximab (or Erbitux®, Eli Lilly and Company, Indianapolis, IN, USA), which originally was designated C225. Cetuximab is a chimeric MoAb directed against EGFR and binds with high affinity to the receptor.17 It can block the binding of both EGF and transforming growth factor-α to their receptors and produces cell cycle arrest and apoptosis. Cetuximab has been effective but not curative for the treatment of EGFR-positive cancers, such as those of the colon and head and neck region, when administered alone and has synergistic effects when combined with ionizing radiation or cytoreductive chemotherapy.18–22
Platinum-containing drugs have been widely used as chemotherapeutic agents for the treatment of a variety of human cancers.23 However, their renal, gastrointestinal, myelo- and neurotoxicity, rapid binding to plasma proteins, and poor penetration of the central nervous system have limited their use for the treatment of brain tumors.24 The present study was based on our previous observations relating to the significant neurotoxicity associated with ic CED of either a liposomal formulation of cis-DDP or the free drug in non-tumor-bearing Fischer rats.25 Both of these agents had significant neurotoxic effects consisting of cerebral necrosis and hemorrhage at 4 days for the former and 7 days for the latter. On the basis of these observations, we hypothesized that if C225 was used as the delivery agent, this might specifically target EGFR-expressing F98 glioma cells (F98EGFR) and could reformat the toxicity profile of cis-DDP. As reported by us now, although there was a reduction in the neurotoxicity of C225-Pt bioconjugates compared to free cis-DDP, they were devoid of in vivo therapeutic activity. Therefore, we hypothesized that if EGFR-targeting, lower molecular weight (MW) peptides were used as the targeting moiety and their chemical linkage to cis-DDP was simplified, these steps might improve their therapeutic efficacy and “reformat” the toxicity profile of cis-DDP. The results of the studies described in this report confirm our hypothesis.
Materials and methods
Preparation of cis-DDP containing EGFR targeting C225-G5-Pt bioconjugates
The antihuman EGFR MoAb cetuximab (C225, MW 152 kDa) was generously provided to us by Dr Daniel Hicklin (formerly of ImClone Systems, Inc., New York, NY, USA). Site-specific attachment of cis-DDP to a precision macromolecule was carried out using a fifth-generation polyamidoamine (PAMAM) dendrimer, designated G5 (Sigma-Aldrich, St Louis, MO, USA), containing 128 terminal amino groups. This was reacted with cis-DDP to yield Pt-containing macromolecules. These in turn were linked to C225 by means of two heterobifunctional reagents, N-succinimidyl 3-(2-pyridyldithio)-propionate and N-(k-maleimido undecanoic acid)-hydrazide, as previously described by us for linking boron-containing G5 to C225.26 The protein content of the bioconjugate was determined spectrophotometrically by means of the Coomassie (Bio-Rad Laboratories Inc., Hercules, CA, USA) blue staining technique, and the Pt content was quantified by means of direct current plasma-atomic emission spectroscopy (DCP-AES), as previously described by us.27 The resulting bioconjugate, designated C225-G5-Pt, was used in the therapy studies described in the section on Evaluation of therapeutic efficacy of PEP-Pt bioconjugates.
An alternative conjugation scheme using another macromolecule, polyglutamic acid (PGA) as a cis-DDP carrier, has been summarized in Figure 1A. PGs with a MW range of 15,000–50,000 Da (Sigma-Aldrich; 2.5 mg, 0.083 mmol) were mixed with cis-DDP (2.5 mg, 8.3 mmol) and incubated at 70°C for 12 hours. The Pt-containing PG (PG-Pt, 0.083 mmol) was then incubated with the heterobifunctional reagent PMPI (N-[p-maleidophenyl] isocyanate), and maleimide groups (PMPI-PG-Pt) were introduced into PG by incubating 0.8 mM of PMPI for 2 hours at pH 9.0. Separately, C225 (4.0 mg, 0.027 mM) was incubated with Traut’s reagent (0.018 mg, 0.133 mM) for 1 hour at ambient temperature, following which it was mixed with PMPI-PG-Pt for 4 hours. The resulting bioconjugate, designated C225-PG-Pt, was purified by Sephacryl S-300 gel filtration column chromatography, and the Pt concentration was determined by DCP-AES.
Figure 1.

(A) Conjugation scheme using PGA as a carrier for cis-DDP. (B) Conjugation of cis-DDP to peptides.
Notes: Briefly summarized, PGA was mixed with cis-DDP and incubated at 70°C for 12 hours, followed by column chromatography. Free drug was removed by passing the conjugate through a PD-10 column and the Pt content was determined by DCP-AES. The PG-Pt then was incubated with PMPI, and maleimide groups were then introduced into PG (PMPI-PG-Pt). C225 was incubated with Traut’s reagent and then mixed with PMPI-PG-Pt for 4 hours. The resulting bioconjugate, designated C225-PG-Pt, was purified by Sephacryl gel filtration. (B) Conjugation of cis-DDP to peptides. Briefly summarized, cis-DDP was added to the peptide and kept at 37°C for 48 hours, followed by passage through a Mini G-10 column to remove unbound cis-DDP. The molar ratio between cis-DDP and peptide was ~2:1, as determined by quantitation of the Pt content of the bioconjugates by means of ICP-OES. The resulting PEP-Pt bioconjugates were water soluble and stable for at least 1 month at 4°C.
Abbreviations: DDP, dichlorodiammineplatinum; PGA, polyglutamic acid; DCP-AES, direct current plasma-atomic emission spectroscopy; PG-Pt, Pt-containing PG; PMPI, N-[p-maleidophenyl]isocyanate; ICP-OES, inductively coupled plasma-optical emission spectroscopy.
Peptide synthesis, characterization, and preparation of PEP-Pt bioconjugates
The EGFR-targeting peptides were synthesized using a 9600 Milligen/Biosearch solid-phase peptide synthesizer (EMD Millipore, Billerica, MA, USA) employing Fmoc/t-But chemistry and a clear amide resin (0.32 mmol/g) (Peptide International, Louisville, KY, USA). Following synthesis, cleavage of the peptides from the resin was carried out as previously described.28 They were then purified by reverse-phase high-pressure liquid chromatography (HPLC) in a gradient system using a C-4 vydac column in water/acetonitrile (0.1% trifluoroacetic acid) on a Waters Breeze System (Waters Corporation, Milford, MA, USA). The purified fractions were then subjected to analytical HPLC, and the fractions of interest were pooled and lyophilized in a solution containing 1% acetic acid. The final purified peptides were then identified using electrospray ionization mass spectrometry (Campus Chemical Instrumentation Center, The Ohio State University, Columbus, OH, USA). Two EGF peptides were selected, a B-cell epitope sequence 382–410, designated PEP382, and the HER-1 epitope sequence 455–470, designated PEP455. The first had an MW of 3,484 Da, and the latter, 2,005 Da, consistent with each peptide sequence and an additional CH3CN solvent molecule from the HPLC purification step. The amino acid sequence for the 30 mer PEP382 was Ac-Leu-Ile-Gln-Ala-Trp-Pro-Glu-Asn-Arg-Thr-Asp-Leu-His-Ala-Phe-Glu-Asn-Leu-Glu-Ile-Ile-Arg-Gly-Arg-Thr-Lys-Gln-His-Gly-NH2. The amino acid sequence of the 19 mer PEP455 was Ac-Lys-Leu-Phe-Gly-Thr-Ser-Gly-Gln-Lys-Thr-Lys-Ile-Ile-Ser-Asn-Arg-Gly-Glu-NH2.28 The binding profiles with EGFR-expressing breast and lung cancer cell lines, MDA-468 and A549, respectively, have been described in detail elsewhere,28 and each bound to specific regions of EGFR.
As shown in Figure 1B, the PEP-Pt bioconjugates were prepared by adding 7.5 mg cis-DDP to 1.0 mg of peptide (50:1 molar ratio) in 1.2 mL of sterile, double-distilled deionized water. The reaction mixture was kept in the dark at 37°C for 48 hours, followed by passage through PD MiniTrap G-10 columns (GE Life Sciences, Pittsburgh, PA, USA) against water to remove unbound cis-DDP. The chemical reaction itself involved the replacement of one of the chlorides of cis-DDP with an electron donor group of the peptide. The amount of Pt attached to the peptide was determined by means of inductively coupled plasma-optical emission spectroscopy, as previously described,11 and the values obtained were multiplied by 1.54 to convert Pt content to the amount of cis-DDP. The molar ratio between cis-DDP and peptide was ~2:1, assuming no loss of peptide during the reaction. The resulting PEP-Pt bioconjugates were water soluble and stable for at least 1 month at 4°C. The 2:1 molar ratio of [Pt]:[peptide] was consistent with the expected coordination sites for the soft Pt metal on the sequences of each peptide. Met, Cys, and His are the three amino acids with the largest affinity for Pt coordination,29 followed by Arg and the terminal NH2 group, as has been previously established.30 Neither peptide used by us possessed Met or Cys residues. The presence of two His amino acids in PEP382 was consistent with the selective coordination of Pt to those sites, resulting in the 2:1 [Pt]:[PEP382] ratio. Although no Met, Cys, or His residues with the highest affinity for Pt were available for metal coordination in PEP455, it has previously been shown30 that Arg and the terminal NH2 groups also exhibit selective binding. It is, therefore, postulated that cis-DDP bound to PEP455 at the single Arg residue and to the terminal NH2, thereby resulting in the 2:1 [Pt]:[PEP455] stoichiometry.
In vitro cellular uptake and cytocidal activity of Pt bioconjugates
F98 wild-type (F98WT) and F98EGFR glioma cells were used for in vitro Pt uptake studies. Dulbecco’s minimal essential medium containing 1.68 mg of C225-G5-Pt (100 μg cis-DDP) was added to T-150 flasks, and the cells were incubated for an additional 2 hours at 37°C. Following this, they were washed three times with phosphate-buffered saline (PBS), pH 7.4, and disaggregated by treatment with 0.5 mmol of ethylenediaminetetraacetic acid for 5 minutes, counted, and sedimented. They were then digested with concentrated sulfuric acid and 50% hydrogen peroxide in an oil bath at 100°C, and Pt uptake subsequently was determined by DCP-AES. On the basis of the atomic weight of Pt (195.1 Da), the concentrations of cis-DDP (300.05 Da) were calculated by multiplying the Pt values by 1.54.
The procedure used to determine the cytocidal activity of the Pt bioconjugates was modified from the method of Pauwels et al31 using the sulforhodamine-B (SRB) assay. F98EGFR or F98WT glioma cells were seeded into 96-well microplates (5,000 or 10,000 cells/well). The following day, incremental concentrations (2 nM–20 μM) of cis-DDP, C225-PGA-Pt-, or PEP-Pt bioconjugates were incubated with the F98EGFR cells for either 4 or 72 hours. Three days after the start of incubation, the cells were fixed with a cold 50% solution of trichloroacetic acid, then washed five times with distilled water, allowed to dry, and stained with 0.4% SRB. Unbound SRB was removed by washing the plates five times with 1% acetic acid. The bound SRB–protein complex was solubilized with Tris base, following which spectrophotometric determination of SRB concentrations was performed using a Dynex MRB II microplate reader (Dynex Technologies Inc., Chantilly, VA, USA) at a wavelength of 490 nm.
F98EGFR glioma model and therapy studies with C225-G5-Pt bioconjugates
The F98 rat glioma has been propagated in vitro and in vivo since 1971. It is a radioresistant tumor that is invariably fatal with an inoculum of as few as 100 cells. As has been described in detail elsewhere,32 it has been used in a wide variety of studies in experimental neurooncology. F98EGFR cells were produced by transfecting F98WT cells with the human gene encoding wild-type EGFR.26,33 The resulting cell line, designated F98EGFR, expressed ~1.60×105 receptor sites of EGFR compared to ~104 EGFR for F98WT cells.26 Furthermore, the receptor was inactive and did not activate a signal transduction cascade when exposed to EGF.34 Most importantly, this cell line was ideally suited for our studies because the number of receptors expressed was below the threshold required to evoke a xenoimmune response.35 Consequently, F98EGFR gliomas were nonimmunogenic when implanted into the brains of syngeneic Fischer rats.36 All animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, and the experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of The Ohio State University, including permission to carry out a survival study based on our previous experience with the F98 glioma model.34,35,37
For the therapy studies with the C225-G5-Pt bioconjugate, 103 F98EGFR glioma cells were implanted stereotactically (David Kopf Instruments, Tujunga, CA, USA) into the right caudate nucleus of syngeneic Fischer rats (weight ~200 g; (Charles River Laboratories International, Inc., Wilmington, MA, USA), using a procedure described in detail elsewhere.39 This procedure has allowed us to reliably and reproducibly implant tumor cells into a precisely defined anatomic region, the caudate nucleus, of the rat brain. Seven days later, the animals were randomized into three groups, namely, 1) untreated controls (four rats); 2) those that received free cis-DDP (seven rats); and 3) C225-G5-Pt bioconjugates (six rats). The test agents were administered stereotactically by means of CED40–42 to the same site that the tumor cells previously had been implanted using a procedure that has been used extensively by us.33–38 Following treatment, all animals were weighed three times per week, and their clinical status was evaluated at the same time. Once the animals had progressively growing tumors, as evidenced by sustained weight loss (20% of body weight over 3 days), they were euthanized to minimize discomfort, as stipulated in our approved IACUC protocol. Mean survival time (MST) and median survival time (MeST) and the percent increase in life span (% ILS) were calculated as previously described.38 On the basis of the lack of in vivo therapeutic efficacy of the C225-G5-Pt bioconjugates (MW >180 kDa), we decided to use the much lower MW PEP455-Pt (MW ~2.2 kDa) for further in vivo studies.
Neurotoxicologic studies of PEP-Pt bioconjugates
The neurotoxicologic effects of the EGFR-targeting peptides were evaluated in non-tumor-bearing Fischer rats that had received free cis-DDP or cis-DDP–containing peptides. The test agents included unmodified PEP382 and PEP455 (138 μM), PEP382-Pt (80 μM), PEP455-Pt (138 μM), and free cis-DDP at concentrations of 1.7 μg/10 μL (low dose) or 16.5 μg/10 μL (high dose). All of the agents were administered stereotactically by means of CED, using a kd5310 Nanoliter Syringe Pump (KD Scientific, Holliston, MA, USA) into the right caudate nucleus at a volume of 10 μL over 30 minutes at a flow rate of 0.33 μL/min. Rats were euthanized at 7 days following administration, their brains were removed, fixed in buffered formalin, cut coronally at 2 mm intervals, and then processed for neuropathologic examination. Following this, a pilot study was initiated in F98EGFR and F98WT glioma-bearing rats.
Evaluation of therapeutic efficacy of PEP-Pt bioconjugates
In contrast to our previous studies to evaluate the therapeutic efficacy of carboplatin alone or in combination with X-irradiation,11,12 where prolongation of survival time was taken as the end point, in our pilot study with PEP-Pt bioconjugates, we chose a different approach where the F98 glioma-bearing rats were euthanized at 42 days following tumor implantation.12 Two weeks following tumor cell implantation, the therapeutic efficacy of PEP455-Pt was evaluated following ic CED to a group of seven rats, four bearing F98EGFR tumors and three bearing F98WT gliomas. Four weeks later, the animals were euthanized and their brains were removed and processed for neuropathologic examination to determine if there were macro- or microscopic deposits of tumor cells. To obtain quantitative data on the relationship of brain tumor size and the time of death of untreated control rats, coronal sections of the brains of F98 glioma-bearing rats were converted to digital slide images using the ScanScope XT by Leica-Aperio (Vista, CA, USA, and scanned at 20× resolution, approximately equivalent to 200× by light microscopy. The format of the scans were of svs files, a proprietary version of JPEG2000, and these were reviewed to confirm clarity and crispness of the images. Image analysis was undertaken using Tissue Studio 3.6.1 software by Definiens (Munich, Germany). Tissue Studio uses a context-based, relational analysis of the component pixels in digital slide images to differentiate between types of tissue, in this study–tumor versus normal brain, and measure the cross-sectional areas of the tumor. An algorithm was developed using a dozen representative sections of the scanned brain tumors to guide assignment of pixels to distinguish between tumor and surrounding normal brain. Size was calculated for the tumor area from the pixel area and the magnification settings of the scanned images and a plot of tumor cross-sectional area as a function of the time of death.
Statistical methods
The statistical analyses were conducted using STATA SE13 (StataCorp LP, College Station, TX, USA). Continuous variables were presented as either mean ± standard deviation (SD) or median and range and compared using one-way analysis of variance (ANOVA). Tumor status of PEP-Pt bioconjugate-treated rats and untreated controls were compared using Fisher’s exact test due to multiple zero cells in the table. Survival times of the animals between groups, listed in Table 1, were compared using log-rank test. Correlation between tumor cross-sectional area and survival time of the tumor-bearing rats was computed. A P-value ≤0.05 was considered statistically significant.
Table 1.
Survival times of F98EGFR glioma-bearing rats following CED of either cisplatin or C225-G5-Pt
| Groupa | Nb | Survival times (days)c
|
Range | % increased life spand
|
||
|---|---|---|---|---|---|---|
| Mean ± SE | Median | Mean | Median | |||
| CED of C225-G5-Pt | 6 | 21.7±6.6 | 19 | 18–35 | 6 | 0 |
| CED of cis-DDPc | 7 | >73.4±16.4 | 76 | 45–>90 (2)c | >259 | 261 |
| Untreated controls | 4 | 20.5±4.1 | 21 | 15–25 | – | – |
Notes:
Animals received 105 F98EGFR cells ic and therapy studies were initiated 7 days later.
N = number of animals per group.
Seven days after tumor implantation, 3 μg of free cis-DDP or C225-G5-Pt bioconjugate containing 46.16 μg of cis-DDP in a volume of 10 μL were administered by CED over 30 minutes.
The difference in MST between rats that received cis-DDP and untreated controls was highly significant (P<0.001).
Abbreviations: ic, intracerebral; CED, convection-enhanced delivery; MST, mean survival time; cis-DDP, cis-diamminedichloroplatinum; SE, standard error.
Results
In vitro cellular uptake and cytocidal activity of Pt bioconjugates
There was almost a 3.4-fold increase in the amount of cis-DDP delivered to the tumor cells by the C225-PG-Pt bioconjugate (14.51 μg/109 cells) compared to the untargeted PGA-Pt bioconjugate (4.47 μg/109 cells), assuming that 109 cells equaled ~1 g. The in vitro cytotoxicity profiles of the Pt bioconjugates are shown in Figure 2. C225 itself and C225-G5-Pt were devoid of in vitro cytotoxic activity (Figure 2A) compared to cis-DDP, which at a concentration of 10 μM produced a 80% reduction in the viability of F98EGFR cells. Up to a concentration of 1,000 μM, C225-PG-Pt was less cytotoxic in vitro than free cis-DDP, suggesting that there was reduced release of the Pt fragment at lower concentrations of the bioconjugate (Figure 2B). PGA-Pt also was cytotoxic, although there was no specific targeting of EGFR. PEP455-Pt and PEP382-Pt showed equivalent toxicity against F98EGFR cells (Figure 2C), and nonplatinated PEP455 was devoid of intrinsic toxicity (Figure 2D), even at the highest concentration. The IC50 values of PEP382-Pt and PEP455-Pt bioconjugates were determined by extrapolating from the 50% cell viability on the y-axis to the corresponding point on the x-axis (Figure 2E and F). These were 0.20 and 0.10, respectively, against F98WT cells, and 0.02 and 0.02, respectively, against F98EGFR cells relative to the undiluted stock solutions of PEP382 (200 μg in 800 μL of PBS) and PEP455 (138.8 μg in 500 μL of PBS; Figure 2C). Finally, as shown in Figure 2E and F, there was specificity for F98EGFR cells vs F98WT, at an intermediate dilution (0.01) for both PEP382-Pt and PEP455-Pt, and this specificity also was evident at the highest concentrations (40% vs 20%). However, for the highest concentrations of PEP455-Pt, which obscured target specificity, the percent cell viability was equivalent for both cell lines.
Figure 2.


In vitro cytotoxicity profiles of EGFR-targeting Pt-containing bioconjugates against F98EGFR cells unless indicated otherwise in the figures.
Notes: Cells were seeded into microplates at a density of 10,000 per well, and the following day, the test agents at varying concentrations were added to each well. After 3 days, the cells were fixed and stained with SRB, and SRB protein concentrations were determined spectrophotometrically and normalized to 100% for untreated cells. (A) C225-G5-Pt; (B) C225-PGA-Pt; (C and D) PEP455-Pt, 4 hours incubation time; (E) PEP382-Pt; and (F) PEP455-Pt, 72 hours incubation time.
Abbreviations: EGFR, epidermal growth factor receptor; SRB, sulforhodamine-B; WT, wild type.
Therapy studies with C225-G5-Pt bioconjugates in F98EGFR glioma-bearing rats
The lack of in vitro cytotoxicity of C225-G5-Pt notwithstanding, we decided to initiate a therapy study in F98EGFR glioma-bearing rats, hypothesizing that prolonged contact of tumor cells with the bioconjugate would result in in vivo release of cis-DDP. The survival data for rats that had received either the C225-G5-Pt bioconjugate or cis-DDP are summarized in Table 1. The MST of rats that received the former was 21.7±6.6 and 73.4±16.4 days for the latter compared to 20.5±4.1 days for untreated controls. Using one-way ANOVA, based on an approximate normal distribution of the MSTs, this difference was highly significant (P<0.001). These data clearly demonstrated the therapeutic efficacy of ic CED of cis-DDP at a dose of 3 μg and the lack of efficacy of the C225-G5-Pt bioconjugate at a Pt dose of 46.16 μg, which resulted in an MST equivalent to that of untreated controls. This suggested that the Pt fragment had not been released from the bioconjugate since, as listed in Table 1, free cis-DDP was therapeutically effective when administered ic by CED. On the basis of the lack of effectiveness of the C225-G5-Pt bioconjugate, a pilot study was initiated with the PEP455-Pt bioconjugate in F98 glioma-bearing rats. This had a much lower MW (~2 kDa), and, based on Fick’s law,43 would have had enhanced diffusion within the brain tumor following ic administration by CED compared to high MW MoAbs.
Neurotoxicologic studies of PEP-Pt bioconjugates
The brains of non-tumor-bearing rats that received free cis-DDP (3 μg) administered by CED and were euthanized 7 days later showed severe hemorrhage and necrosis (Figure 3A), indicating that the free drug was highly neurotoxic. The brains of rats that received unmodified PEP382 and PEP455 showed evidence of mild intrinsic neurotoxicity, as evidenced by microscopic foci of reactive inflammatory cells consisting of macrophages and occasional lymphocytes. In several of the rat brains, there were evolving foci of necrosis at the presumptive site of administration of the peptides (Figure 3B). The PEP-Pt bioconjugates produced necrosis, moderate-to-intense infiltrates of macrophages, and focal hemorrhage, suggesting that free cis-DDP had been released (Figure 3C). However, these changes were mild compared to those seen in rats that had received free cis-DDP (Figure 3A). In contrast, Figure 3D shows a representative section of brain of an F98EGFR glioma-bearing rat that received the PEP455-Pt bioconjugate and was euthanized at 42 days, with a focus of reactivity consisting of mild necrosis and prominent vacuolization. On the basis of these findings, we selected the PEP455-Pt bioconjugate for our in vivo therapy studies.
Figure 3.

Neurotoxicologic and neuropathologic properties of cis-DDP and PEP-Pt bioconjugates.
Notes: Neuropathologic correlates (A)–(D) are coronal sections of H&E-stained sections of brains from non-tumor-bearing Fischer rats that were euthanized 7 days after administration of the test agent. (A) Approximately 3 μg of cis-DDP ic by CED. There is severe focal hemorrhage (h) and necrosis (n) at the site of administration (400×). (B) ic PEP455 by CED. There is mild, focal edema (e) and evolving necrosis (n), and a dense focal infiltrate of hemosiderin-laden macrophages (m) (200×). (C) ic PEP-Pt-382 by CED. The changes are similar to those seen in (B) except larger numbers of hm (200×) are noted, suggesting previous hemorrhage. (D) ic PEP455-Pt by CED. There is vacuolization (v) of white matter, mild necrosis (n), and a mci consisting of macrophages and lymphocytes (200×). (E) F98EGFR glioma-bearing rat that received PEP455-Pt containing 3.3 μg of cis-DDP on day 14 after tumor cell implantation and euthanized on day 42. There is a focus of reactivity consisting of mild necrosis (n) and prominent vacuolization (v) of white matter, but no inflammatory cell infiltrates nor evidence of tumor (100×). (F) Brain of an untreated Fischer rat that died on day 27 following implantation, at which time the tumor measured 22.78 mm2. The histology is characteristic of the F98 glioma with a highly invasive pattern of growth, with itc at varying distances from the main tumor mass (200×). At lower magnification, central zones of necrosis were seen.
Abbreviations: H&E, hematoxylin and eosin; itc, infiltrating tumor cells; hm, hemosiderin-laden macrophages; mci, mononuclear cellular infiltrate; CED, convection-enhanced delivery; cis-DDP, cis-diamminedichloroplatinum; ic, intracerebral; mtc, main tumor cells.
Therapy studies of PEP455-Pt bioconjugate in F98EGFR glioma-bearing rats
On the basis of the enhanced in vitro cytotoxicity of the PEP455-Pt bioconjugate, which recognized the HER-1 epitope, we elected to carry out a pilot study to assess its therapeutic efficacy. As recently reported by us,12 a study was carried out to assess the therapeutic effects of combined treatment with carboplatin and X-irradiation in F98WT glioma-bearing rats. A component of this study was to assess the histopathologic changes associated with this treatment. Groups consisting of eight F98WT and F98EGFR glioma-bearing rats each were euthanized sequentially at 1–4 weeks following treatment. Those animals, which had been euthanized at 4 weeks after treatment, corresponded to the 4-week posttreatment group in this study. Histopathologic examination of the brains of F98EGFR and F98WT glioma-bearing rats that had received the PEP455-Pt bioconjugate (3.31 μg of cis-DDP) at 2 weeks following tumor cell implantation and were euthanized 4 weeks later indicated that there had been a therapeutic response (Figure 3E) compared to untreated control rats (Figure 3F). As summarized in Table 2, two of the four F98EGFR glioma-bearing rats showed no evidence of tumor, one had a microscopic tumor, and one had a 6 mm tumor in the white matter, which indicated a treatment failure, most likely related to failure of the PEP-Pt bioconjugate to attain a sufficient concentration at the site of the tumor. Two of the three rats bearing F98WT gliomas had no evidence of tumors, and one had a macroscopic tumor, suggesting if there was a high enough concentration of the Pt bioconjugate at the site of the tumor, EGFR targeting was not essential for in vivo tumoricidal activity. In contrast, as determined by quantitative image analysis, the untreated control rats had tumor cross-sectional areas ranging from 7.49 to 32.79 mm2 at the times of their deaths, which ranged from 21 to 34 days, and there was a linear inverse relationship (Figure 4) between the former and the latter (R=0.996). It may seem slightly counterintuitive that rats with smaller tumors died earlier than those with larger tumors, but this suggests that cerebral edema, which we could not measure, was greater in the former compared to the latter. Furthermore, these findings provide strong evidence for a therapeutic effect since the treated animals were euthanized at 42 days. This already was 60% greater than the MST of 26.3±2.5 days for untreated control rats, all of which had macroscopic tumors. As determined by the log-rank test, this difference was significant at the level P<0.01. On the basis of our previously reported study on the therapeutic effects of CED of carboplatin in F98WT glioma-bearing rats,11 we hypothesize that the MST of the PEP455-Pt-treated rats could have been in the range of 52–62+ days.
Table 2.
Therapeutic effects of PEP-Pt bioconjugates in F98 gliomas-bearing rats
| Test agenta | Number of rats | Tumor status (n [%])b,c
|
||
|---|---|---|---|---|
| Free | Micro | Macro | ||
| PEP455-Pta | 7 | 4 (57) | 1 (14) | 2 (29) |
| None (untreated controls) | 6 | 0 (0) | 0 (0) | 6 (100)c |
Notes:
Animals received PEP455-Pt bioconjugates by CED containing 3.31 μg of cis-DDP at 2 weeks following tumor cell implantation. Three rats had F98WT gliomas, and four had F98EGFR tumors.
Animals were euthanized at 4 weeks following administration of the bioconjugate and 6 weeks following tumor cell implantation.
Untreated control rats had an MST ± SE of 26.3±2.5 days. Even though survival was censored at 42 days, the difference in the survival time between untreated controls and PEP455-Pt was statistically significant (P<0.01).
Abbreviations: CED, convection-enhanced delivery; MST, mean survival time; SE, standard error; WT, wild type; cis-DDP, cis-diamminedichloroplatinum.
Figure 4.
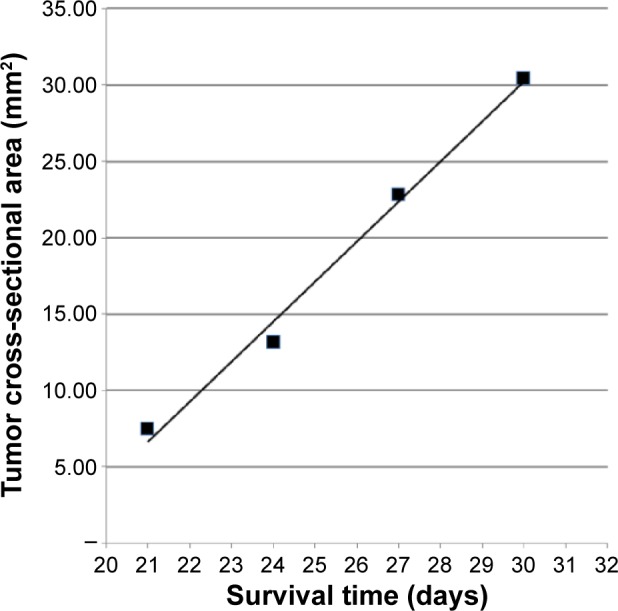
Plot of tumor cross-sectional areas in mm2 as a function of survival time in days following stereotactic intracerebral implantation of 10,000 F98EGFR tumor cells into the caudate nucleus of Fischer rats.
Notes: Microsoft® Excel was used to calculate the linear regression and generate the line, which had a correlation coefficient of R=0.996. Two additional rats’ tumors were outliers and therefore not included in this plot. Cross-sectional areas ranged from 7.49 to 32.79 mm2 at the times of the rats’ deaths, which ranged from 21 to 34 days.
Abbreviation: EGFR, epidermal growth factor receptor.
Discussion
We have previously carried out extensive studies on the use of C225 as an EGFR-targeting agent for the delivery of a heavily boronated PAMAM dendrimer for boron neutron capture therapy of F98EGFR glioma-bearing rats.26,33,36 Here, there was no need for either pharmacologic activity or release of boron-10 from the bioconjugates for them to be therapeutically effective. In a paradigm shift, we have turned our attention to a completely different chemoradiotherapeutic approach combining ic CED of carboplatin with external beam photon irradiation using the F98 rat glioma model.9–12 Here, there was a requirement for release and pharmacologic activity of the Pt. We had hoped to use cis-DDP, which has enhanced tumoricidal activity compared to that of carboplatin,23 but we found that ic CED of cis-DDP was highly neurotoxic in non-tumor-bearing Fischer rats.25 On the basis of these observations, we decided to determine if the therapeutic efficacy and neurotoxicity profile of cis-DDP following ic administration by CED could be reformatted by linking it to C225. Although we succeeded in reducing its neurotoxicity,44 the C225-G5-Pt bioconjugate had no therapeutic effect (Table 1), most likely due to its failure to release Pt. This lack of release of the drug payload was very similar to that which we had previously observed with another C225-based bioconjugate that contained methotrexate linked to a fifth-generation PAMAM dendrimer.37 This bioconjugate, designated C225-G5-MTX, demonstrated specific in vivo molecular targeting of EGFR following ic CED to F98EGFR glioma-bearing rats but were devoid of therapeutic efficacy.37 This study demonstrated that molecular targeting was but one of several requirements that must be fulfilled if an antibody-targeted drug is to be therapeutically effective. It also has been previously reported that the release of cis-DDP bound to antibody-PAMAM conjugates is slow, and these kinetics result in lower in vitro and in vivo activity.45
We decided not to carry out any in vivo therapy studies with C225-PG-Pt bioconjugates based on the fact that they had reduced cytotoxicity at a concentration of 400 μM against F98EGFR target cells compared to cis-DDP (Figure 2B). We then hypothesized that lower MW EGFR-targeting peptides might have significant advantages over cetuximab.46 First, it was possible to directly link cis-DDP to them by a simple metal coordination chemical reaction in contrast to the more complicated synthetic schemes that were used to prepare C225-G5-Pt and C225-PG-Pt bioconjugates (Figure 1A). Second, assuming an average of two molecules of cis-DDP could be attached per molecule of the peptide, the MWs of PEP382-Pt and PEP455-Pt bioconjugates would have had MWs of ~3,744 and 2,271 Da, respectively, which was approximately two orders of magnitude less than that of the C225 bioconjugate (~200 kDa). We hypothesized that this would result in improved tumor uptake and microdistribution of the bioconjugates following ic CED. The neurotoxicologic studies with PEP382-Pt (Figure 3C) and PEP455-Pt (Figure 3D) bioconjugates demonstrated that they had reduced neurotoxicity compared to free cis-DDP (Figure 3A). On the basis of this, we decided to initiate a pilot study to determine if PEP455-Pt bioconjugates had in vivo efficacy, as indicated by the presence or absence of macro-or microscopic tumor at 4 weeks following treatment and 6 weeks following tumor implantation. All of the treated rats were alive at 42 days, which was 60% greater than the MST of 26.3±2.5 days for untreated controls with a P-value =0.03 using Fisher’s exact test (Table 2). Furthermore, four of seven rats had no evidence of any tumor, one had a microscopic tumor, and two rats had macroscopic tumors at day 42. Since our survival data were censored at 42 days, we hypothesize that if we had used survival time as the end point of the study, the MST could have exceeded the 55.2±7.8 days that we had previously observed in carboplatin-treated rats.11 In this pilot study, the PEP455-Pt bioconjugate appeared to be equally effective against both F98EGFR and F98WT cells. This is explained in part by the in vitro cell viability data shown in Figure 2F. Although there was a difference in tumoricidal activity against F98EGFR and F98WT cells at a lower concentration of PEP455-Pt (0.01 μM), at the highest concentration (~1 μM), it was equally cytotoxic against both F98EGFR and F98WT cells. This provides a possible explanation for the equivalent therapeutic efficacy against F98EGFR and F98WT tumors in vivo. First, the concentration of the drug was so high due to the fact that it was delivered directly to the site of the tumor that there was no difference between targeted and nontargeted delivery. Second, the enhanced permeability and retention effect47 within the tumor itself, due to abnormal molecular and fluid transport dynamics, also could have contributed to the high intratumoral concentration of Pt. On the basis of this, we hypothesize that if lower concentrations of PEP455-Pt were administered to F98 glioma-bearing rats then we could have seen a difference in therapeutic effectiveness between animals bearing F98EGFR and F98WT tumors. Be that as it may, we have succeeded in reducing the neurotoxicity of cis-DDP by linking the drug to PEP455.
The in vitro and in vivo activity of the peptide-Pt bioconjugates can be explained in terms of ligand exchange chemistry that is typical of these and related systems.48,49 Inside the cell, the peptide group bound to the Pt metal is substituted for a water molecule to release cis-[Pt(NH3)2 (H2O)Cl]+, as shown in Figure 5.50 This molecular species is identical to that produced following the first aquation step of cis-DDP. It undergoes a second ligand exchange to generate activated cis-DDP, cis-[Pt(NH3)2(H2O)2]2+ (Figure 5), which covalently bonds to the N7 position of purine bases to form intra- and interstrand DNA cross-links.50 The lower activity of PEP382-Pt compared to PEP455-Pt can be explained by the stronger His-Pt bonds in the former, resulting in slower release of the drug from the peptide.29,30
Figure 5.

Schematic representation of the stepwise equation of peptide–Pt bioconjugates to generated activated cis-DDP.
Notes: This molecular species is identical to that produced following the first equation step of cis-DDP. It undergoes a second ligand exchange to generate activated cisplatin, cis-[Pt(NH3)2(H2O)2]2+, as shown in the figure, which covalently bonds to the N7 position of purine bases to form intra- and interstrand DNA cross-links.
Abbreviation: cis-DDP, cis-diamminedichloroplatinum.
On the basis of the in vitro and in vivo data from this study, we have concluded that the EGFR-targeting PEP-Pt bioconjugates were superior to those employing C225 as the targeting moiety. The problem of release of the therapeutic payload is essential for effectiveness, and it is one that investigators have grappled with for several decades. Although MoAbs have become an important addition to the cancer armentarium,51 only a small number of drug bioconjugates52–56 currently are in clinical use, and none for the treatment of brain tumors. One of the most important reasons for this is the inability of these high MW bioconjugates to cross the BBB. For this reason, we and others have employed ic CED to maximize the amount of the therapeutic agents that reaches the brain tumor.11,12,37 For example, we observed that, to obtain the same tumor concentration of 10.4 μg of carboplatin per g tumor following ic administration of 20 μg by CED to F98 glioma-bearing rats, it was necessary to administer a dose of 20 mg (20,000 μg) by intravenous (iv) injection, which would have been lethal.11
Several recent reports have described the development of peptide–drug bioconjugates.57–59 One of these, designated ANG4043, which crosses the BBB, was conjugated to anti-HER2 MoAb using a two-step conjugation procedure employing copper-free click chemistry.59 In vivo studies in nude mice bearing intracranial implants of the human BT-474 breast cancer cell line demonstrated that the MeST of ANG4043-treated mice was 60 days vs 47 days for anti-HER2 alone. Based on these observations, it would be of interest to determine if angiopep-2 could be linked to PEP455-Pt, thereby allowing it to traverse the BBB following iv administration.
Conclusion
We have demonstrated that the PEP455-Pt bioconjugate was therapeutically effective and that it had reduced neurotoxicity compared to free cis-DDP. Further studies are warranted to assess the therapeutic potential of PEP-Pt bioconjugates as part of the expanding role of antibody and peptide drug conjugates in cancer treatment.60
Acknowledgments
We thank Dr Hamza Numan Gokozan for assistance in taking photomicrographs and Loretta Bahn for assistance in the preparation of this manuscript. The authors dedicate this paper to Professor Emeritus Albert Soloway in celebration of his 91st birthday. The studies described in this report were supported in part by the National Institutes of Health grant 1R01 CA098945, Voices Against Brain Cancer, and the Kevin J Mullin Memorial Fund for Brain Tumor Research.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Bi WL, Beroukhim R. Beating the odds: extreme long-term survival with glioblastoma. Neuro Oncol. 2014;16(9):1159–1160. doi: 10.1093/neuonc/nou166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Olson JJ, Nayak L, Ormond DR, Wen PY, Kalkanis SN. The role of cytotoxic chemotherapy in the management of progressive glioblastoma: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2014;118(3):501–555. doi: 10.1007/s11060-013-1338-5. [DOI] [PubMed] [Google Scholar]
- 4.Huse JT, Aldape KD. The evolving role of molecular markers in the diagnosis and management of diffuse glioma. Clin Cancer Res. 2014;20(22):5601–5611. doi: 10.1158/1078-0432.CCR-14-0831. [DOI] [PubMed] [Google Scholar]
- 5.Toporkiewicz M, Meissner J, Matusewicz L, Czogalla A, Sikorski AF. Toward a magic or imaginary bullet? Ligands for drug targeting to cancer cells: principles, hopes, and challenges. Int J Nanomedicine. 2015;10:1399–1414. doi: 10.2147/IJN.S74514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Firer MA, Gellerman G. Targeted drug delivery for cancer therapy: the other side of antibodies. J Hematol Oncol. 2012;5:70. doi: 10.1186/1756-8722-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oberoi RK, Parrish KE, Sio TT, Mittapalli RK, Elmquist WF, Sarkaria JN. Strategies to improve delivery of anticancer drugs across the blood- brain barrier to treat glioblastoma. Neuro Oncol. 2016;18(1):27–36. doi: 10.1093/neuonc/nov164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biston MC, Joubert A, Adam JF, et al. Cure of Fisher rats bearing radioresistant F98 glioma treated with cis-platinum and irradiated with monochromatic synchrotron X-rays. Cancer Res. 2004;64(7):2317–2323. doi: 10.1158/0008-5472.can-03-3600. [DOI] [PubMed] [Google Scholar]
- 9.Rousseau J, Boudou C, Barth RF, Balosso J, Esteve F, Elleaume H. Enhanced survival and cure of F98 glioma-bearing rats following intracerebral delivery of carboplatin in combination with photon irradiation. Clin Cancer Res. 2007;13(17):5195–5201. doi: 10.1158/1078-0432.CCR-07-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rousseau J, Barth RF, Moeschberger ML, Elleaume H. Efficacy of intracerebral delivery of carboplatin in combination with photon irradiation for treatment of F98 glioma-bearing rats. Int J Radiat Oncol Biol Phys. 2009;73(2):530–536. doi: 10.1016/j.ijrobp.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Yang W, Huo T, Barth RF, et al. Convection enhanced delivery of carboplatin in combination with radiotherapy for the treatment of brain tumors. J Neurooncol. 2011;101(3):379–390. doi: 10.1007/s11060-010-0272-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang W, Barth RF, Huo T, et al. Radiation therapy combined with intracerebral administration of carboplatin for the treatment of brain tumors. Radiat Oncol. 2014;9:25. doi: 10.1186/1748-717X-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lonser RR, Sarntinoranont M, Morrison PF, Oldfield EH. Convection-enhanced delivery to the central nervous system. J Neurosurg. 2015;122(3):697–706. doi: 10.3171/2014.10.JNS14229. [DOI] [PubMed] [Google Scholar]
- 14.Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25(3):282–303. doi: 10.1016/j.ccr.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loew S, Schmidt U, Unterberg A, Halatsch ME. The epidermal growth factor receptor as a therapeutic target in glioblastoma multiforme and other malignant neoplasms. Anticancer Agents Med Chem. 2009;9(6):703–715. doi: 10.2174/187152009788680019. [DOI] [PubMed] [Google Scholar]
- 16.Taylor TE, Furnari FB, Cavenee WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets. 2012;12(3):197–209. doi: 10.2174/156800912799277557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harding J, Burtness B. Cetuximab: an epidermal growth factor receptor chemeric human-murine monoclonal antibody. Drugs Today (Barc) 2005;41(2):107–127. doi: 10.1358/dot.2005.41.2.882662. [DOI] [PubMed] [Google Scholar]
- 18.Berezowska S, Schlegel J. Targeting ErbB receptors in high-grade glioma. Curr Pharm Des. 2011;17(23):2468–2487. doi: 10.2174/138161211797249233. [DOI] [PubMed] [Google Scholar]
- 19.Broadbridge VT, Karapetis CS, Price TJ. Cetuximab in metastatic colorectal cancer. Expert Rev Anticancer Ther. 2012;12(5):555–565. doi: 10.1586/era.12.25. [DOI] [PubMed] [Google Scholar]
- 20.Frampton JE. Cetuximab: a review of its use in squamous cell carcinoma of the head and neck. Drugs. 2010;70(15):1987–2010. doi: 10.2165/11205010-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 21.Vale CL, Tierney JF, Fisher D, et al. Does anti-EGFR therapy improve outcome in advanced colorectal cancer? A systematic review and meta-analysis. Cancer Treat Rev. 2012;38(6):618–625. doi: 10.1016/j.ctrv.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Garrett CR, Eng C. Cetuximab in the treatment of patients with colorectal cancer. Expert Opin Biol Ther. 2011;11(7):937–949. doi: 10.1517/14712598.2011.582464. [DOI] [PubMed] [Google Scholar]
- 23.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 24.Roci E, Cakani B, Brace G, et al. Platinum-based chemotherapy in recurrent high-grade glioma patients: retrospective study. Med Arch. 2014;68(2):140–143. doi: 10.5455/medarh.2014.68.140-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huo T, Barth RF, Yang W, et al. Preparation, biodistribution and neurotoxicity of liposomal cisplatin following convection enhanced delivery in normal and F98 glioma bearing rats. PLoS One. 2012;7(11):e48752. doi: 10.1371/journal.pone.0048752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu G, Barth RF, Yang W, et al. Site-specific conjugation of boron-containing dendrimers to anti-EGF receptor monoclonal antibody cetuximab (IMC-C225) and its evaluation as a potential delivery agent for neutron capture therapy. Bioconjug Chem. 2004;15(1):185–194. doi: 10.1021/bc0341674. [DOI] [PubMed] [Google Scholar]
- 27.Barth RF, Adams DM, Soloway AH, Mechetner EB, Alam F, Anisuzzaman AK. Determination of boron in tissues and cells using direct-current plasma atomic emission spectroscopy. Anal Chem. 1991;63(9):890–893. doi: 10.1021/ac00009a010. [DOI] [PubMed] [Google Scholar]
- 28.Foy KC, Wygle RM, Miller MJ, Overholser JP, Bekaii-Saab T, Kaumaya PT. Peptide vaccines and peptidomimetics of EGFR (HER-1) ligand binding domain inhibit cancer cell growth in vitro and in vivo. J Immunol. 2013;191(1):217–227. doi: 10.4049/jimmunol.1300231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Zhao Y, Phillips HI, et al. Mass spectrometry evidence for cisplatin as a protein cross-linking reagent. Anal Chem. 2011;83(13):5369–5376. doi: 10.1021/ac200861k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Snelling JR, Barrow MP, Scrivens JH, Sadler PJ, O’Connor PB. Mass spectrometric strategies to improve the identification of Pt(II)-modification sites on peptides and proteins. J Am Soc Mass Spectrom. 2014;25(7):1217–1227. doi: 10.1007/s13361-014-0877-0. [DOI] [PubMed] [Google Scholar]
- 31.Pauwels B, Korst AE, de Pooter CM, et al. Comparison of the sulforhodamine B assay and the clonogenic assay for in vitro chemoradiation studies. Cancer Chemother Pharmacol. 2003;51(3):221–226. doi: 10.1007/s00280-002-0557-9. [DOI] [PubMed] [Google Scholar]
- 32.Barth RF, Kaur B. Rat brain tumor models in experimental neuro-oncology: the C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J Neurooncol. 2009;94(3):299–312. doi: 10.1007/s11060-009-9875-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barth RF, Yang W, Adams DM, et al. Molecular targeting of the epidermal growth factor receptor for neutron capture therapy of gliomas. Cancer Res. 2002;62(11):3159–3166. [PubMed] [Google Scholar]
- 34.Yang W, Barth RF, Wu G, et al. Development of a syngeneic rat brain tumor model expressing EGFRvIII and its use for molecular targeting studies with monoclonal antibody L8A4. Clin Cancer Res. 2005;11(1):341–350. [PubMed] [Google Scholar]
- 35.Yang W, Wu G, Barth RF, et al. Molecular targeting and treatment of composite EGFR and EGFRvIII-positive gliomas using boronated monoclonal antibodies. Clin Cancer Res. 2008;14(3):883–891. doi: 10.1158/1078-0432.CCR-07-1968. [DOI] [PubMed] [Google Scholar]
- 36.Wu G, Yang W, Barth RF, et al. Molecular targeting and treatment of an epidermal growth factor receptor-positive glioma using boronated cetuximab. Clin Cancer Res. 2007;13(4):1260–1268. doi: 10.1158/1078-0432.CCR-06-2399. [DOI] [PubMed] [Google Scholar]
- 37.Wu G, Barth RF, Yang W, Kawabata S, Zhang L, Green-Church K. Targeted delivery of methotrexate to epidermal growth factor receptor-positive brain tumors by means of cetuximab (IMC-C225) dendrimer bioconjugates. Mol Cancer Ther. 2006;5(1):52–59. doi: 10.1158/1535-7163.MCT-05-0325. [DOI] [PubMed] [Google Scholar]
- 38.Yang W, Barth RF, Wu G, et al. Molecular targeting and treatment of EGFRvIII-positive gliomas using boronated monoclonal antibody L8A4. Clin Cancer Res. 2006;12(12):3792–3802. doi: 10.1158/1078-0432.CCR-06-0141. [DOI] [PubMed] [Google Scholar]
- 39.Goodman JH, McGregor JM, Clendenon NR, et al. Ultrastructural microvascular response to boron neutron capture therapy in an experimental model. Neurosurgery. 1989;24(5):701–708. doi: 10.1227/00006123-198905000-00007. [DOI] [PubMed] [Google Scholar]
- 40.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A. 1994;91(6):2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nduom EK, Walbridge S, Lonser RR. Comparison of pulsed versus continuous convective flow for central nervous system tissue perfusion: laboratory investigation. J Neurosurg. 2012;117(6):1150–1154. doi: 10.3171/2012.9.JNS12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vogelbaum MA, Aghi MK. Convection-enhanced delivery for the treatment of glioblastoma. Neuro Oncol. 2015;17(Suppl 2):ii3–ii8. doi: 10.1093/neuonc/nou354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolak DJ, Thorne RG. Diffusion of macromolecules in the brain: implications for drug delivery. Mol Pharm. 2013;10(5):1492–1504. doi: 10.1021/mp300495e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McWhinney SR, Goldberg RM, McLeod HL. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009;8(1):10–16. doi: 10.1158/1535-7163.MCT-08-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yellepeddi V, Vangara K, Palakurthi S. Poly(amido)amine (PAMAM) dendrimer–cisplatin complexes for chemotherapy of cisplatin-resistant ovarian cancer cells. J Nanopart Res. 2013;15(9):1–15. [Google Scholar]
- 46.Ruoslahti E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv Mater. 2012;24(28):3747–3756. doi: 10.1002/adma.201200454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164(2):138–144. doi: 10.1016/j.jconrel.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 48.Reedijk J. New clues for platinum antitumor chemistry: kinetically controlled metal binding to DNA. Proc Natl Acad Sci U S A. 2003;100(7):3611–3616. doi: 10.1073/pnas.0737293100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Callari M, Aldrich-Wright JR, de Souza PL, Stenzel MH. Polymers with platinum drugs and other macromolecular metal complexes for cancer treatment. Prog Poly Sci. 2014;39(9):1614–1643. [Google Scholar]
- 50.Paraskar AS, Soni S, Chin KT, et al. Harnessing structure-activity relationship to engineer a cisplatin nanoparticle for enhanced antitumor efficacy. Proc Natl Acad Sci U S A. 2010;107(28):12435–12440. doi: 10.1073/pnas.1007026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shuptrine CW, Surana R, Weiner LM. Monoclonal antibodies for the treatment of cancer. Semin Cancer Biol. 2012;22(1):3–13. doi: 10.1016/j.semcancer.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sapra P, Shor B. Monoclonal antibody-based therapies in cancer: advances and challenges. Pharmacol Ther. 2013;138(3):452–469. doi: 10.1016/j.pharmthera.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 53.Teicher BA, Chari RV. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res. 2011;17(20):6389–6397. doi: 10.1158/1078-0432.CCR-11-1417. [DOI] [PubMed] [Google Scholar]
- 54.Trail A. Antibody drug conjugates as cancer therapeutics. Antibodies (Basel) 2013;2:113. [Google Scholar]
- 55.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 56.Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21(1):5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 57.Tai W, Shukla RS, Qin B, Li B, Cheng K. Development of a peptide-drug conjugate for prostate cancer therapy. Mol Pharm. 2011;8(3):901–912. doi: 10.1021/mp200007b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang P, Cheetham AG, Lock LL, Cui H. Cellular uptake and cytotoxicity of drug-peptide conjugates regulated by conjugation site. Bioconjug Chem. 2013;24(4):604–613. doi: 10.1021/bc300585h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Regina A, Demeule M, Tripathy S, et al. ANG4043, a novel brain-penetrant peptide-mAb conjugate, is efficacious against HER2-positive intracranial tumors in mice. Mol Cancer Ther. 2015;14(1):129–140. doi: 10.1158/1535-7163.MCT-14-0399. [DOI] [PubMed] [Google Scholar]
- 60.Gharpure KM, Wu SY, Li C, Lopez-Berestein G, Sood AK. Nanotechnology: Future of Oncotherapy. Clin Cancer Res. 2015;21(14):3121–3130. doi: 10.1158/1078-0432.CCR-14-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]