

Abstract
Aim
To compare different variables among (S)CID patients diagnosed in the USA and Kuwait.
Methods
Review of patients registered in The US Immune Deficiency Network registry or Kuwait National PID Registry between 2004 and 2014.
Results
Totals of 98 and 69 (S)CID patients were registered during the study period in the USIDNET registry and the KNPIDR, respectively. The average annual incidence rate for the period 2004–2014 of (S)CID in children in Kuwait was 13.01/100,000 children, with an estimated occurrence of 1/7500 live births. There were differences between the two countries in the following variables: age at onset and diagnosis, family history of (S)CID, parental consanguinity, and outcome. More than 14% of (S)CID patients from USIDNET registry were diagnosed through newborn screening.
Conclusions
Patients’ characteristics and molecular causes of S(CID) are different between USA and Kuwait. NBS for SCID should be started in countries where the incidence of (S)CID is high.
Keywords: Combined immunodeficiency Registry, Epidemiology, Prevalence, Incidence, Genetics, Newborn screening
1. Introduction
Combined immunodeficiencies (CID) are characterized by defects in T-lymphocyte differentiation or function and variably associated with defects of B- or NK-lymphocytes. Over 40 different molecular defects can result in CID [1] and the list is growing due to availability of next generation sequencing (NGS) and advances made in functional assays. CID are characterized by a high level of genetic, immunologic and clinical heterogeneity. Patients with severe combined immune deficiency (SCID) present very early in life with interstitial pneumonia, failure to thrive, candidiasis and chronic diarrhea [2]. However, atypical SCID and CID are often characterized by delayed clinical presentation (beyond 1 year with recurrent infections, autoimmunity, granuloma, skin manifestations, lymphoproliferation and increase risk of malignancies) [3–5]. These patients often harbor hypomorphic mutations in SCID-causing genes, or have defects in gene less critical for T-lymphocyte development and/or function. In many cases, the genetic defect remains unknown. Early diagnosis of CID is of critical importance for prompt medical intervention, including prophylactic antibiotics, avoidance of live vaccines and non-irradiated blood products, and initiation of immunoglobulin replacement therapy. However, immune reconstitution can only be achieved with allogeneic hematopoietic stem cell transplantation (HSCT) or gene therapy [6], and in the case of adenosine deaminase (ADA) deficiency with enzyme replacement therapy.
Based on newborn screening (NBS) results in eleven of the United States of America (USA), typical and atypical SCID were found to affect 1 in 58,000 newborns [7]. Registry reports of several countries and regions show wide variations in geographical and racial prevalence as well as the frequencies of different types of PID [8–15]. Because most of (S)CID-causing gene defects are inherited in an autosomal recessive pattern, it is expected that (S)CID would be more prevalent in countries in which consanguineous marriages are common.
The aim of this study is to compare different variables among (S)CID patients diagnosed in the USA and Kuwait.
2. Method
Patients included in this study were diagnosed between January 2004 and December 2014 and registered in the United States Immune Deficiency Network (USIDNET) registry or Kuwait National Primary Immunodeficiency Disorders Registry (KNPIDR). Details about both registries can be found elsewhere [11,15]. The registries were queried for the following patients’ data: gender, age at onset, age at diagnosis, family history of CID, parental consanguinity, molecular diagnosis, outcome (alive/dead) at the time of data retrieval and whether the patient was diagnosed through NBS. NBS in Kuwait was limited to the use of flow cytometry for patients who had a family member affected by (S)CID. The same was applied in the United States until 2008 when NBS was started in Wisconsin by quantifying levels of T-cell receptor excision circles (TRECs) in dried blood spots collected at birth followed by flow cytometry for confirmation. In the United States, TREC assay is currently implemented in 30 states, the District of Columbia and the Navajo Nation. For patients who were treated with HSCT, age at transplant and type of donor were also queried.
Statistical analysis was performed using the Statistical Package for Social Sciences (SPSS version 22, IBM Corp., Armonk, NY, USA). Pearson’s Chi-square test was used to assess the association or significant differences between two qualitative variables, while Z-test was applied to compare two proportions. Two-sample non-parametric Kolmogorov–Smirnov test was used for the quantitative variables. A probability value of p < 0.05 was considered as the cut-off level for statistical significance.
3. Results
Totals of 98 and 69 (S)CID patients were registered during the study period in the USIDNET registry and the KNPIDR, respectively. There were missing data from USIDNET registry as shown in Supplementary Table 1. Patients from the USIDNET registry presented at an earlier age [mean: 2.78 months, standard deviation (SD): 2.65, standard error (SE): 0.41] compared to those from KNPIDR [mean: 7.99 months, SD: 18.21, SE: 2.20], p: <001. They were also diagnosed at an earlier age [mean: 3.06 months, SD: 3.07, SE: 0.527] compared to patients from KNPIDR [mean: 20.71, SD: 35.28, SE: 4.247], p: <001. Patients’ characteristics from both registries are shown in Fig. 1. There were differences between the two countries in the following variables: family history of (S)CID, parental consanguinity, and outcome. More than 14% of (S)CID patients from USIDNET registry were diagnosed through NBS. They constituted 23% of patients who were diagnosed in 2008 onward.
Fig. 1.
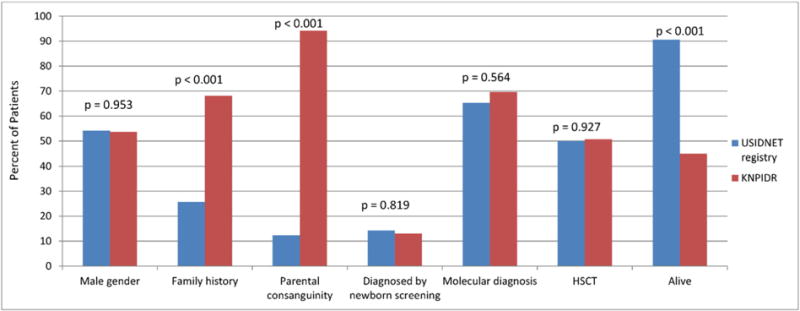
Characteristics of S(CID) registered in the United States Immune Deficiency Network (USIDNET) registry and Kuwait National Primary Immunodeficiency Disorders Registry (KNPIDR).
While there were no statistical differences in the frequency of molecularly-confirmed diagnosis in the two registries, there were important differences in the distribution of individual genotypes. In particular, most of the (S)CID patients from the USIDNET registry had adenosine deaminase (ADA) or γc deficiencies, while those from KNPIDR had recombinase activating gene (RAG) 1 or 2, major histocompatibility complex (MHC) II and dedicator of cytokinesis 8 (DOCK8) deficiencies (Fig. 2). The frequency of X-linked disease-causing gene defects (γc and CD40L) in USIDNET registry was 28% compared to none in KNPIDR (p: <001).
Fig. 2.
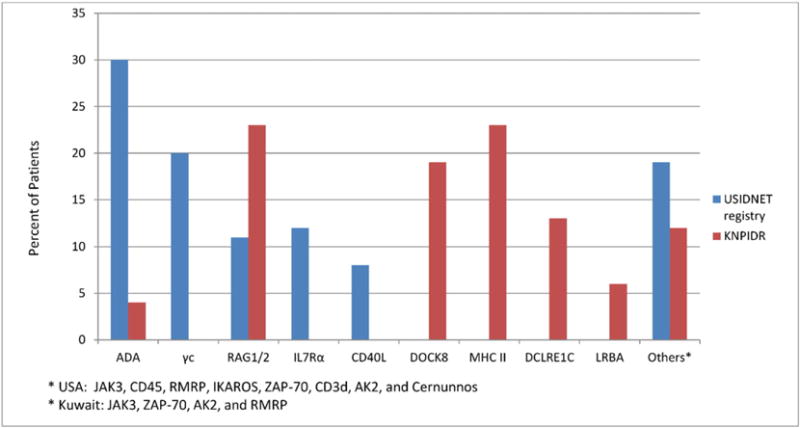
Molecular diagnosis of S(CID) registered in the United States Immune Deficiency Network (USIDNET) registry and Kuwait National Primary Immunodeficiency Disorders Registry (KNPIDR).
The frequency of HSCT performance in both registries was comparable (Fig. 1). However, most of the transplants in USIDNET registry were from haploidentical donors, whereas the majority of transplants in the KNPIDR were from matched related donors (Table 1), p: <001.
Table 1.
Type of donors for (S)CID patients who received HSCT.
| Donor type | USIDNET registry (n = 49) | KNPIDR (n = 35) |
|---|---|---|
| Matched related | 4 (8%) | 18 (51%) |
| Matched unrelated | 13 (27%) | 6 (17%) |
| Partially matched unrelated | 6 (12%) | 5 (15%) |
| Haploidentical | 25 (51%) | 6 (17%) |
| Unknown | 1 (2%) | 0 |
4. Discussion
In this report, we have compared patients’ characteristics and molecular profile of (S)CID patients diagnosed in the USA and Kuwait between 2004 and 2014. The average annual incidence rate for the study period was 13.01/100,000 children born in Kuwait, with an estimated occurrence of 1/7500 live births. This is almost 8-fold higher than the reported incidence of 1 in 58,000 infants in the USA [7]. This high incidence is most probably due to the common practice of consanguineous marriages in Kuwait (94% in the current report). A similar high incidence of CID (1 per 2000 births) was documented in the Navajo Nation [16] and in Konya, a city in central Turkey, where the incidence of CID was found to be 1 per 10,000 live births [17], demonstrating that the high incidence of CID can also be caused by other variables such as ethnic, genetic (founder effect, genetic isolates) and geographical factors. Consanguineous marriages not only increase the frequency of CID, but also affect the distribution and the type of genetic defects causing these diseases as apparent by the fact that none of the presented CID patients from KNPIDR suffered from X-linked forms of (S)CID, whereas they accounted for 28% of all cases in the USIDNET registry. A recent review of published CID-related data in PubMed showed that deficiencies in MHC II and RAG1/2, which are transmitted as autosomal recessive traits, are the most common causes of CID in the Middle East, while γc deficiency, which is responsible for X-linked SCID, represents the most common cause of SCID in other geographical areas and ethnicities [18]. Most of the (S)CID patients from the USIDNET in the current study were caused by ADA deficiency (30%) followed by γc deficiency (20%). This seems to contradict previous reports from the USA where >50% of SCID cases were caused by γc deficiency [19–21]. Several reasons may account for this discrepancy, including differences in single center series vs. nationwide registries and inadequate reporting of cases to the USIDNET registry. Universal newborn screening would provide a more accurate description of the frequency of the individual genotypes. In a recent report, among 52 consecutive cases of (S)CID identified through newborn screening in the United States, 10 (19.2%) had γc deficiency, 9 (17.3%) had RAG1/2 deficiency, 6 (11.5%) had interleukin-7 receptor (IL7R) deficiency, and 5 (9.6%) had ADA deficiency [7]. Interestingly, γc deficiency was most common (21.4%) among infants with typical SCID, whereas RAG1 deficiency was the most common cause of atypical SCID, accounting for 40% of these cases [7].
Similar to previous reports from the USA [5,19], the current study shows that CID patients registered in USIDNET presented earlier in life (mean of 2.78 months) compared to patients registered in KNPIDR (mean of 7.99 months). This difference may be contributed by a higher incidence of typical forms of SCID in the USA as compared to more frequent occurrence of “atypical SCID” in Kuwait. Another possible cause of such difference is that several SCID patients in Kuwait died before being diagnosed. However, these data need to be interpreted with caution, since data about the onset age was missing in 58% of the (S)CID patients registered in the USIDNET. A similar finding of missing data about age at diagnosis was also noted previously in the USIDNET registry [15].
Despite the fact that almost 70% of CID patients in Kuwait had a family history suggestive of CID, only 13% were diagnosed by flow cytometry at birth. This finding is consistent with an earlier study where the diagnosis of about two-thirds of patients with X-linked agammaglobulinemia who could have been diagnosed at birth based on a positive family history, were diagnosed only later in life when they developed recurrent infections [22]. Another study showed that the majority of, but not all, families with a previous case of a SCID-affected child, had subsequently born children tested for SCID either prenatally or at birth [19]. This indicates that genetic counseling and family education were not fully effective and should be an integral part of the patients’ care.
While only 23% of the (S)CID patients reported to the USIDNET registry in 2008 and later were diagnosed through NBS, it can be expected that this percentage will gradually rise, as NBS is being implemented in a growing number of states in the country. The effectiveness of NBS for SCID in the USA strongly suggests that a similar program should be started in countries where the prevalence of (S)CID is much higher, as in Kuwait. However, it is important to recognize that the TREC assay would not identify all babies with CID. In particular, genetic forms of CID that are permissive for significant residual intrathymic T cell development (such as ZAP-70 and MHC class II deficiencies) are not identified by NBS. Many of these disorders have a higher prevalence in the Middle East. Accordingly, additional or alternative systems may be required for NBS in countries like Kuwait to allow early recognition of such patients.
A similar proportion of patients in both registries received HSCT as a definitive form of treatment. However, most of the donors in the USIDNET registry were haploidentical parents as compared to more than 50% of transplants being from matched related donors in the KNPIDR. This is consistent with previous findings that the probability of finding an HLA-identical related donor for HSCT was about 30% in Western countries [23], compared to 70% in some areas of Pakistan [24] and 65% in Jordan [25]. This finding can be attributed to large family sizes, high population growth and frequent consanguineous marriages in these areas.
Collection of clinical, laboratory, and outcome data about primary immunodeficiencies (PID) through national and regional registries is regarded as a crucial element to address the needs of patients, healthcare providers and policy makers. Despite the limitations of poor documentations, under reporting, accuracy, funding and sustainability, these registries allow evaluation of epidemiologic trends in PID and clinical presentation. The current study has proven the strength of such registries by enabling the exchange of information and comparison between different centers and regions. In addition, such registries provide investigators the opportunity to perform epidemiologic, molecular and functional studies to better define disease pathophysiology and outcome in spite of the rarity of these diseases.
Supplementary Material
Acknowledgments
We would like to thank Tara Caulder USIDNET Registry Manager for providing subjects information.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.clim.2015.07.013.
References
- 1.Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, Etzioni A, Franco JL, Gaspar HB, Holland SM, Klein C, Nonoyama S, Ochs HD, Oksenhendler E, Picard C, Puck JM, Sullivan K, Tang ML. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol (5) 2014;162(22) doi: 10.3389/fimmu.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McWilliams LM, Railey MD, Buckley RH. Positive family history, infection, low absolute lymphocyte count (ALC) and absent thymic shadow: diagnostic clues for severe combined immunodeficiency (SCID) J Allergy Clin Immunol Pract. 2015;3:585–591. doi: 10.1016/j.jaip.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, Avcin T, Qasim W, Davies EG, Niehues T, Ehl S. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol. 2011;141(1):73–82. doi: 10.1016/j.clim.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, Grunebaum E. Defining combined immunodeficiency. J Allergy Clin Immunol. 2012;130(1):177–183. doi: 10.1016/j.jaci.2012.04.029. [DOI] [PubMed] [Google Scholar]
- 5.Dvorak CC, Cowan MJ, Logan BR, Notarangelo LD, Griffith LM, Puck JM, Kohn DB, Shearer WT, O’Reilly RJ, Fleisher TA, Pai SY, Hanson IC, Pulsipher MA, Fuleihan R, Filipovich A, Goldman F, Kapoor N, Small T, Smith A, Chan KW, Cuvelier G, Heimall J, Knutsen A, Loechelt B, Moore T, Buckley RH. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. J Clin Immunol. 2013;33(7):1156–1164. doi: 10.1007/s10875-013-9917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffith LM, Cowan MJ, Notarangelo LD, Puck JM, Buckley RH, Candotti F, Conley ME, Fleisher TA, Gaspar HB, Kohn DB, Ochs HD, O’Reilly RJ, Rizzo JD, Roifman CM, Small TN, Shearer WT. Workshop participants. Improving cellular therapy for primary immune deficiency diseases: recognition, diagnosis, and management. J Allergy Clin Immunol. 2009;124(6):1152–1160. doi: 10.1016/j.jaci.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, Baker M, Ballow M, Bartoshesky LE, Bonilla FA, Brokopp C, Brooks E, Caggana M, Celestin J, Church JA, Comeau AM, Connelly JA, Cowan MJ, Cunningham-Rundles C, Dasu T, Dave N, De La Morena MT, Duffner U, Fong CT, Forbes L, Freedenberg D, Gelfand EW, Hale JE, Hanson IC, Hay BN, Hu D, Infante A, Johnson D, Kapoor N, Kay DM, Kohn DB, Lee R, Lehman H, Lin Z, Lorey F, Abdel-Mageed A, Manning A, McGhee S, Moore TB, Naides SJ, Notarangelo LD, Orange JS, Pai SY, Porteus M, Rodriguez R, Romberg N, Routes J, Ruehle M, Rubenstein A, Saavedra-Matiz CA, Scott G, Scott PM, Secord E, Seroogy C, Shearer WT, Siegel S, Silvers SK, Stiehm ER, Sugerman RW, Sullivan JL, Tanksley S, Tierce ML, IV, Verbsky J, Vogel B, Walker R, Walkovich K, Walter JE, Wasserman RL, Watson MS, Weinberg GA, Weiner LB, Wood H, Yates AB, Puck JM, Bonagura VR. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312(7):729–738. doi: 10.1001/jama.2014.9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aghamohammadi A, Mohammadinejad P, Abolhassani H, Mirminachi B, Movahedi M, Gharagozlou M, Parvaneh N, Zeiaee V, Mirsaeed-Ghazi B, Chavoushzadeh Z, Mahdaviani A, Mansouri M, Yousefzadegan S, Sharifi B, Zandieh F, Hedayat E, Nadjafi A, Sherkat R, Shakerian B, Sadeghi-Shabestari M, Hosseini RF, Jabbari-Azad F, Ahanchian H, Behmanesh F, Zandkarimi M, Shirkani A, Cheraghi T, Fayezi A, Mohammadzadeh I, Amin R, Aleyasin S, Moghtaderi M, Ghaffari J, Arshi S, Javahertrash N, Nabavi M, Bemanian MH, Shafiei A, Kalantari N, Ahmadiafshar A, Khazaei HA, Atarod L, Rezaei N. Primary immunodeficiency disorders in Iran: update and new insights from the third report of the national registry. J Clin Immunol. 2014;34(4):478–490. doi: 10.1007/s10875-014-0001-z. [DOI] [PubMed] [Google Scholar]
- 9.Grimbacher B, ESID Registry Working Party The European Society for Immunodeficiencies (ESID) registry 2014. Clin Exp Immunol. 2014;178(Suppl. 1):18–20. doi: 10.1111/cei.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gathmann B, Goldacker S, Klima M, Belohradsky BH, Notheis G, Ehl S, Ritterbusch H, Baumann U, Meyer-Bahlburg A, Witte T, Schmidt R, Borte M, Borte S, Linde R, Schubert R, Bienemann K, Laws HJ, Dueckers G, Roesler J, Rothoeft T, Krüger R, Scharbatke EC, Masjosthusmann K, Wasmuth JC, Moser O, Kaiser P, Groß-Wieltsch U, Classen CF, Horneff G, Reiser V, Binder N, El-Helou SM, Klein C, Grimbacher B, Kindle G. The German National Registry for primary immunodeficiencies (PID) Clin Exp Immunol. 2013;173(2):372–380. doi: 10.1111/cei.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Herz W. Primary immunodeficiency disorders in Kuwait: first report from Kuwait National Primary Immunodeficiency Registry (2004–2006) J Clin Immunol. 2008;28(2):186–193. doi: 10.1007/s10875-007-9144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Tamemi S, Elnour I, Dennison D. Primary immunodeficiency diseases in Oman: five years’ experience at Sultan Qaboos University Hospital. World Allergy Organ J. 2012;5(5):52–56. doi: 10.1097/WOX.0b013e318258830f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishimura M, Takada H, Doi T, Imai K, Sasahara Y, Kanegane H, Nishikomori R, Morio T, Heike T, Kobayashi M, Ariga T, Tsuchiya S, Nonoyama S, Miyawaki T, Hara T. Nationwide survey of patients with primary immunodeficiency diseases in Japan. J Clin Immunol. 2011;31(6):968–976. doi: 10.1007/s10875-011-9594-7. [DOI] [PubMed] [Google Scholar]
- 14.Bousfiha AA, Jeddane L, El Hafidi N, Benajiba N, Rada N, El Bakkouri J, Kili A, Benmiloud S, Benhsaien I, Faiz I, Maataoui O, Aadam Z, Aglaguel A, Baba LA, Jouhadi Z, Abilkassem R, Bouskraoui M, Hida M, Najib J, Alj HS, Ailal F. Moroccan Society for Primary Immunodeficiencies (MSPID), First report on the Moroccan registry of primary immunodeficiencies: 15 years of experience (1998–2012) J Clin Immunol. 2014;34(4):459–468. doi: 10.1007/s10875-014-0005-8. [DOI] [PubMed] [Google Scholar]
- 15.Sullivan KE, Puck JM, Notarangelo LD, Fuleihan R, Caulder T, Wang C, Boyle M, Cunningham-Rundles C. USIDNET: a strategy to build a community of clinical immunologists. J Clin Immunol. 2014;34(4):428–435. doi: 10.1007/s10875-014-0028-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwan A, Hu D, Song M, Gomes H, Brown DR, Bourque T, Gonzalez-Espinosa D, Lin Z, Cowan MJ, Puck JM. Successful newborn screening for SCID in the Navajo Nation. Clin Immunol. 2015;158(1):29–34. doi: 10.1016/j.clim.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Azarsiz E, Gulez N, Edeer Karaca N, Aksu G, Kutukculer N. Consanguinity rate and delay in diagnosis in Turkish patients with combined immunodeficiencies: a single-center study. J Clin Immunol. 2011;31(1):106–111. doi: 10.1007/s10875-010-9472-8. [DOI] [PubMed] [Google Scholar]
- 18.Al-Herz W, Al-Mousa H. Combined immunodeficiency: the Middle East experience. J Allergy Clin Immunol. 2013;131(3):658–660. doi: 10.1016/j.jaci.2012.11.033. [DOI] [PubMed] [Google Scholar]
- 19.Chan A, Scalchunes C, Boyle M, Puck JM. Early vs. delayed diagnosis of severe combined immunodeficiency: a family perspective survey. Clin Immunol. 2011;138(1):3–8. doi: 10.1016/j.clim.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Railey MD, Lokhnygina Y, Buckley RH. Long-term clinical outcome of patients with severe combined immunodeficiency who received related donor bone marrow transplants without pretransplant chemotherapy or post-transplant GVHD prophylaxis. J Pediatr. 2009;155:834–840. doi: 10.1016/j.jpeds.2009.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Modell F, Puente D, Modell V. From genotype to phenotype. Further studies measuring the impact of a Physician Education and Public Awareness Campaign on early diagnosis and management of primary immunodeficiencies. Immunol Res. 2009;44:132–149. doi: 10.1007/s12026-008-8092-3. [DOI] [PubMed] [Google Scholar]
- 22.Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, Conley ME, Cunningham-Rundles C, Ochs HD. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine. 2006;85:193–202. doi: 10.1097/01.md.0000229482.27398.ad. [DOI] [PubMed] [Google Scholar]
- 23.Rosenmayr A, Pointner-Prager M, Mitterschiffthaler A, Bozic L, Pelzmann B, Tüchler H, Fae I, Fischer GF, Greinix HT, Peters Ch, Kalhs P, Krieger O, Linkesch W, Nachbaur D, Urban Ch, Posch U, Lanzer G, Gabriel Ch, Schennach H, Mayr WR. What are a patient’s current chances of finding a matched unrelated donor? Twenty years’ central search experience in a small country. Bone Marrow Transplant. 2012;47:172–180. doi: 10.1038/bmt.2011.67. [DOI] [PubMed] [Google Scholar]
- 24.Shamsi Ts, Hashmi K, Adil S, Ahmad P, Irfan M, Raza S, Masood N, Shaikh U, Satti T, Farzana T, Ansari S. The stem cell transplant program in Pakistan—the first decade. Bone Marrow Transplant. 2008;42:S114–S117. doi: 10.1038/bmt.2008.137. [DOI] [PubMed] [Google Scholar]
- 25.Elbjeirami WM, Abdel-Rahman F, Hussein AA. Probability of finding an HLA-matched donor in immediate and extended families: the Jordanian experience. Biol Blood Marrow Transplant. 2013;19(2):221–226. doi: 10.1016/j.bbmt.2012.09.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.