

Abstract
ERG (ETS-related gene) is a member of the ETS (erythroblast transformation-specific) family of transcription factors. Overexpression of the ERG transcription factor is observed in half of all prostate tumors and is an underlying cause of this disease. However, the mechanisms involved in the functions of ERG are still not fully understood. In the present study, we showed that ERG can directly bind to KDM4A (also known as JMJD2A), a histone demethylase that particularly demethylates lysine 9 on histone H3. ERG and KDM4A cooperated in upregulating the promoter of Yes-associated protein 1 (YAP1), a downstream effector in the Hippo signaling pathway and crucial growth regulator. Multiple ERG binding sites within the human YAP1 gene promoter were identified and their impact on transcription was determined through mutational analysis. Furthermore, we found that ERG expression reduced histone H3 lysine 9 trimethylation at the YAP1 gene promoter, consistent with its epigenetic regulation through the ERG interaction partner, KDM4A. Finally, downregulation of YAP1 phenocopied the growth-retarding effect of ERG or KDM4A depletion in human VCaP prostate cancer cells. Collectively, these results elucidated a novel mechanism - ERG promotes prostate tumorigenesis together with KDM4A through the upregulation of YAP1. A corollary is that KDM4A as well as YAP1 inhibitors may prove beneficial for the therapy of ERG-overexpressing prostate tumors.
Keywords: ERG, ETS protein, histone demethylase, JMJD2A, KDM4A, prostate cancer, YAP1
Introduction
Prostate cancer is among the most common neoplasias in men and a leading cause of death (1). Although the majority of prostate tumor patients are diagnosed with localized disease that can be efficiently treated with surgery and radiation therapy, men presenting with metastasized prostate tumors have a much bleaker survival chance. Thus, there is a critical need to develop new avenues of therapy, which may arise from a better understanding of the molecular changes in prostate cancer cells (2).
In approximately half of all human prostate tumors, the gene encoding the ETS transcription factor, ERG, becomes translocated, resulting in the overexpression of the ERG protein (3,4). Mimicking this overexpression in the prostates of mice led to the development of the precursor of prostate carcinomas, prostatic intraepithelial neoplasia (5,6). One research group even reported that very high prostate-specific expression of ERG induced carcinoma formation in approximately half of the respective transgenic mice at old age (7). Furthermore, when combined with knockout of the tumor-suppressor phosphatase and tensin homolog (PTEN), ERG overexpression accelerated prostate cancer development (8–11). These data implicate a causal role for ERG in the development of prostate tumors. However, the molecular details of how ERG overexpression contributes to the neoplastic transformation of prostate cells are far from resolved.
In the present study, we identified lysine demethylase 4A (KDM4A), also known as Jumonji C domain-containing protein 2A (JMJD2A), as a novel interaction partner of ERG. Furthermore, we found that ERG and KDM4A cooperated in regulating the transcription of the Yes-associated protein 1 (YAP1) gene, which is a downstream effector in the Hippo signaling pathway that plays important roles in development, homeostasis and cancer (12,13).
Materials and methods
Luciferase assays
Human VCaP prostate cancer cells were grown in 6-wells at 37°C in a humidified atmosphere containing 5% CO2 and were transfected utilizing 8 µg polyethylenimine. In general, 1,500 ng pBluescript KS+ as a carrier and 500 ng of indicated luciferase reporter gene constructs, which were based on the pGL2-Basic plasmid (Promega) and contained human YAP1 promoter fragments (amplified out of LNCaP prostate cancer cells) cloned between the SmaI and HindIII sites, were used for transfection. In addition, indicated amounts of ERG expression plasmid, empty vector pEV3S and Flag-tagged KDM4A (or its H188A mutant) expression vector were co-transfected. Cells were lysed 36 h after transfection and luciferase activity was measured as previously reported (14). In the case of human BPH-1 normal prostate cells, they were grown in 12-wells and transfected with 500 ng pBluescript KS+ and 500 ng YAP1 (−390/+22) luciferase reporter construct utilizing 2 µg polyethylenimine. Similarly, human LAP-C4 prostate cancer cells were grown in 12-wells and transfected with 750 ng pBluescript KS+ and 250 ng YAP1 (−390/+22) luciferase reporter plasmid also using 2 µg polyethylenimine, whereas 200 ng luciferase reporter construct, 800 ng pBluescript KS+, 1 ng ERG expression plasmid or pEV3S, and 2.5 µg polyethylenimine were employed in the case of human embryonic kidney 293T cells.
Preparation of protein extracts
Human 293T cells were seeded onto poly-L-lysine-coated 6-cm dishes (15) and transiently transfected by the calcium phosphate coprecipitation method (16) with 4.5 µg pBluescript KS+ and either 4.5 µg empty vector pEV3S or ERG-Myc-Flag expression plasmid. Thirty-six hours after transfection, cells were washed once with phosphate-buffered saline and cells were detached by a 5-min incubation in 40 mM HEPES (pH 7.4), 150 mM NaCl, 10 mM EDTA, after which cells were sprayed off by pipetting. Then, cells were collected by centrifugation and resuspended in 150 µl of 10 mM Tris, 30 mM Na4P2O7 (pH 7.1), 175 mM NaCl, 50 mM NaF, 2 mM dithiothreitol, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 2 µg/ml aprotinin, 1 µg/ml pepstatin, lysed for 30 min on ice and debris was removed by centrifugation (17). Extracts were frozen in liquid nitrogen and then stored at −80°C before use in DNA-binding assays. The presence of ERG in these extracts was assessed by western blotting (18) utilizing rabbit monoclonal ERG antibody (EPR3864; ab92513; Abcam), while total actin was detected with a rabbit polyclonal antibody (A2066; Sigma).
Electrophoretic mobility shift assay
Wild-type or mutated E74 oligonucleotides, which were previously described (19) or the below listed pairs of DNA oligonucleotides were hybridized to obtain double-stranded oligonucleotides. Then, they were radioactively labeled with 32P-dATP by filling in 5′-overhanging ends with Klenow DNA polymerase (20). Binding of 0.1 µl protein extract to ~0.25 ng 32P-labeled oligonucleotides occurred in 10 µl of 20 mM HEPES (pH 7.4), 25 mM NaCl, 0.5 mM EDTA, 0.1 µg/µl bovine serum albumin, 0.05 µg/µl poly(dI-dC)•poly(dI-dC), 2 mM dithiothreitol, 0.01% Tween-20 and 12% glycerol (21). As indicated, 0.05 µl of anti-Myc (9E10 mouse monoclonal antibody; M4439; Sigma) or anti-HA (12CA5 mouse monoclonal antibody; ab16918; Abcam) antibody was added. For competition experiments, 0.05 µl unlabeled oligonucleotide (12.5 ng) was additionally added. After a 30-min incubation on ice, the binding reactions were electrophoresed at 4°C on 4% native acrylamide gels as previously described (19). After drying, the gels were exposed to film at −80°C (20). The sequence of the oligonucleotide pairs used was as follows: '1/2', 5′-AGCGGAGCGGAAGAACTTCCTGCAGCCA-3′ and 5′-CTTGGCTGCAGGAAGTTCTTC CGCTCCGCT-3′; 'm1/2', 5′-AGCGGAGCGGTAGAACTTC CTGCAGCCA-3′ and 5′-CTTGGCTGCAGGAAGTTCTACC GCTCCGCT-3′; '1/m2', 5′-AGCGGAGCGGAAGAACTACCTGCAGCCA-3′ and 5′-CTTGGCTGCAGGTAGTTCTTCCGCTCCGCT-3′; '3', 5′-GTTCGGACCCGGATTGGACCC-3′ and 5′-GATGGGTCCAATCCGGGTCCGA-3′; '4', 5′-AGTGTGCAGGAATGTAGCA-3′ and 5′-AGTTGCTACATTCCTGCAC-3′; '5', 5′-CTTGCAGCGAAAAGTTTCCCT GCGCTG-3′ and 5′-CAGCGCAGGGAAACTTTTCGCT GCA-3′; '6/7', 5′-GCGCAGAGGAAGGAAGAGCCGAG-3′ and 5′-CTCTCGGCTCTTCCTTCCTCTGCGC-3′; 'm6/7', 5′-GCGCAGACGAAGGAAGAGCCGAG-3′ and 5′-CTCTCGGCTCTTCCTTCGTCTGCGC-3′; '6/m7', 5′-GCGCAGAGGAAGGACGAGCCGAG-3′ and 5′-CTCTCGGCTCGTCCTTCCTCTGCGC-3′; '8', 5′-GCCGCCAGGGAAAAGAA-3′ and 5′-CTTTCTTTTCCCTGGCGGC-3′.
Protein binding assays
ERG was fused C-terminally of GST (glutathione S transferase) and was expressed in Escherichia coli (22). The resulting GST-ERG fusion protein was purified by employing glutathione agarose beads, and then was dialyzed against 20 mM HEPES (pH 7.4), 50 mM NaCl, 10% glycerol, 0.2 mM phenylmethylsulfonyl fluoride and 1 mM dithiothreitol (23). To produce human KDM4A protein, its cDNA was cloned into a derivative of pFastBac™ 1 (Invitrogen, Carlsbad, CA, USA), which added a combined Flag/6His-tag onto the KDM4A N-terminus. The Bac-to-Bac system (Invitrogen) was used to generate KDM4A recombinant baculovirus according to the recommendations of the manufacturers, and Sf9 insect cells were infected with this virus and subsequently grown at 27°C in a spinner culture for 4 days. The His-tagged KDM4A protein was then affinity-purified with the help of Ni2+-nitrilotriacetic acid agarose (Qiagen) and dialyzed as previously described (24). Then, binding reactions were set up in 600 µl of 25 mM HEPES (pH 7.4), 25 mM NaCl, 0.01% Tween-20, 1 mM dithiothreitol, 0.2 mM phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 2 µg/ml aprotinin, 1 µg/ml pepstatin by first binding GST or GST-ERG to ~20 µl of glutathione agarose beads followed by challenge with purified Flag/6His-KDM4A. After three washes in binding buffer, bound proteins were boiled off with Laemmli sample buffer, subjected to SDS polyacrylamide gel electrophoresis and then revealed by either Coomassie staining or anti-Flag (M2 mouse monoclonal antibody; F3165; Sigma) western blotting (25).
Coimmunoprecipitation
Human embryonic kidney 293T cells, which were grown in 6-cm dishes, were transfected using the calcium phosphate coprecipitation method (26) with expression plasmids encoding Myc-tagged ERG and HA-tagged KDM4A. Thirty-six hours after transfection, cells were lysed and immunoprecipitations with anti-Myc mouse monoclonal antibodies (9E10; M4439; Sigma) were performed as previously described (27). Thereafter, the immunoprecipitates were subjected to SDS polyacrylamide gel electrophoresis. Then, western blotting was performed using anti-HA mouse monoclonal antibodies (12CA5; ab16918; Abcam) for detection of coprecipitated KDM4A (28).
Chromatin immunoprecipitation assay
Human 293T cells were grown in 10-cm dishes and transiently transfected with 3 µg YAP1 (−496/+22) luciferase reporter gene, 25 µg pBluescript KS+ and 0, 10 or 50 ng ERG expression plasmid using the calcium phosphate coprecipitation method (29). Two days after transfection, the cells were treated with 1% formaldehyde for 12 min at room temperature (30). Lysis of cells, sonication of resultant extracts and chromatin immunoprecipitations were then performed as previously described (31). The following rabbit polyclonal antibodies were employed: H3K4me3 (2.4 µg; ab8580; Abcam), H3K9me3 (3 µg; 07-442), H3K27me3 (4 µg; 07-449) (both from Upstate) and H3K36me3 (2 µg; ab9050; Abcam). Immunoprecipitated DNA fragments were amplified by PCR using the GoTaq DNA polymerase kit (M3008; Promega, Fitchburg, WI, USA) according to the manufacturer's recommendation and with the following temperature program (32): 2 min at 98°C; 8 cycles at 98°C for 25 sec, 65°C (−1°C/cycle) for 25 sec, 72°C for 25 sec; 25 cycles (or 20 cycles for input DNA) at 98°C for 25 sec, 57°C for 25 sec, 72°C for 25 sec (+1 sec/cycle); 72°C for 4 min as a final additional extension step. Primers used were: YAP1-2561-f (5′-GGCGAACTGGAAGCGCCTTTCC-3′) and YAP1-2989-r (5′-GAGACAGAAACTCGCCTCAAACGC-3′), yielding a 429-bp PCR product. Please note that these two primers can potentially amplify both the endogenous YAP1 promoter as well as the YAP1 promoter fragment in the YAP1 (−496/+22) luciferase reporter; however the utilized PCR cycle number was too low to detect any endogenous YAP1 promoter signals. In case of input DNA, the alternative primers: pGL2, sense (5′-CACTGCATTCTAGTTGTGGTTTGTCC-3′) and YAP1-2845-r (5′-CGCTGCAAGTTGCTACATTCCTGC-3′) were utilized that yielded a 419-bp PCR product. All PCR products were separated on 1.5% agarose gels and visualized with ethidium bromide staining (33).
Knockdown experiments
Oligonucleotides encoding shRNAs were inserted into the pSIREN-RetroQ (Clontech, Mountain View, CA, USA) retroviral vector and targeted the following human sequences: ERG #1 (5′-GCAGCTACATGGAGGAG AA-3′), ERG #3 (5′-GGGAAGGAACTGTGCAAGA-3′), KDM4A #3 (5′-GTTGAGGATGGTCTTACCT-3′), KDM4A #4 (5′-CACAGTTATTGACCATACT-3′), YAP1 #2 (5′-GCTTATAAGGCATGAGACA-3′) and YAP1 #3 (5′-AGT AATAGTTGGTTGTGAA-3′). Retrovirus was generated as previously described (34) and used to thrice infect VCaP cells followed by selection with 1 µg/ml puromycin (35). Cell growth was then measured with the PrestoBlue cell viability kit (Invitrogen) according to the recommendations of the manufacturer. For this, cells were seeded into 96-well plates, grown for 1–5 days, treated with PrestoBlue reagent for 1 h, excited with 530 nm light and fluorescence was measured at 590 nm.
Statistical analysis
Averages with standard deviations of at least three experiments were calculated, and statistical significance was assessed by performing an unpaired, two-tailed t-test. P≤0.05 was considered to indicate statistical significance.
Results
Activation of the human YAP1 promoter by ERG
Previously, it was shown that the ETS transcription factor GABP can bind to and stimulate the promoter of the mouse YAP1 gene, whereas ERG seemingly was incapable of doing so (36). In contrast, a recent report indicated that ERG promotes transcription of the human YAP1 gene (7). To clarify this discrepancy, we employed a luciferase reporter gene controlled by the human YAP1 promoter and transfected it into three different human cell lines: VCaP, a prostate cancer cell line characterized by the TMPRSS2-ERG translocation (3), another prostate cancer cell line (LAP-C4) that is devoid of such a translocation (37), and benign BPH-1 prostate cells. In all three cell lines, we observed a dose-dependent activation of the YAP1 gene promoter by ERG (Fig. 1). In VCaP and BPH-1 cells, 5 ng ERG expression vector was sufficient to elicit a maximal response, and 25 ng expression vector led to a slight reduction of promoter activity. In contrast, LAP-C4 cells displayed the highest YAP1 promoter activity at 1 ng ERG expression vector, and larger amounts led progressively to greatly reduced luciferase activities. Such a behavior is likely due to squelching, the titration of limiting cofactors by an abundance of ERG. Regardless, our data showed that ERG is capable of stimulating the human YAP1 gene promoter in various prostate cell lines.
Figure 1.

Activation of the human YAP1 gene promoter by ERG. Increasing amounts of ERG expression vector were transfected into cancerous VCaP and LAP-C4 or benign BPH-1 prostate cells and the activity of a cotransfected YAP1 (−390/+22) luciferase reporter construct was determined. Shown increases in luciferase activities (averages with standard deviations) upon ERG expression were normalized to the respective amount of transfected empty vector.
Direct binding of ERG to ETS sites within the human YAP1 promoter
Although ERG was found to interact with the human YAP1 promoter in chromatin immunoprecipitation assays (7), it has remained unresolved whether this is due to direct DNA-binding of ERG. Analysis of the human YAP1 promoter revealed the presence of eight potential ETS binding sites (Fig. 2A) that are characterized by a 5′-GGAA/T-3′ core sequence (38) and may be bound by ERG. Thus, we expressed Myc-tagged ERG in human 293T cells that have no detectable endogenous ERG (Fig. 2B), and prepared cell extracts to probe for a potential binding of ERG to the ETS sites in the YAP1 gene promoter. To this end, we generated 32P-labeled oligonucleotides encompassing these ETS sites and incubated them with control lysate or lysate from ERG-transfected 293T cells. In preliminary experiments (data not shown, but see also Fig. 2D), we observed that ERG alone did not bind to any of the ETS sites within the YAP1 promoter. However, it is known that ERG DNA-binding is auto-inhibited and this inhibition may be relieved by interaction with other proteins (39). Since our ERG expression construct contained a C-terminal Myc-tag, we employed anti-Myc antibodies to emulate such a protein-protein interaction and indeed, this resulted into noticeable DNA-binding (Fig. 2C); please note that we cannot exclude other explanations why the anti-Myc antibodies promoted DNA-binding of ERG, such as the disruption of binding of an ERG inhibitor that is present in lysates from 293T cells. In particular, we observed binding to oligonucleotides encompassing the juxtaposed ETS sites 1 and 2 as well as ETS sites 6 and 7. Binding to ETS sites 6 and 7 appeared to be stronger than to ETS sites 1 and 2 (Fig. 2C; and more visible in the shorter exposures of autoradiograms shown in Fig. 2D). No binding to ETS sites 3, 4, 5 and 8 was detected (Fig. 2C), consistent with those four ETS sites being very divergent to the ERG consensus site of 5′-ACCGGAAGT-3′ (40). In addition, as a positive control, we observed DNA-binding to the E74 site, a paradigmatic ETS binding site that was shown to interact with various ETS proteins (19,41,42).
Figure 2.

In vitro binding of ERG to the YAP1 promoter. (A) Sequence of the eight putative ETS binding sites within the human YAP1 gene promoter from −390 to +22. Also shown is the consensus DNA-binding sequence for ERG. (B) Western blotting shows the degree of ERG expression in the transfected 293T cells. Actin levels serve as a control. (C) In vitro binding of ERG to 32P-labeled oligonucleotides encompassing the indicated ETS sites or the E74 oligonucleotide. Mutation of ETS site 1, 2, 6 or 7 is marked by the prefix 'm̓. Asterisks denote ERG, DNA complexes. (D) DNA-binding assays with indicated radioactively labeled oligonucleotides. Inclusion of antibodies (anti-Myc or anti-HA) and unlabeled competitor oligonucleotides (E74 or the mutated mE74) is indicated.
To determine whether ERG binds to both ETS sites 1 and 2, we mutated each one individually in the '1/2' oligonucleotide. Mutation of either ETS site 1 or 2 resulted in similarly reduced ERG binding (Fig. 2C), indicating that ERG can interact with ETS sites 1 and 2 with comparable affinity. Likewise, we observed that mutation of either ETS site 6 or 7 reduced ERG binding to the 32P-labeled '6/7' oligonucleotide (Fig. 2C). However, whereas mutation of ETS site 7 somewhat reduced DNA-binding, mutation of ETS site 6 completely abolished DNA-binding, suggesting that ERG binding to ETS site 7 is dependent on the integrity of ETS site 6. Lastly, we assessed the specificity of the observed DNA-binding. To this end, we made use of the unlabeled E74 oligonucleotide. An excess of this oligonucleotide suppressed binding to the 32P-labeled '1/2' and '6/7' oligonucleotides (Fig. 2D). In contrast, a mutated E74 oligonucleotide that no longer binds to ETS proteins was unable to compete for binding. In conclusion, our data show that ERG can directly bind to several ETS sites within the human YAP1 gene promoter.
Importance of ETS sites 6 and 7
Next, we started to evaluate which of the ERG binding sites in the YAP1 promoter are crucial for its activity. First, we employed promoter truncations. The −180/+22 truncation, in which ETS sites 1–3 become deleted, and the −145/+22 truncation, in which additionally ETS sites 4 and 5 become removed, were at least as active as the longest YAP1 promoter (−390/+22) fragment in the absence or presence of ectopic ERG in the VCaP prostate cancer cells (Fig. 3A), suggesting that ETS sites 1–5 are not important for ERG-dependent YAP1 promoter upregulation. When ETS sites 1–7 were removed in the −62/+22 promoter construct, promoter activity was vastly reduced, suggesting that ETS sites 6 and 7 are crucial for YAP1 promoter activity. This would be consistent with the fact that ETS sites 6 and 7 were most avidly bound by ERG as shown above.
Figure 3.

Impact of ERG binding sites on YAP1 promoter activity. (A) Indicated YAP1 gene promoter luciferase constructs were cotransfected with 5 ng empty vector or ERG expression plasmid into VCaP prostate cancer cells and luciferase activities were determined (averages with standard deviations). The top shows a scheme of the YAP1 promoter with the location of the eight ETS sites. (B) VCaP cells transfected with the wild-type YAP1 (−390/+22) luciferase reporter construct or indicated ETS site mutations thereof. Luciferase activities (averages with standard deviations) in the presence of 5 ng cotransfected empty vector or ERG expression plasmid are presented.
To corroborate this, we mutated ETS sites in the −390/+22 YAP1 promoter construct. Neither mutation of ETS site 1 or 2 resulted in decreased promoter activity, and even joint mutation of these two ETS sites had no significant effect (Fig. 3B). In contrast, mutation of ETS site 6 or 7 reduced YAP1 promoter activity. Joint mutation of ETS site 6 and 7 resulted in even more reduction of transcription: ERG was only able to increase luciferase activity by ~1/3, compared to 2.5-fold for the wild-type promoter. Lastly, joint mutation of ETS sites 1, 2, 6 and 7 was no different from mutation of ETS sites 6 and 7 (Fig. 3B). We conclude that ETS sites 6 and 7 are crucial for ERG-dependent stimulation of YAP1 promoter transcription.
Cooperation between ERG and KDM4A
Since our laboratory is interested in the (de)methylation of histone H3 on lysine residues, we analyzed how ERG would affect trimethylation on four prominent H3 lysine residues. For this, we utilized human embryonic kidney 293T cells, since: i) they can be efficiently transfected in contrast to VCaP cells; and ii) they also display activation of the YAP1 promoter by ERG (Fig. 4A). Moreover, we observed that ETS sites 6 and 7 were crucial for ERG-dependent transcription in 293T cells, while mutation of ETS sites 1 and 2 had, in contrast to VCaP cells (compare to Fig. 3B), also a small effect (Fig. 4A). This suggests that in some cell lines, ETS sites 1 and 2 may contribute to ERG-dependent YAP1 upregulation. Utilizing chromatin immunoprecipitation assays, we observed that expression of ERG led to no significant changes of trimethylation on histone H3 lysines K4, K27 and K36, but K9me3 levels were reduced on the YAP1 luciferase reporter (Fig. 4B). H3K9me3 is normally a marker for transcriptional repression (43), thus, it would be consistent that its removal contributes to ERG-mediated transcriptional activation.
Figure 4.
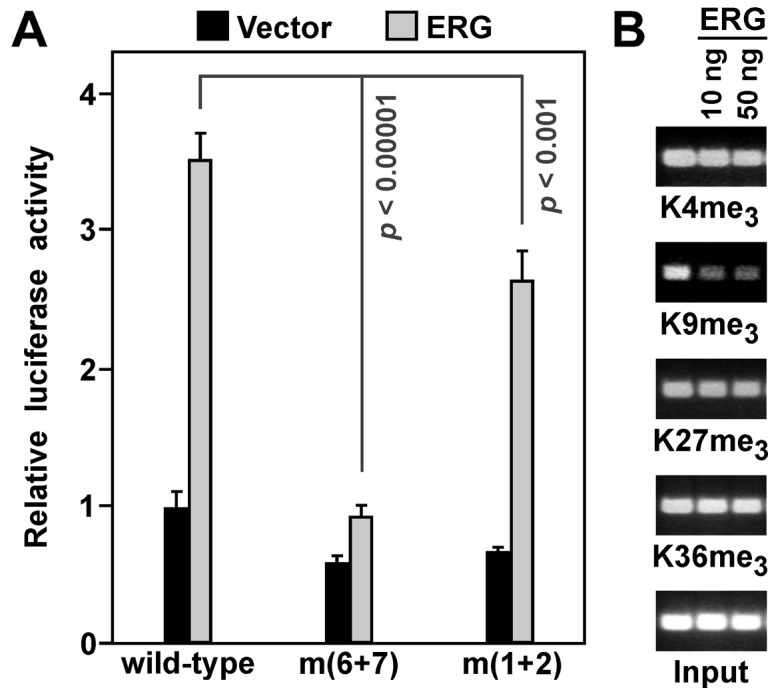
(A) Activation of wild-type or mutated YAP1 (−390/+22) luciferase reporter constructs by ERG in 293T cells. Shown are averages of luciferase activities with standard deviations. (B) Chromatin immunoprecipitation assay with 293T cells transiently transfected with ERG and a YAP1 luciferase reporter plasmid.
This result led us to speculate that ERG may recruit a histone demethylase targeting trimethylated H3K9. Only one subclass of histone demethylases, the KDM4 proteins, is known for demethylating H3K9me3 (44). Hence, we focused on the protagonist of this family, KDM4A (45), and tested whether it would interact with ERG. To this end, we coexpressed Myc-tagged ERG and HA-tagged KDM4A in 293T cells and observed that KDM4A coprecipitated with ERG (Fig. 5A). Moreover, we produced a GST-ERG fusion protein in bacteria and challenged it with KDM4A purified from baculovirus. Whereas GST-ERG bound KDM4A, GST did not (Fig. 5B), indicating that ERG and KDM4A can directly bind to each other.
Figure 5.

Interaction of KDM4A with ERG. (A) Myc-tagged ERG and HA-tagged KDM4A were coexpressed in 293T cells. After anti-Myc immunoprecipitation (IP), any coprecipitated KDM4A was revealed by anti-HA western blotting (top panel). The bottom two panels show input levels of HA-KDM4A and Myc-ERG. (B) Binding of purified Flag-tagged KDM4A to purified GST-ERG. Top panels show retention of Flag-KDM4A on glutathione beads loaded with GST or GST-ERG, whereas bottom panels demonstrate the purity and amount of utilized proteins by Coomassie staining of protein gels.
We then determined whether KDM4A would cooperate with ERG in activating YAP1 gene transcription. On its own, KDM4A had a modest impact on YAP1 luciferase activity in VCaP cells, but combined with ERG it caused a synergistic activation of the YAP1 promoter (Fig. 6). We also employed a catalytically inactive KDM4A protein, the H188A point mutant (46,47). This mutant was less active compared to wild-type KDM4A, yet still significantly raised ERG-mediated YAP1 luciferase activity (Fig. 6). These data suggest that KDM4A is a coactivator that stimulates ERG in a manner dependent and independent of its catalytic activity.
Figure 6.

Transcriptional cooperation between ERG and KDM4A. VCaP prostate cancer cells were transfected with the YAP1 (−390/+22) luciferase reporter construct, 1 ng vector or ERG expression plasmid, and 5 ng empty vector pEV3S or Flag-tagged KDM4A. Shown luciferase activities are averages with standard deviations.
Relationship between ERG/KDM4A and YAP1 in VCaP cells
Next, we wished to confirm that YAP1 is a target gene of ERG and KDM4A in VCaP prostate cancer cells. To this end, we downregulated either ERG or KDM4A with two different shRNAs and observed that YAP1 protein levels were reduced (Fig. 7A and B, top panels). This suggested that both ERG and KDM4A are required for maximal YAP1 gene transcription in VCaP cells.
Figure 7.

Impact of ERG, KDM4A and YAP1 on VCaP prostate cancer cells. (A) Cell growth (averages with standard deviations) upon downregulation of ERG with two different shRNAs. The top panels show western blot analyses of ERG, actin or YAP1. (B and C) Analogous for downregulation of KDM4A or YAP1. *p<0.05; **p<0.01; #p<0.005; ##p<0.001.
Furthermore, we assayed VCaP cell growth upon ERG and KDM4A downregulation. As previously reported (48,49), ERG knockdown led to a robust decrease in VCaP cell growth (Fig. 7A, bottom panel). Notably, the same was observed upon KDM4A knockdown (Fig. 7B, bottom panel), highlighting a role of KDM4A in cell proliferation. We then reasoned that whether YAP1 is a seminal downstream target of both ERG and KDM4A, its downregulation should phenocopy the observed reduction in cell growth upon ERG/KDM4A knockdown. In addition, indeed, we observed that YAP1 was required for maximal VCaP cell proliferation (Fig. 7C).
Discussion
In the present study, we uncovered a new mechanism by which ERG may exert its oncogenic function. This mechanism involves a physical interaction of ERG with the histone demethylase KDM4A that could lead to pleiotropic changes in the transcriptome, including an upregulation of YAP1 gene transcription. Since ERG overexpression is found in approximately half of all prostate tumor patients (4), our findings particularly pertain to prostatic malignancies.
YAP1 is a transcriptional cofactor that can be recruited to chromatin by several DNA-binding proteins. Frequently, YAP1 expression is enhanced in various human tumors and may correlate with poor prognosis, and its oncogenic potential was confirmed both in vitro as well as in transgenic mouse models (12,13). However, recent studies suggest that YAP1 may also exert growth suppressive actions in the colon and hematological cancers (50,51), suggesting that YAP1 context-dependently acts as an oncogene or tumor suppressor. However, the fact that YAP1 is overexpressed in human prostate tumors (52) indicates that it functions as an oncogene in this organ, which is consistent with prostate-specific overexpression of YAP1 leading to the development of prostatic neoplasias in mice (7). All this stresses that YAP1 may serve as a target for therapy particularly in ERG-overexpressing prostate tumors. Notably, small molecules as well as a peptide that suppress YAP1 function have been identified (53,54), which could be harnessed for future avenues of therapeutic interference. A caveat is that our report does not establish whether YAP1 is the only crucial downstream effector of ERG. Given that ERG downregulation seems to be more detrimental to VCaP cell proliferation than YAP1 downregulation (see Fig. 7), it is likely that YAP1 upregulation is not the sole reason why ERG overexpression induces prostate tumors. Yet, even partially blunting ERG's oncogenic potential through YAP1 inhibition would still have therapeutic value.
KDM4A is the protagonist of the KDM4 family of histone demethylases that are encoded by six different genes in the human genome (45,55). It is particularly competent in demethylating trimethylated lysine 9 on histone H3 and lysine 26 on histone H1.4 that are regarded as repressive chromatin marks (46,47,56,57). Accordingly, KDM4A may function as a transcriptional coactivator at least in part by removing these repressive marks. However, we observed that catalytically inactive KDM4A was still capable, albeit at a much reduced rate compared to wild-type KDM4A, to cooperate with ERG in stimulating the YAP1 promoter. This suggests that KDM4A coactivates ERG both in a manner dependent on and independent of its catalytic activity. Likewise, Drosophila KDM4A has been shown to often affect gene transcription independent of its catalytic activity (58) and also mammalian KDM4A can impact DNA repair without involving its catalytic activity (59), corroborating that KDM4A may act both as an enzyme and in non-enzymatic ways.
However, in case of stimulating ERG, our data suggest that KDM4A is mostly acting through its enzymatic activity. If so, inhibition of its catalytic center may prove beneficial in the treatment of prostate cancer patients that are afflicted by an ERG chromosomal translocation. Several small molecules have been uncovered that can inhibit KDM4A enzymatic activity (60–66). However, the specificity of these inhibitors, their selectivity for suppressing tumor vs. normal cells, their toxicity, pharmacokinetics and pharmacodynamics need to be further explored before any of these inhibitors can enter clinical trials.
Similar to ERG, KDM4A seems to be overexpressed in prostate tumors (67), which would be alike to breast and lung tumors that display overexpression of KDM4A (68–71). This may suggest that KDM4A is oncogenic in its own right in the prostate, breast or lung. Furthermore, it is unlikely that KDM4A exclusively promotes prostate tumorigenesis as a coactivator of ERG. For instance, KDM4A can also stimulate the androgen receptor or repress the p53 tumor suppressor thereby leading to abnormal cell growth (72,73). Moreover, KDM4A is capable of inducing copy number gains in cells, which may represent another mechanism by which it contributes to the development of cancer (74).
In conclusion, the present study has provided more mechanistic insight into how ERG overexpression due to chromosomal translocations can induce prostate cancer formation. Despite its obvious validity as a drug target in prostate cancer, no effective ERG inhibitors have surfaced in the clinic, which may be due to the difficulty of targeting a DNA-binding transcription factor. The present study suggests two alternative targets to blunt the ERG oncogenic activity, KDM4A and YAP1, both of which can in principal be inhibited by small molecules and may therefore merit more research.
Acknowledgments
The present study was in part funded by a grant from the National Institutes of Health/National Cancer Institute (R01 CA154745) to R.J. The content is solely the responsibility of the authors and does not necessarily represent the official views of the granting agencies.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Ryan CJ, Tindall DJ. Androgen receptor rediscovered: The new biology and targeting the androgen receptor therapeutically. J Clin Oncol. 2011;29:3651–3658. doi: 10.1200/JCO.2011.35.2005. [DOI] [PubMed] [Google Scholar]
- 3.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 4.Clark JP, Cooper CS. ETS gene fusions in prostate cancer. Nat Rev Urol. 2009;6:429–439. doi: 10.1038/nrurol.2009.127. [DOI] [PubMed] [Google Scholar]
- 5.Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, True LD, Nelson PS, Vasioukhin V. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci USA. 2008;105:2105–2110. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, Cao Q, Prensner JR, Rubin MA, Shah RB, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–188. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen LT, Tretiakova MS, Silvis MR, Lucas J, Klezovitch O, Coleman I, Bolouri H, Kutyavin VI, Morrissey C, True LD, et al. ERG activates the YAP1 transcriptional program and induces the development of age-related prostate tumors. Cancer Cell. 2015;27:797–808. doi: 10.1016/j.ccell.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, Alimonti A, Nardella C, Varmeh S, Scardino PT, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–624. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, Taylor BS, Sander C, Cardiff RD, Couto SS, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–526. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y, Kim J, Nguyen M, Zhang X, Godinho FJ, et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013;27:683–698. doi: 10.1101/gad.211011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, Gao D, Sirota I, Carver BS, Wongvipat J, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med. 2013;19:1023–1029. doi: 10.1038/nm.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 13.Varelas X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development. 2014;141:1614–1626. doi: 10.1242/dev.102376. [DOI] [PubMed] [Google Scholar]
- 14.Dowdy SC, Mariani A, Janknecht R. HER2/Neu- and TAK1-mediated up-regulation of the transforming growth factor beta inhibitor Smad7 via the ETS protein ER81. J Biol Chem. 2003;278:44377–44384. doi: 10.1074/jbc.M307202200. [DOI] [PubMed] [Google Scholar]
- 15.Mooney SM, Grande JP, Salisbury JL, Janknecht R. Sumoylation of p68 and p72 RNA helicases affects protein stability and transactivation potential. Biochemistry. 2010;49:1–10. doi: 10.1021/bi901263m. [DOI] [PubMed] [Google Scholar]
- 16.Oh S, Shin S, Lightfoot SA, Janknecht R. 14-3-3 proteins modulate the ETS transcription factor ETV1 in prostate cancer. Cancer Res. 2013;73:5110–5119. doi: 10.1158/0008-5472.CAN-13-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Janknecht R. Regulation of the ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1 and protein kinase A. J Biol Chem. 2002;277:42669–42679. doi: 10.1074/jbc.M205501200. [DOI] [PubMed] [Google Scholar]
- 18.Papoutsopoulou S, Janknecht R. Phosphorylation of ETS transcription factor ER81 in a complex with its coactivators CREB-binding protein and p300. Mol Cell Biol. 2000;20:7300–7310. doi: 10.1128/MCB.20.19.7300-7310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Haro L, Janknecht R. Cloning of the murine ER71 gene (Etsrp71) and initial characterization of its promoter. Genomics. 2005;85:493–502. doi: 10.1016/j.ygeno.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 20.DiTacchio L, Bowles J, Shin S, Lim DS, Koopman P, Janknecht R. Transcription factors ER71/ETV2 and SOX9 participate in a positive feedback loop in fetal and adult mouse testis. J Biol Chem. 2012;287:23657–23666. doi: 10.1074/jbc.M111.320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Haro L, Janknecht R. Functional analysis of the transcription factor ER71 and its activation of the matrix metal-loproteinase-1 promoter. Nucleic Acids Res. 2002;30:2972–2979. doi: 10.1093/nar/gkf390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mooney SM, Goel A, D'Assoro AB, Salisbury JL, Janknecht R. Pleiotropic effects of p300-mediated acetylation on p68 and p72 RNA helicase. J Biol Chem. 2010;285:30443–30452. doi: 10.1074/jbc.M110.143792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knebel J, De Haro L, Janknecht R. Repression of transcription by TSGA/Jmjd1a, a novel interaction partner of the ETS protein ER71. J Cell Biochem. 2006;99:319–329. doi: 10.1002/jcb.20945. [DOI] [PubMed] [Google Scholar]
- 24.Shin S, Janknecht R. Concerted activation of the Mdm2 promoter by p72 RNA helicase and the coactivators p300 and P/CAF. J Cell Biochem. 2007;101:1252–1265. doi: 10.1002/jcb.21250. [DOI] [PubMed] [Google Scholar]
- 25.Goel A, Janknecht R. Acetylation-mediated transcriptional activation of the ETS protein ER81 by p300, P/CAF, and HER2/Neu. Mol Cell Biol. 2003;23:6243–6254. doi: 10.1128/MCB.23.17.6243-6254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goel A, Janknecht R. Concerted activation of ETS protein ER81 by p160 coactivators, the acetyltransferase p300 and the receptor tyrosine kinase HER2/Neu. J Biol Chem. 2004;279:14909–14916. doi: 10.1074/jbc.M400036200. [DOI] [PubMed] [Google Scholar]
- 27.Berry WL, Kim TD, Janknecht R. Stimulation of β-catenin and colon cancer cell growth by the KDM4B histone demethylase. Int J Oncol. 2014;44:1341–1348. doi: 10.3892/ijo.2014.2279. [DOI] [PubMed] [Google Scholar]
- 28.Janknecht R. Regulation of the ER81 transcription factor and its coactivators by mitogen- and stress-activated protein kinase 1 (MSK1) Oncogene. 2003;22:746–755. doi: 10.1038/sj.onc.1206185. [DOI] [PubMed] [Google Scholar]
- 29.Shin S, Bosc DG, Ingle JN, Spelsberg TC, Janknecht R. Rcl is a novel ETV1/ER81 target gene upregulated in breast tumors. J Cell Biochem. 2008;105:866–874. doi: 10.1002/jcb.21884. [DOI] [PubMed] [Google Scholar]
- 30.Goueli BS, Janknecht R. Regulation of telomerase reverse transcriptase gene activity by upstream stimulatory factor. Oncogene. 2003;22:8042–8047. doi: 10.1038/sj.onc.1206847. [DOI] [PubMed] [Google Scholar]
- 31.Shin S, Rossow KL, Grande JP, Janknecht R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res. 2007;67:7572–7578. doi: 10.1158/0008-5472.CAN-06-4652. [DOI] [PubMed] [Google Scholar]
- 32.Shin S, Oh S, An S, Janknecht R. ETS variant 1 regulates matrix metalloproteinase-7 transcription in LNCaP prostate cancer cells. Oncol Rep. 2013;29:306–314. doi: 10.3892/or.2012.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goueli BS, Janknecht R. Upregulation of the catalytic telomerase subunit by the transcription factor ER81 and oncogenic HER2/Neu, Ras, or Raf. Mol Cell Biol. 2004;24:25–35. doi: 10.1128/MCB.24.1.25-35.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shin S, Kim TD, Jin F, van Deursen JM, Dehm SM, Tindall DJ, Grande JP, Munz JM, Vasmatzis G, Janknecht R. Induction of prostatic intraepithelial neoplasia and modulation of androgen receptor by ETS variant 1/ETS-related protein 81. Cancer Res. 2009;69:8102–8110. doi: 10.1158/0008-5472.CAN-09-0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim TD, Oh S, Shin S, Janknecht R. Regulation of tumor suppressor p53 and HCT116 cell physiology by histone demethylase JMJD2D/KDM4D. PLoS One. 2012;7:e34618. doi: 10.1371/journal.pone.0034618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu H, Xiao Y, Zhang S, Ji S, Wei L, Fan F, Geng J, Tian J, Sun X, Qin F, et al. The Ets transcription factor GABP is a component of the hippo pathway essential for growth and antioxidant defense. Cell Reports. 2013;3:1663–1677. doi: 10.1016/j.celrep.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668–675. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hollenhorst PC, McIntosh LP, Graves BJ. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu Rev Biochem. 2011;80:437–471. doi: 10.1146/annurev.biochem.79.081507.103945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Regan MC, Horanyi PS, Pryor EE, Jr, Sarver JL, Cafiso DS, Bushweller JH. Structural and dynamic studies of the transcription factor ERG reveal DNA binding is allosterically autoinhibited. Proc Natl Acad Sci USA. 2013;110:13374–13379. doi: 10.1073/pnas.1301726110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010;29:2147–2160. doi: 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bosc DG, Goueli BS, Janknecht R. HER2/Neu-mediated activation of the ETS transcription factor ER81 and its target gene MMP-1. Oncogene. 2001;20:6215–6224. doi: 10.1038/sj.onc.1204820. [DOI] [PubMed] [Google Scholar]
- 42.Janknecht R, Monté D, Baert JL, de Launoit Y. The ETS-related transcription factor ERM is a nuclear target of signaling cascades involving MAPK and PKA. Oncogene. 1996;13:1745–1754. [PubMed] [Google Scholar]
- 43.Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol Cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- 45.Berry WL, Janknecht R. KDM4/JMJD2 histone demeth-ylases: Epigenetic regulators in cancer cells. Cancer Res. 2013;73:2936–2942. doi: 10.1158/0008-5472.CAN-12-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 47.Shin S, Janknecht R. Diversity within the JMJD2 histone demethylase family. Biochem Biophys Res Commun. 2007;353:973–977. doi: 10.1016/j.bbrc.2006.12.147. [DOI] [PubMed] [Google Scholar]
- 48.Sun C, Dobi A, Mohamed A, Li H, Thangapazham RL, Furusato B, Shaheduzzaman S, Tan SH, Vaidyanathan G, Whitman E, et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene. 2008;27:5348–5353. doi: 10.1038/onc.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Cai Y, Yu W, Ren C, Spencer DM, Ittmann M. Pleiotropic biological activities of alternatively spliced TMPRSS2/ERG fusion gene transcripts. Cancer Res. 2008;68:8516–8524. doi: 10.1158/0008-5472.CAN-08-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature. 2013;493:106–110. doi: 10.1038/nature11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cottini F, Hideshima T, Xu C, Sattler M, Dori M, Agnelli L, ten Hacken E, Bertilaccio MT, Antonini E, Neri A, et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat Med. 2014;20:599–606. doi: 10.1038/nm.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. 2014;25:166–180. doi: 10.1016/j.ccr.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 55.Labbé RM, Holowatyj A, Yang ZQ. Histone lysine demeth-ylase (KDM) subfamily 4: Structures, functions and therapeutic potential. Am J Transl Res. 2013;6:1–15. [PMC free article] [PubMed] [Google Scholar]
- 56.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 57.Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the Jumonji domain-containing JMJD2/KDM4 proteins. J Biol Chem. 2009;284:8395–8405. doi: 10.1074/jbc.M807818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crona F, Dahlberg O, Lundberg LE, Larsson J, Mannervik M. Gene regulation by the lysine demethylase KDM4A in Drosophila. Dev Biol. 2013;373:453–463. doi: 10.1016/j.ydbio.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 59.Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, Richard S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31:1865–1878. doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hamada S, Kim TD, Suzuki T, Itoh Y, Tsumoto H, Nakagawa H, Janknecht R, Miyata N. Synthesis and activity of N-oxalylglycine and its derivatives as Jumonji C-domain-containing histone lysine demethylase inhibitors. Bioorg Med Chem Lett. 2009;19:2852–2855. doi: 10.1016/j.bmcl.2009.03.098. [DOI] [PubMed] [Google Scholar]
- 61.Hamada S, Suzuki T, Mino K, Koseki K, Oehme F, Flamme I, Ozasa H, Itoh Y, Ogasawara D, Komaarashi H, et al. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of jumonji domain-containing protein 2 histone demethylase inhibitors. J Med Chem. 2010;53:5629–5638. doi: 10.1021/jm1003655. [DOI] [PubMed] [Google Scholar]
- 62.Rose NR, Woon EC, Kingham GL, King ON, Mecinović J, Clifton IJ, Ng SS, Talib-Hardy J, Oppermann U, McDonough MA, et al. Selective inhibitors of the JMJD2 histone demethylases: Combined nondenaturing mass spectrometric screening and crystallographic approaches. J Med Chem. 2010;53:1810–1818. doi: 10.1021/jm901680b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.King ON, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, Quinn AM, Rai G, Mott BT, Beswick P, et al. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS One. 2010;5:e15535. doi: 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo X, Liu Y, Kubicek S, Myllyharju J, Tumber A, Ng S, Che KH, Podoll J, Heightman TD, Oppermann U, et al. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J Am Chem Soc. 2011;133:9451–9456. doi: 10.1021/ja201597b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, Ruiz J, Bruick R, Peña-Llopis S, Xu J, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. 2013;4:2035. doi: 10.1038/ncomms3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim TD, Fuchs JR, Schwartz E, Abdelhamid D, Etter J, Berry WL, Li C, Ihnat MA, Li PK, Janknecht R. Pro-growth role of the JMJD2C histone demethylase in HCT-116 colon cancer cells and identification of curcuminoids as JMJD2 inhibitors. Am J Transl Res. 2014;6:236–247. [PMC free article] [PubMed] [Google Scholar]
- 67.Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 68.Patani N, Jiang WG, Newbold RF, Mokbel K. Histone-modifier gene expression profiles are associated with pathological and clinical outcomes in human breast cancer. Anticancer Res. 2011;31:4115–4125. [PubMed] [Google Scholar]
- 69.Berry WL, Shin S, Lightfoot SA, Janknecht R. Oncogenic features of the JMJD2A histone demethylase in breast cancer. Int J Oncol. 2012;41:1701–1706. doi: 10.3892/ijo.2012.1618. [DOI] [PubMed] [Google Scholar]
- 70.Slee RB, Steiner CM, Herbert BS, Vance GH, Hickey RJ, Schwarz T, Christan S, Radovich M, Schneider BP, Schindelhauer D, et al. Cancer-associated alteration of pericentromeric heterochromatin may contribute to chromosome instability. Oncogene. 2012;31:3244–3253. doi: 10.1038/onc.2011.502. [DOI] [PubMed] [Google Scholar]
- 71.Mallette FA, Richard S. JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell Reports. 2012;2:1233–1243. doi: 10.1016/j.celrep.2012.09.033. [DOI] [PubMed] [Google Scholar]
- 72.Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun. 2007;359:742–746. doi: 10.1016/j.bbrc.2007.05.179. [DOI] [PubMed] [Google Scholar]
- 73.Kim TD, Shin S, Berry WL, Oh S, Janknecht R. The JMJD2A demethylase regulates apoptosis and proliferation in colon cancer cells. J Cell Biochem. 2012;113:1368–1376. doi: 10.1002/jcb.24009. [DOI] [PubMed] [Google Scholar]
- 74.Black JC, Manning AL, Van Rechem C, Kim J, Ladd B, Cho J, Pineda CM, Murphy N, Daniels DL, Montagna C, et al. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell. 2013;154:541–555. doi: 10.1016/j.cell.2013.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]