

Abstract
Allergic disease affects millions. Despite many advances in our understanding of the immune system in the past century, the physiologic underpinning for the existence of allergy remains largely mysterious. Food allergies, in particular, have increased dramatically in recent years, adding a new sense of urgency to unraveling this mystery. The concurrence of significant lifestyle changes in Western societies with increasing disease prevalence implies a causal link. Demographic variables that influence the composition and function of the commensal microbiota early in life seem to be most important. Identifying the evolutionary and physiologic foundations of allergic disease and defining what about our modern environment is responsible for its increased incidence will provide insights critical to the development of new approaches to prevention and treatment.
Introduction
The prevalence of allergic disease has climbed steadily during the past fifty years (Asher et al., 2006; Okada et al., 2010). Its clinical presentation often follows an ordered developmental progression (atopic dermatitis, food allergy, asthma, allergic rhinitis) referred to as the allergic march (Alduraywish et al., 2015). Epidemic increases in asthma prevalence were the first to gain notice (Eder et al., 2006; Masoli et al., 2004). More recently, potentially life-threatening allergic responses to food have become an important public health concern (Prescott and Allen, 2011; Sicherer and Sampson, 2014). Nut-free classrooms, virtually unheard of in earlier generations, are now commonplace. In developed countries worldwide, as many as 10% of preschool children currently suffer from food allergies (Prescott et al., 2013). Recent reports estimate that there are 15 million children and adults with food allergies in the United States alone (Branum and Lukacs, 2008; Jackson et al., 2013). While hundreds of foods can elicit an allergic response (Hefle et al., 1996), eight in particular—namely, milk, eggs, peanuts, tree nuts, wheat, soy, fish and shellfish—account for most cases (Sicherer and Sampson, 2014). Genetic susceptibility cannot explain a marked increase in prevalence in such a short time frame, suggesting that something about our modern environment is promoting allergic disease.
We begin this review with a brief history of allergy, focusing on IgE-mediated hypersensitivity, and describe emerging concepts regarding its potential physiologic roles. We then discuss recent data that point to early life as an important time where environmental context—particularly with regard to the microbiota—influences susceptibility to allergic disease. We end with some remarks on how recent findings will inform the development of novel therapeutic strategies to prevent or treat food allergy and other allergic diseases.
Why does allergy exist?
The term “allergy” was coined in 1906 by the Viennese pediatrician Baron Clemens von Pirquet from the Greek “allos” meaning other or altered and “ergon” meaning works or reaction. Von Pirquet observed that changes in reactivity occurred on subsequent exposures to an antigen. In some instances, re-exposure resulted in diminished reactivity whereas in others reactivity increased (Igea, 2013). A major advance in the understanding of food allergy occurred in 1921, when Prausnitz and Kustner determined that hypersensitivity to an antigen could be passively transferred with serum from one individual—namely, Kustner, who was allergic to fish—to another—namely, Prausnitz, who was not allergic to fish—by intradermal injection. When Prausnitz subsequently ate fish, the injection site became hot, red, and swollen (Prausnitz and Kustner, 1921). The antigens responsible for this reaction were called atopens, and the plasma factor that conferred sensitivity was known as atopic reagin (Paul, 2013). In 1966, Teruko and Kimishige Ishizaka showed that a novel class of immunoglobulin (Ig), which they designated γE-globulin, or IgE, was responsible for reaginic activity (Ishizaka and Ishizaka, 1966).
Since the discovery of IgE, much about the molecular and cellular mechanisms by which allergens elicit clinical symptoms has been elucidated. However, little insight has been gained into why allergies exist in the first place. One hypothesis prevalent in the current literature suggests that Th2 immunity evolved to facilitate the elimination of worms and parasites and that allergy represents a misfiring of this response to otherwise innocuous substances (Fitzsimmons and Dunne, 2009; Pulendran and Artis, 2012; Stetson et al., 2004). The overwhelming majority of allergens are not, however, helminths or their products. The major allergens are a diverse group for which no one structural or biological activity appears to dominate, and include proteins associated with shellfish, nuts, venoms, pollens, animal dander, and penicillin (Erwin and Platts-Mills, 2005). It is difficult to conceptualize, that within an immune system with both innate and adaptive components that can respond with astonishing precision, a subprogram exists where nearly all of the reactivity is off-target or misguided to common environmental substances. Profet articulated a more comprehensive and intriguing hypothesis that she termed the toxin hypothesis of allergy (Profet, 1991); a similar perspective was offered by Medzhitov and colleagues in 2012 (Palm et al., 2012). This hypothesis states that allergic responses are targeted to expel, and/or reduce the potential damage incurred by noxious substances, such as toxins and venoms, in addition to parasites and worms. In this view, many, if not all, allergens have noxious potential, either directly or by proxy (Palm et al., 2012). This theory predicts that the manifestation of allergy in some individuals involves excessive, and potentially tissue damaging, immune reactivity against the host. Allergic hypersensitivity may represent the immunologic cost of protection from noxious damage similar to the way that the pathology associated with severe pneumonia or sepsis is a consequence of immunologic responses against infection with microbial pathogens—both are intended to protect their hosts, but may be deadly if excessive. In each case, symptoms occur when subclinical mechanisms of pathogen or noxious substance avoidance or clearance mechanisms fail. Studies demonstrating that phospholipase A2 in bee venom assists in orchestrating Th2 immunity (Palm et al., 2013), and that anti-venom IgE provides a survival advantage against subsequent venom exposures (Marichal et al., 2013) have provided recent support for the toxin hypothesis of allergy.
While the evolutionary selection pressures at the root of allergic hypersensitivity remain to be fully explained, a deeper understanding of the physiologic role of allergy may provide insight into underlying variations in disease susceptibility on both the genetic and environmental levels. A barrier regulation hypothesis of allergy suggests that diverse barrier mechanisms including allergen exclusion and deactivation may underlie this variance. In the context of food allergy, non-food-allergic individuals may have relatively more effectual barrier immunity—thus leaving the allergic response untriggered. Allergen penetration of barriers may induce perturbations that lead to epithelial stress, which can set the stage for a Th2 response (Pulendran and Artis, 2012; Strid et al., 2011). That Th2 immunity, in turn, sets tissue repair pathways in motion provides further evidence for its tight coupling to barrier integrity (Pulendran and Artis, 2012). While it is not clear what barrier mechanisms may be defective in allergic individuals, our modern environment harbors clues. In this context, the rising prevalence of food allergies in westernized societies parallels that of other allergic and inflammatory disorders such as asthma, inflammatory bowel disease and diabetes (Thorburn et al., 2014). These diseases, and others, are increasingly associated with demographic variables uncommon in earlier generations that have become widespread in the 21st century including Caesarean birth, formula feeding, repeated exposure to antibiotics and consumption of a diet of processed foods high in fat and sugar, and low in dietary fiber (Cho and Blaser, 2012; Feehley et al., 2012). What these lifestyle practices have in common is their ability to alter the composition of the bacterial populations that live in and on our bodies.
Acquisition of a commensal microbiota
Changes that occur during infancy and early childhood seem to be particularly important. The last ten years has witnessed an explosion of information about microbial symbionts, which outnumber the cells of their eukaryotic hosts by at least ten fold (Savage, 1977). Commensal bacteria are among the best characterized, but other microbial populations including viruses, bacteriophage and fungi abound. It is clear that the influence of these microbial inhabitants on their hosts’ physiology is profound. Nowhere is this more apparent than in the regulation of immunity at the mucosal barriers that form the body’s interface with the external environment (Cho and Blaser, 2012). Characteristic commensal bacteria occupy different anatomic sites (Cho and Blaser, 2012; The Human Microbiome Project Consortium, 2012). Maintenance of homeostasis is a particular challenge in the intestines, the lumen of which contains, in addition to the many potential antigens present in food, trillions of bacteria that increase in density from its proximal to distal end (Donaldson et al., 2015). Initial colonization occurs at birth and, during natural delivery, bacteria derived from the vagina and feces provide the founder population for the neonate (Koenig et al., 2011; Pantoja-Feliciano et al., 2013). Breast milk further shapes the diversification of the microbiota by providing a rich source of secretory IgA, as well as prebiotic glycans that promote the expansion of species adapted to utilize this food source (Barile and Rastall, 2013; Coppa et al., 2004; Rogier et al., 2014). Caesarean birth and formula feeding disturb this co-evolved host–microbe developmental strategy. In infants born by Caesarean section, bacterial populations derived from the skin of the mother or caregiver predominate (Dominguez-Bello et al., 2010; Mueller et al., 2015). Formula feeding may exacerbate this effect; since our knowledge of all of the factors that influence the emergence of the neonatal microbiota is incomplete, pre or probiotic supplemented infant formulas may not fully replicate the effect of breast milk on neonatal microbial succession and stability (Guaraldi and Salvatori, 2012). The composition of intestinal bacterial communities is plastic during infancy and early childhood and continues to change rapidly in response to environmental interventions, including invasion by pathogenic microorganisms, antibiotic treatment and diet (Dominguez-Bello et al., 2011), which may, in turn, affect immune homeostasis. Not surprisingly, antibiotic use profoundly impacts these developing microbial communities; emerging epidemiological data links pre-natal and early post-natal antibiotic use to the subsequent development of atopic dermatitis (Lee et al., 2014) and cow’s milk allergy (Metsala et al., 2013).
The early life window of opportunity
Perturbations in the composition of the commensal microbiota during a neonatal window of opportunity around the time of weaning have been associated with the development of atopy in various murine models (Bashir et al., 2004; Hill et al., 2012; Olszak et al., 2012; Russell et al., 2012; Stefka et al., 2014). Recent analysis of a large prospective birth cohort supports the concept that gut dysbiosis during the first 100 days of life can influence the subsequent development of allergic disease (Arrieta et al., 2015). Bacteria in the genera Lachnospira, Veillonella, Faecalibacterium and Rothia, normally abundant, were selectively depleted in samples obtained at three months of age in infants who exhibited wheezing and positive allergen skin prick testing at twelve months when compared to controls that didn’t exhibit this clinical phenotype. This dysbiosis was no longer detectable in fecal samples obtained at the twelve-month time point (Arrieta et al., 2015; Dominguez-Bello and Blaser, 2015). Studies that predate the advent of high throughput sequencing methodologies have associated commensal dysbiosis with the pathogenesis of allergic disease (Bjorksten et al., 1999; Bjorksten et al., 2001; Kalliomaki et al., 2001). More recent work has confirmed and extended these earlier findings (Nakayama et al., 2011; Thompson-Chagoyan et al., 2011; Thompson-Chagoyan et al., 2010) (Abrahamsson et al., 2012; Azad et al., 2015; Ling et al., 2014; Nylund et al., 2015; Penders et al., 2013). Berni Canani et al showed that the intestinal microbiota of infants allergic to cow’s milk is significantly more diverse than that of healthy age-matched controls when obtained at the time of diagnosis at 4–5 months of age (Berni Canani et al., 2015). Early life changes in fecal bacterial composition and diversity present a “chicken and egg” conundrum. Do environmentally induced changes in the intestinal microbiota drive allergic disease by limiting microbial diversity and depleting populations of bacteria with barrier-protective function? Or is intestinal dysbiosis itself the consequence of allergic inflammation, as some murine model work seems to suggest (Noval Rivas et al., 2013)? These two scenarios are not mutually exclusive as they may operate at different points in space and time. In this context, the intestinal microbiota is likely to have multiple complex roles in initiating, regulating and promoting allergic sensitization.
The skin microbiome and epicutaneous sensitization
The allergic march typically manifests first in the skin, suggesting that this site may have either a particular role in initiating allergic sensitization or the lowest threshold to manifest symptoms. The skin has a site-specific microbiome that varies from region to region; different niche-specific communities populate moist or dry areas (Belkaid and Segre, 2014). In patients with atopic dermatitis, marked reductions in skin microbial diversity occur during disease flares; effective treatment restores diversity to the skin bacterial community (Kong et al., 2012). Flares are characterized by an increased abundance of pathogenic S. aureus (which accounts for the overall drop in microbial diversity). In mice, the release of δ-toxin by S. aureus induces mast cell degranulation and exacerbates allergic sensitization to a model antigen applied to tape-stripped skin (Nakamura et al., 2013). Mutations in filaggrin—a gene product required for skin epithelial barrier function—are strongly associated with atopic dermatitis in genome wide association studies (Baurecht et al., 2007; Brown et al., 2008; Filipiak-Pittroff et al., 2011; McAleer and Irvine, 2013; Palmer et al., 2006; Palmer et al., 2007; Weidinger et al., 2006). Accordingly, atopic-like skin lesions develop, and allergen priming is facilitated, in mice homozygous for a spontaneous deactivating mutation in the gene encoding filaggrin (designated Flgft/ft for flaky tail mutant) (Fallon et al., 2009). In addition, while spontaneous dermatitis appears to develop to the same degree in germ free (GF) and specific pathogen-free (SPF) Flgft/ft mice, commensal bacteria are important for IL17A expression as well as eosinophil and neutrophil recruitment to sites of inflammation in these animals (Hoff et al., 2015). In the setting of impaired epithelial barrier function, environmentally induced perturbations in skin microbial communities (particularly those that increase S. aureus abundance) may influence allergic sensitization. Clinical sensitization to peanut may be primed via the skin through the use of peanut-containing oils (Lack et al., 2003) or environmental exposure to peanut allergens in house dust (Brough et al., 2015; Brough et al., 2013). In this regard, future work promises to determine the degree to which molecular and clinical sensitization (Box 1) occurs parenterally, and is expected to identify the extent to which microbial communities influence this process.
Box 1. What is Sensitization?
Treating and curing food allergy will likely require experts in multiple fields of clinical and basic sciences. A clear language can enhance communication and facilitate scientific advances. Over the past decades, words such as sensitization, desensitization and tolerance have become confusing in the setting of dialogue between clinicians and scientists. As an attempt to align language in a mechanistic context and to foster clarity between fields, we propose that the following clarifications to the nomenclature be considered.
Molecular sensitization
The presence of IgE specific for a food allergen. Molecular sensitization is not sufficient for clinical sensitization.
Clinical sensitization
A state where allergic symptoms are expected to appear upon exposure to the allergen based on history or testing. This is established by a clinical history of unambiguous association and/or by oral food challenge in allergist’s office. This term is similar to what some refer to as being clinically reactive.
Clinical desensitization
The loss of clinical reactivity to an allergen for which one was previously clinically sensitized. In food allergy, this often occurs in the setting of an experimental desensitization program with escalating doses of the particular food. Clinical desensitization often occurs without molecular desensitization, but the desensitized state, if achieved, may require continued allergen exposure.
Molecular desensitization
The loss of measureable allergen-specific IgE.
Sustained unresponsiveness
Acquisition of persistent non-reactivity after clinical desensitization.
Tolerance
Systemic non-responsiveness to dietary antigen mediated by an immunological process that actively maintains mucosal homeostasis. Maintenance dosing is not required.
Mechanisms regulating non-responsiveness to dietary antigen
Oral tolerance can be defined as the induction of mucosal and systemic non-responsiveness to dietary antigen (Iweala and Nagler, 2006; Pabst and Mowat, 2012). In experimental models of oral tolerance, the ability of intragastric administration of soluble protein antigens to induce systemic non-responsiveness is revealed by subsequent peripheral immunization with the same antigen in adjuvant. As such, these experimental models are imperfect surrogates for the physiologic process that induces tolerance to food that they are intended to represent. The accepted paradigm has been that oral tolerance is mediated primarily by the induction of antigen specific Foxp3+ regulatory T cells (Tregs) (Pabst and Mowat, 2012). Specialized populations of dendritic cells (DC) are thought to carry antigens that are able to cross the epithelial barrier to the mesenteric lymph nodes (MLN) that drain the gut associated lymphoid tissue (GALT). In the MLN, antigen presentation to naïve T cells in the presence of TGF–β and retinoic acid induces upregulation of Foxp3 and expression of the gut homing receptors α4β7 and CCR9, allowing these newly differentiated Tregs to home back to the intestinal lamina propria (LP) to protect against subsequent allergen challenge (Hadis et al., 2011).
Restoration of tolerance has been the guiding principle behind oral (Wood and Sampson, 2014) and epicutaneous (Mondoulet et al., 2015) approaches to immunotherapy, in which small increasing doses of allergen are administered via ingestion or skin patch to patients with food allergies. The clinical efficacy of either approach is variable, for unknown reasons. Moreover there is little evidence for the ability of either mode of immunotherapy to induce long-lasting tolerance (as opposed to transient desensitization) in the absence of ongoing allergen administration (Wood and Sampson, 2014). The timing of antigen administration is clearly important; introduction of allergenic foods early in life seems to be optimal. An increasing prevalence of peanut allergy was already apparent by the year 2000, prompting the American Academy of Pediatrics to caution parents to withhold peanuts from children with a family history of atopy until age 3 (Gruchalla and Sampson, 2015). Yet, the prevalence of peanut allergy continued to rise (Branum and Lukacs, 2008; Jackson et al., 2013). A recent large-scale randomized trial (LEAP study) showed that early introduction of peanuts to high-risk infants who were not yet clinically reactive greatly reduced the incidence of peanut allergy in this population as determined by oral food challenge at five years of age (Du Toit et al., 2015). Allergy prevention by early life dietary exposure may be mechanistically distinct from oral or epicutaneous desensitization protocols administered to patients who are already clinically sensitized. While recent peanut avoidance practices in certain countries may have contributed to increased clinical sensitization to peanut, it is important to note that the delayed introduction of potentially allergenic foods cannot explain the full magnitude of a generational increase in food allergies overall.
Recent work has proposed that, in addition to the induction of antigen specific Tregs, the maintenance of tolerance to dietary antigen requires an epithelial barrier-protective response mediated by a class of mucosa-associated commensal anaerobes (Cao et al., 2014; Stefka et al., 2014). Atarashi et al originally reported that anaerobic Firmicutes, in particular the Clostridia class, are critical for the induction of Foxp3+Tregs in the intestinal LP (Atarashi et al., 2011). Both immunological and physical adaptations are required to fortify the single-cell layered intestinal epithelium from continuous bombardment by the wide variety of dietary and microbial antigens continuously present in its lumen (Cao et al., 2014; Iweala and Nagler, 2006; Nagler-Anderson, 2001). Mouse studies suggest that many of these specialized physical adaptations are regulated by IL-22; its production by RORγt+ innate lymphoid cells (ILC3) controls enterocyte proliferation, the production of mucus and secretion of the anti-microbial peptides (AMPs), such as RegIIIβ and RegIIIγ (Sabat et al., 2014). Microarray analysis of epithelial cells isolated from differentially colonized gnotobiotic mice demonstrated that Clostridia colonization selectively induces the expression of the anti-microbial peptide Reg3b, implicating activation of the IL-22/IL-23 axis (Stefka et al., 2014). Treatment of mice with neutralizing antibodies to IL-22 reduced the expression of Reg3b/g and increased Clostridia abundance (Stefka et al., 2014).
How does innate immune signaling by Clostridia regulate allergic sensitization to food? When dysbiosis was induced by antibiotic treatment of neonatal mice high concentrations of the peanut allergens Ara h 2 and Ara h 6 were detected in serum upon oral food challenge at one week post weaning (Stefka et al., 2014). Clostridia-induced IL-22 was necessary and sufficient to significantly reduce serum peanut allergen levels. Detection of Ara h 2 and Ara h 6 by capture ELISA indicated that these proteins reached the systemic circulation with their secondary structure (B cell epitopes) intact. Resistance to degradation may, in fact, be a distinguishing feature of food allergens (Valenta et al., 2015). The relative roles of antigen specific and bacteria induced Foxp3+Tregs in the intestinal LP has not yet been examined. The repertoire of T cell receptors detectable on Tregs in the gut is distinct from that found in peripheral sites; at least some bacteria induced Tregs are likely to recognize bacterial antigens (Lathrop et al., 2011). Other mucosa-associated bacterial populations may also stimulate innate immune signaling pathways in the host through direct cell-to-cell contact or by their secretion of metabolites. Several reports have shown that bacteria-produced short chain fatty acids (SCFAs) regulate both the proportions and functional capabilities of intestinal Tregs (Arpaia et al., 2013; Furusawa et al., 2013; Smith et al., 2013). In this context, both antigen-specific and bacteria-induced Tregs cooperate with ILC-produced IL-22 to maintain barrier homeostasis.
Education of B cells in the intestinal mucosa
IgE specific for food allergens is required (though not sufficient, see Box 1) for the development of an anaphylactic response to food. But the mechanisms for development of Ig against dietary substances, and the morphing of food protein-reactive Ig into IgE, are poorly understood. The pre-immune Ig repertoire is generated in developing bone marrow (BM) B cells through RAG-dependent DNA assembly of V, (D), and J gene segments resulting in production of IgM, which serves as the B cell receptor (BCR). Immature B cells undergo receptor editing, where continued RAG expression and V(D)J recombination leads to changes in BCR specificity, which are thought to facilitate the development of B cell tolerance (Nemazee, 2000; Yurasov and Nussenzweig, 2007). As essentially all studies examining BCR editing have utilized BCR transgenic or knock in mice—which artificially and severely limit the Ig repertoire—its role in regulating the natural repertoire is not clear. Several other poorly defined selection events also play a significant role in shaping the primary Ig repertoire during maturation through the transitional-to-mature B cell stages (Levine et al., 2000). Identifying the extent to which luminal antigens (both microbial and food) regulate these primary Ig repertoire-modifying B cell selection events may help shed light on mechanisms of molecular sensitization.
After the primary Ig repertoire is established, activated mature B cells can undergo secondary Ig diversification events, namely, IgH class switch recombination (CSR), and Ig variable region exon somatic hypermutation (SHM). The IgH constant region exons (CHs) are arranged in tandem, beginning with Cμ (that encodes the IgM constant region) at the 5′ end. CSR replaces initially expressed IgM with other isotypes such as IgG, IgE, or IgA by targeted repositioning of the alternative IgH locus constant region exons (e.g. Cγ, Cɛ, Cα), which results in deletion of intervening CHs. SHM can lead to affinity maturation through iterative rounds of mutation and selection in germinal centers (Tong and Wesemann, 2015). Allergen-reactive B cells that undergo IgH CSR to IgE have the potential to elicit symptomatic allergy. However, if these B cells remain unswitched, or undergo IgH CSR to IgG or IgA instead of IgE, molecular sensitization does not occur. IgG+ B cells may therefore house a reservoir of memory B cell specificities which can sequentially switch to IgE (He et al., 2015). Unlike IgG, CSR to IgA in mice (or IgA2 in humans), precludes that particular clone from future switching to IgE, or any other IgH isotype, because the exons encoding IgA (mice) and IgA2 (humans) occupy the 3′ most part of the IgH locus.
In terms of food allergy, the degree to which SHM is required for the development of allergen reactive B cells undergoing CSR to IgE is not known. While allergen sensitization may occur by cutaneous exposure as mentioned above, most food allergen contact likely occurs in the gut mucosa where commensal microbes are required to generate an environment that favors B cell CSR to IgA. Accordingly, GF mice have minimal IgA production, and this appears to license abnormally high levels of mucosal B cell CSR to IgE (Cahenzli et al., 2013). In this regard, an environment permissive for IgE may potentially result from insufficient stimuli from gut microbes. A non-mutually exclusive alternative is that defective barrier function may allow increased allergen access to peripheral lymphoid tissue where cognate B cell CSR to IgE may take place.
In addition to the lack of clarity regarding where IgE CSR occurs, how food allergen-reactive specificities appear within the Ig repertoire in the first place is not understood. As mentioned above, recent work supports the idea that the intestinal microbiota influences lymphocytes and their receptor repertoires (Chai et al., 2014; Lathrop et al., 2011; Wesemann et al., 2013). The first hint that contents of the gut lumen influence the primary Ig repertoire came from studies in chickens, where B cell development and diversification takes place in the bursa of Fabricius, an outpouching of epithelium within the avian hindgut (Weill and Reynaud, 1987). More recent work in chicks (Davani et al., 2014), newborn rabbits (Rhee et al., 2005), and lambs (Mutwiri et al., 1999) shows that the B cell repertoire in the GALT is positively selected toward gut luminal content early in life. The mouse small intestinal LP harbors early developing B cells that peak around weaning age, when V(D)J recombination and receptor editing processes modulate primary LP Ig repertoires (Wesemann et al., 2013). Consistent with LP-specific receptor editing, RAG-expressing LP B-lineage cells have similar VH repertoires, but significantly different Vκ repertoires, when compared to those of Rag2-expressing BM counterparts. Colonization of GF mice results in an increased ratio of Igλ- versus Igκ-expressing B cells specifically in the LP, indicating that microbial antigens and/or their products may drive early mucosal B cell selection (Wesemann et al., 2013). The degree to which positive versus negative selection takes place at the receptor editing stage in the setting of a polyclonal repertoire remains to be determined. In addition, the role of dietary versus microbial antigens in influencing selection events awaits future elucidation. In humans, B cell processes in neonatal intestine have not yet been examined, but precursor B cells have been observed in fetal intestine (Golby et al., 2002), and transitional B cells have also been seen in human adult gut tissue (Vossenkamper et al., 2013).
IgA at the Mucosal Barrier
While IgE and the IgG isotype subclasses largely function within host circulation and tissues, both IgA and IgM can bind to immunoglobulin J (joining) chain, which enables transport across the epithelium and release into the lumen (Macpherson et al., 2008). IgA is dominant when sheer amount is taken into consideration. More IgA is produced than all other IgH isotypes combined (Conley and Delacroix, 1987), and most of this IgA is secreted from mucosal tissues. Until relatively recently IgA was thought to function primarily by binding to luminal content and limiting antigen access to host tissues (referred to as immune exclusion (Pabst, 2012)). IgA may also play a role in shaping the composition of the commensal microbiota and governing the establishment of its mutualistic relationship with its host (Macpherson et al., 2015; Peterson et al., 2007). The aberrant expansion of pathobionts in mice deficient in activation induced activation-induced cytidine deaminase (which cannot undergo CSR) provided the first clue that IgA and SHM may be required to maintain commensal homeostasis (Fagarasan et al., 2002; Suzuki et al., 2004; Wei et al., 2011). Interestingly, Foxp3+ Tregs contribute to IgA selection, forming a coordinated axis to regulate and potentially diversify the composition of the microbiota in the gut (Cong et al., 2009; Kawamoto et al., 2014; Tsuji et al., 2009).
The physiologic advantage of early life B cell selection in the proximity of gut content, as discussed in the animal examples above, is not yet clear. It is possible that gut microbe-selected primary (IgM) repertoires may remain localized in the GALT and serve as a foundation for both CD4 T cell (T) independent and T dependent IgA production. Commensal microbes can elicit T independent IgA production with antibacterial specificity (Macpherson et al., 2000; Shroff et al., 1995) with contributions from both conventional (B2) and innate (B1b) B cell lineages (Bunker et al., 2015). T independent production of innate-like, polyreactive IgA may facilitate non-invasive commensal bacterial uptake into Peyer’s patches in the small intestine, enabling the production of T dependent, commensal specific IgA (Fransen et al., 2015). T independent IgA responses are also elicited by ILC3-derived lymphotoxin (LT)-α3, linking ILC3 to both innate and adaptive barrier responses (Kruglov et al., 2013). Gut microbe-primed primary Ig repertoires may thus provide a reservoir of luminal content-reactive B lineage cells as T-independent IgA precursors, distinct from the T-dependent IgA induced by toxins and invasive organisms (Pabst, 2012; Palm et al., 2014).
In terms of dietary allergen reactivity, non-IgE isotypes may also have a regulatory function. This could be particularly relevant for IgA against food proteins that resist degradation in early digestion processes. IgA coating may reduce the chance that allergens gain access to the epithelium, slowing movement in the mucus layer (Lai et al., 2009; Olmsted et al., 2001), thus inhibiting allergen contact and entry. Clinically, however, selective IgA-deficiency is a relatively common immune deficiency and is rarely associated with symptomatic disease (Hammarstrom et al., 2000), possibly due to compensating secreted IgM (Macpherson and McCoy, 2015). Although several studies do not find an association of IgA deficiency and atopy (Franco et al., 2011; Kanok et al., 1978; Plebani et al., 1987), a study of 2423 Swedish children, reported that 33% of those identified with IgA deficiency had food allergy when compared to 12% of their IgA-sufficient counterparts at 4 years of age (Janzi et al., 2009). Larger studies will be required to identify the potential role of mucosal IgA and IgM in the regulation of food allergen sensitization.
The barrier regulation hypothesis of allergic sensitization to food
The immune system can be viewed broadly as an essential component of our ability to co-exist with an environment that is abundant in both innocuous exposures and infectious or noxious threats. A complicated, multiply redundant, system with both innate and adaptive components is required to maintain homeostasis at barrier surfaces to eliminate potentially harmful foreign substances while avoiding pathological responses to commensal microbes or food. Emerging evidence is consistent with a barrier regulation hypothesis of allergic sensitization (Figure 1) which views allergy as an additional layer of protection against alien elements that somehow penetrate other layers of this system. In the healthy state, a microbiome replete with mucosa-associated taxa (like the Clostridia) that regulate the production of IL-22 stimulates the intestinal mucosa to produce a protective mucous layer and AMPs that titrate the abundance of this bacterial community. These bacteria-induced barrier protective functions reduce the ability of food allergens to cross the epithelial barrier and gain access to the systemic circulation. Early life luminal content induces an IgM and IgA response that shapes the repertoire to aid in immune exclusion. Perhaps IgA targeting of mucin degrading bacteria is also important early in life to protect the developing mucus barrier of the small intestine and limit uptake of dietary allergens. Potentially noxious (or noxious by proxy) food proteins that evade digestion may be excluded by either (or both) of these barrier mechanisms. Defects in innate or adaptive bacteria-induced barrier protective responses may exacerbate genetic predispositions (e.g. mutations in filaggrin) that render the host susceptible to allergen contact and entry and elicit direct or indirect stress in the epithelial barrier, particularly in the skin or intestinal mucosa.
Figure 1. The barrier regulation hypothesis of allergic disease.
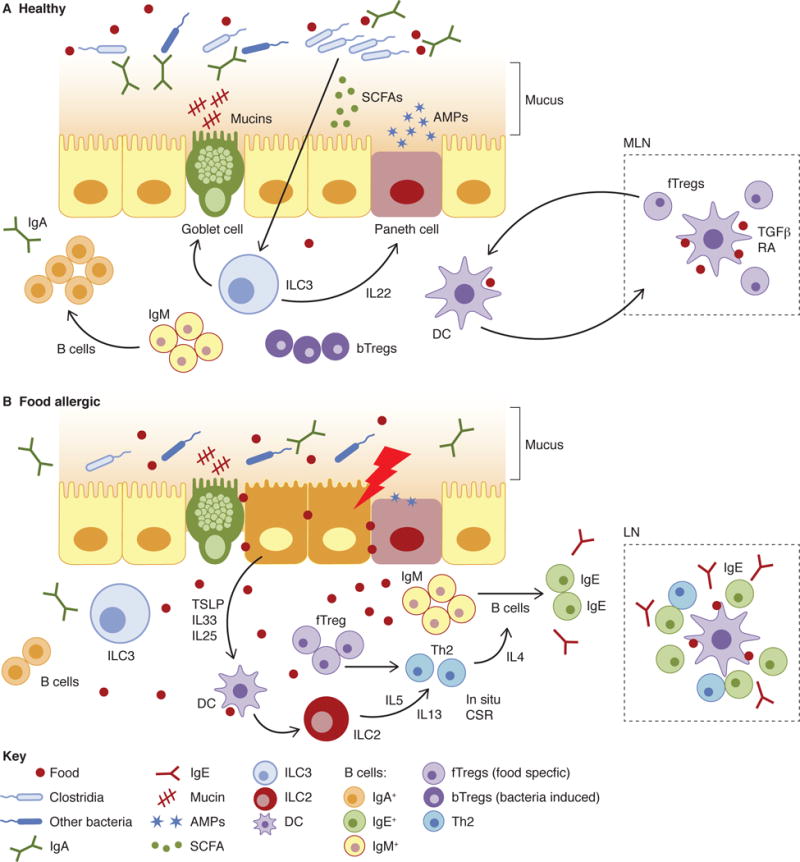
(A) In healthy individuals, both food allergen specific and bacteria-induced Tregs cooperate with ILC-derived IL-22 dependent effector functions (e.g. mucus secretion, induction of AMPs) to maintain barrier integrity and mucosal homeostasis. Allergy protective bacterial populations residing in the proximal colon may secrete metabolites or influence cellular migration from that site to regulate allergen uptake in the small intestine. (B) In food allergic subjects, the loss of these allergy protective bacterial populations impairs barrier integrity. Increased allergen contact and depletion of metabolites like SCFAs stress the epithelial layer. Both DC and ILC2 are primed by epithelial derived cytokines to induce a Th2 response and CSR to IgE, both locally and in peripheral LN.
Barrier surfaces are necessarily poised in an anti-inflammatory state. Upon allergen-mediated activation epithelial cells secrete cytokines including TSLP, IL-33, and IL-25 that educate dendritic cells and ILC2 to promote Th2 immunity (Peterson and Artis, 2014). In this context, Tregs are “reprogrammed” to the Th2 lineage (Noval Rivas et al., 2015) and ILC2 secrete IL-5 and IL-13 (Lee et al., 2015), promoting the induction of an allergic response—including B cell IgH CSR to IgE, which may occur in situ (Cahenzli et al., 2013) in addition to organized lymphoid tissue, where most CSR to IgE likely takes place (Figure 1). Allergic disease can be viewed, in this context, as a consequence of environmentally induced dysregulation of the epithelial mucosal barrier (Cahenzli et al., 2013; Hill et al., 2012; Prioult and Nagler-Anderson, 2005).
The barrier regulation hypothesis suggests that the increased amounts of food-allergen specific IgG and IgA (in addition to IgE) detectable in atopic individuals is a reflection of reduced barrier exclusion that results in increased allergen availability in the systemic circulation (Shek et al., 2005). In the absence of commensal dependent barrier regulation, GF mice have reduced mucosal production of IgA and concomitant increases in IgE production (Cahenzli et al., 2013; Herbst et al., 2011; Hill et al., 2012; McCoy et al., 2006; Stefka et al., 2014) and exhibit exaggerated responses to both aero (Herbst et al., 2011) and food (Stefka et al., 2014) allergens. Reducing the intestinal bacterial load in wild-type mice by treatment with broad-spectrum antibiotics raises the concentration of serum IgE (Hill et al., 2012). Microbial exposure early, but not late, in life is required for regulating IgE levels (Cahenzil et al., 2013) consistent with an early life window of time when luminal content maximally exerts its influences on the immune system.
Concluding Remarks and Future Perspectives
The dramatically increased incidence of allergic disease over a few decades suggests that allergic susceptibility is influenced by environmental factors—which offers new hope for effective treatments. This environmental component to sensitization indicates that the allergic state is plastic and can thus be modulated. As the factors that contribute to sensitization are discovered, innovative modifications can be engineered to reduce the burden of allergic disease. Some of the outstanding questions and challenges include the identification of the factors that license molecular sensitization to become clinical sensitization, how to achieve sustained unresponsiveness rather than transient desensitization, the effect of the environment on Ig selection, as well as IgE production, maintenance, and memory, and what regulates the allergic march.
The microbiota is one target already identified that has the potential to modulate responses at multiple points along the continuum of allergic disease. Microbiome-modulating strategies may be efficacious as either a preventive therapy (to restore functionality in the context of environmentally induced dysbiosis) or as an adjunctive treatment co-administered with orally administered allergens. Ingestion of health-promoting bacterial preparations, or probiotics, has a long history of use and study in the context of treatment of both allergic and inflammatory disease (Hill et al., 2014). Evidence for efficacy has largely been confined to early infancy; a meta-analysis of clinical trials determined that prenatal, or early post-natal, administration of probiotics reduced total IgE levels and the risk of atopic sensitization, but not asthma or wheezing (Elazab et al., 2013; Fiocchi et al., 2015). Counteracting the dysbiosis-associated dysregulation and other potential barrier-related defects that have contributed to recent increases in allergic disease is an attractive prospect for the development of a new class of microbiome-modulating therapeutic approaches (Olle, 2013).
Acknowledgments
The authors thank T. Feehley, J. Hwang, J. Boyce and A. Granato for critical review of this manuscript. The Nagler laboratory is supported by the National Institutes of Health (AI 106302), the Sunshine Foundation, UChicago Center for Translational Medicine, UChicago Digestive Diseases Research Core Center (DK42086) and F.A.R.E. (Food Allergy Research and Education). The Wesemann laboratory is supported by the National Institutes of Health (AI121394, and AI1113217). D.R.W. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and is a New Investigator Award from F.A.R.E.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahamsson TR, Jakobsson HE, Andersson AF, Bjorksten B, Engstrand L, Jenmalm MC. Low diversity of the gut microbiota in infants with atopic eczema. The Journal of allergy and clinical immunology. 2012;129:434–440. doi: 10.1016/j.jaci.2011.10.025. [DOI] [PubMed] [Google Scholar]
- Alduraywish SA, Lodge CJ, Campbell B, Allen KJ, Erbas B, Lowe AJ, Dharmage SC. The march from early life food sensitization to allergic disease: a systematic review and meta-analyses of birth cohort studies. Allergy. 2015 doi: 10.1111/all.12784. [DOI] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AS. Metabolites produced by commensal bacteria promote peripheral regulatory T cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, Kuzeljevic B, Gold MJ, Britton HM, Lefebvre DL, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Science translational medicine. 2015;7:307ra152. doi: 10.1126/scitranslmed.aab2271. [DOI] [PubMed] [Google Scholar]
- Asher MI, Montefort S, Bjorksten B, Lai CK, Strachan DP, Weiland SK, Williams H. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet. 2006;368:733–743. doi: 10.1016/S0140-6736(06)69283-0. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad MB, Konya T, Guttman DS, Field CJ, Sears MR, HayGlass KT, Mandhane PJ, Turvey SE, Subbarao P, Becker AB, et al. Infant gut microbiota and food sensitization: associations in the first year of life. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2015;45:632–643. doi: 10.1111/cea.12487. [DOI] [PubMed] [Google Scholar]
- Barile D, Rastall RA. Human milk and related oligosaccharides as prebiotics. Current opinion in biotechnology. 2013;24:214–219. doi: 10.1016/j.copbio.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Bashir ME, Louie S, Shi HN, Nagler-Anderson C. Toll-like receptor 4 signaling by intestinal microbes influences susceptibility to food allergy. J Immunol. 2004;172:6978–6987. doi: 10.4049/jimmunol.172.11.6978. [DOI] [PubMed] [Google Scholar]
- Baurecht H, Irvine AD, Novak N, Illig T, Buhler B, Ring J, Wagenpfeil S, Weidinger S. Toward a major risk factor for atopic eczema: meta-analysis of filaggrin polymorphism data. The Journal of allergy and clinical immunology. 2007;120:1406–1412. doi: 10.1016/j.jaci.2007.08.067. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science. 2014;346:954–959. doi: 10.1126/science.1260144. [DOI] [PubMed] [Google Scholar]
- Berni Canani R, Sangwan N, Stefka AT, Khan AA, Nocerino R, Aitoro R, Calignano A, Meli R, Mattace Raso G, Simeoli R, et al. Lactobacillus rhamnosus GG-supplemented formula expands butyrate producing bacteria in cow’s milk allergic infants. The ISME journal 2015. 2015 doi: 10.1038/ismej.2015.151. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorksten B, Naaber P, Sepp E, Mikelsaar M. The intestinal microflora in allergic Estonian and Swedish 2-year-old children. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 1999;29:342–346. doi: 10.1046/j.1365-2222.1999.00560.x. [DOI] [PubMed] [Google Scholar]
- Bjorksten B, Sepp E, Julge K, Voor T, Mikelsaar M. Allergy development and the intestinal microflora during the first year of life. The Journal of allergy and clinical immunology. 2001;108:516–520. doi: 10.1067/mai.2001.118130. [DOI] [PubMed] [Google Scholar]
- Branum AM, Lukacs SL. Food allergy among U.S. children: trends in prevalence and hospitalizations. NCHS data brief. 2008:1–8. [PubMed] [Google Scholar]
- Brough HA, Liu AH, Sicherer S, Makinson K, Douiri A, Brown SJ, Stephens AC, Irwin McLean WH, Turcanu V, Wood RA, et al. Atopic dermatitis increases the effect of exposure to peanut antigen in dust on peanut sensitization and likely peanut allergy. The Journal of allergy and clinical immunology. 2015;135:164–170. doi: 10.1016/j.jaci.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brough HA, Santos AF, Makinson K, Penagos M, Stephens AC, Douiri A, Fox AT, Du Toit G, Turcanu V, Lack G. Peanut protein in household dust is related to household peanut consumption and is biologically active. The Journal of allergy and clinical immunology. 2013;132:630–638. doi: 10.1016/j.jaci.2013.02.034. [DOI] [PubMed] [Google Scholar]
- Brown SJ, Relton CL, Liao H, Zhao Y, Sandilands A, Wilson IJ, Burn J, Reynolds NJ, McLean WH, Cordell HJ. Filaggrin null mutations and childhood atopic eczema: a population-based case-control study. The Journal of allergy and clinical immunology. 2008;121:940–946. doi: 10.1016/j.jaci.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunker JJ, Flynn TM, Koval JC, Shaw DG, Meisel M, McDonald BD, Ishizuka IE, Dent AL, Wilson PC, Jabri B, et al. Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity. 2015;43:541–553. doi: 10.1016/j.immuni.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahenzli J, Köller Y, Wyss M, Geuking M, McCoy K. Intestinal Microbial Diversity during Early-Life Colonization Shapes Long-Term IgE Levels. Cell Host & Microbe. 2013;14:559–570. doi: 10.1016/j.chom.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Feehley TJ, Nagler CR. The role of commensal bacteria in the regulation of sensitization to food allergens. FEBS letters. 2014;588:4258–4266. doi: 10.1016/j.febslet.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai JN, Zhou YW, Hsieh CS. T cells and intestinal commensal bacteria–ignorance, rejection, and acceptance. FEBS letters. 2014;588:4167–4175. doi: 10.1016/j.febslet.2014.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nature reviews. Genetics. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19256–19261. doi: 10.1073/pnas.0812681106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley ME, Delacroix DL. Intravascular and mucosal immunoglobulin A: two separate but related systems of immune defense? Ann Intern Med. 1987;106:892–899. doi: 10.7326/0003-4819-106-6-892. [DOI] [PubMed] [Google Scholar]
- Coppa GV, Bruni S, Morelli L, Soldi S, Gabrielli O. The first prebiotics in humans: human milk oligosaccharides. Journal of clinical gastroenterology. 2004;38:S80–83. doi: 10.1097/01.mcg.0000128926.14285.25. [DOI] [PubMed] [Google Scholar]
- Davani D, Pancer Z, Ratcliffe MJ. Ligation of surface Ig by gut-derived antigen positively selects chicken bursal and peripheral B cells. J Immunol. 2014;192:3218–3227. doi: 10.4049/jimmunol.1302395. [DOI] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Blaser MJ. Asthma: Undoing millions of years of coevolution in early life? Science translational medicine. 2015;7:307fs339. doi: 10.1126/scitranslmed.aad2741. [DOI] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Blaser MJ, Ley RE, Knight R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology. 2011;140:1713–1719. doi: 10.1053/j.gastro.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nature reviews Microbiology. 2016;14:20–32. doi: 10.1038/nrmicro3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Toit G, Roberts G, Sayre PH, Bahnson HT, Radulovic S, Santos AF, Brough HA, Phippard D, Basting M, Feeney M, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. The New England journal of medicine. 2015;372:803–813. doi: 10.1056/NEJMoa1414850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder W, Ege MJ, von Mutius E. The asthma epidemic. The New England journal of medicine. 2006;355:2226–2235. doi: 10.1056/NEJMra054308. [DOI] [PubMed] [Google Scholar]
- Elazab N, Mendy A, Gasana J, Vieira ER, Quizon A, Forno E. Probiotic administration in early life, atopy, and asthma: a meta-analysis of clinical trials. Pediatrics. 2013;132:e666–676. doi: 10.1542/peds.2013-0246. [DOI] [PubMed] [Google Scholar]
- Erwin EA, Platts-Mills TA. Allergens. Immunology and allergy clinics of North America. 2005;25:1–14. doi: 10.1016/j.iac.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. 2002;298:1424–1427. doi: 10.1126/science.1077336. [DOI] [PubMed] [Google Scholar]
- Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NE, Callanan JJ, Kawasaki H, Shiohama A, Kubo A, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nature genetics. 2009;41:602–608. doi: 10.1038/ng.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feehley T, Stefka AT, Cao S, Nagler CR. Microbial regulation of allergic responses to food. Seminars in immunopathology. 2012;34:671–688. doi: 10.1007/s00281-012-0337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipiak-Pittroff B, Schnopp C, Berdel D, Naumann A, Sedlmeier S, Onken A, Rodriguez E, Folster-Holst R, Baurecht H, Ollert M, et al. Predictive value of food sensitization and filaggrin mutations in children with eczema. The Journal of allergy and clinical immunology. 2011;128:1235–1241. doi: 10.1016/j.jaci.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Fiocchi A, Pawankar R, Cuello-Garcia C, Ahn K, Al-Hammadi S, Agarwal A, Beyer K, Burks W, Canonica GW, Ebisawa M, et al. World Allergy Organization-McMaster University Guidelines for Allergic Disease Prevention (GLAD-P): Probiotics. The World Allergy Organization journal. 2015;8:4. doi: 10.1186/s40413-015-0055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimmons CM, Dunne DW. Survival of the fittest: allergology or parasitology? Trends in parasitology. 2009;25:447–451. doi: 10.1016/j.pt.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Franco A, Parrella R, Murru F, Ames PR, Martucci F, Rotiroti G, Manfroni PV, Cioffi D, Tommasino C, Esposito V. Lack of association between IgA deficiency and respiratory atopy in young male adults. In Vivo. 2011;25:829–832. [PubMed] [Google Scholar]
- Fransen F, Zagato E, Mazzini E, Fosso B, Manzari C, El Aidy S, Chiavelli A, D’Erchia AM, Sethi MK, Pabst O, et al. BALB/c and C57BL/6 Mice Differ in Polyreactive IgA Abundance, which Impacts the Generation of Antigen-Specific IgA and Microbiota Diversity. Immunity. 2015;43:527–540. doi: 10.1016/j.immuni.2015.08.011. [DOI] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. Commensal microbe-derived butyrate induces differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- Gruchalla RS, Sampson HA. Preventing peanut allergy through early consumption–ready for prime time? The New England journal of medicine. 2015;372:875–877. doi: 10.1056/NEJMe1500186. [DOI] [PubMed] [Google Scholar]
- Guaraldi F, Salvatori G. Effect of breast and formula feeding on gut microbiota shaping in newborns. Frontiers in cellular and infection microbiology. 2012;2:94. doi: 10.3389/fcimb.2012.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, Muller W, Sparwasser T, Forster R, Pabst O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity. 2011;34:237–246. doi: 10.1016/j.immuni.2011.01.016. [DOI] [PubMed] [Google Scholar]
- Hammarstrom L, Vorechovsky I, Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID) Clinical and experimental immunology. 2000;120:225–231. doi: 10.1046/j.1365-2249.2000.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He JS, Narayanan S, Subramaniam S, Ho WQ, Lafaille JJ, Curotto de Lafaille MA. Biology of IgE production: IgE cell differentiation and the memory of IgE responses. Curr Top Microbiol Immunol. 2015;388:1–19. doi: 10.1007/978-3-319-13725-4_1. [DOI] [PubMed] [Google Scholar]
- Hefle SL, Nordlee JA, Taylor SL. Allergenic foods. Crit Rev Food Sci Nutr. 1996;36(Suppl):S69–89. doi: 10.1080/10408399609527760. [DOI] [PubMed] [Google Scholar]
- Herbst T, Sichelstiel A, Schar C, Yadava K, Burki K, Cahenzli J, McCoy K, Marsland BJ, Harris NL. Dysregulation of allergic airway inflammation in the absence of microbial colonization. American journal of respiratory and critical care medicine. 2011;184:198–205. doi: 10.1164/rccm.201010-1574OC. [DOI] [PubMed] [Google Scholar]
- Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, Morelli L, Canani RB, Flint HJ, Salminen S, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nature reviews Gastroenterology & hepatology. 2014;11:506–514. doi: 10.1038/nrgastro.2014.66. [DOI] [PubMed] [Google Scholar]
- Hill DA, Siracusa MC, Abt MC, Kim BS, Kobuley D, Kubo M, Kambayashi T, Larosa DF, Renner ED, Orange JS, et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nature medicine. 2012;18:538–546. doi: 10.1038/nm.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff S, Oyoshi MK, Macpherson A, Geha RS. The microbiota is important for IL-17A expression and neutrophil infiltration in lesional skin of Flg(ft/ft) mice. Clin Immunol. 2015;156:128–130. doi: 10.1016/j.clim.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igea JM. The history of the idea of allergy. Allergy. 2013;68:966–973. doi: 10.1111/all.12174. [DOI] [PubMed] [Google Scholar]
- Ishizaka K, Ishizaka T. Physicochemical properties of reaginic antibody. 1. Association of reaginic activity with an immunoglobulin other than gammaA- or gammaG-globulin. The Journal of allergy. 1966;37:169–185. doi: 10.1016/0021-8707(66)90091-8. [DOI] [PubMed] [Google Scholar]
- Iweala OI, Nagler CR. Immune privilege in the gut: the establishment and maintenance of non-responsiveness to dietary antigens and commensal flora. Immunol Rev. 2006;213:82–100. doi: 10.1111/j.1600-065X.2006.00431.x. [DOI] [PubMed] [Google Scholar]
- Jackson KD, Howie LD, Akinbami LJ. Trends in allergic conditions among children: United States, 1997–2011. NCHS data brief. 2013:1–8. [PubMed] [Google Scholar]
- Janzi M, Kull I, Sjoberg R, Wan J, Melen E, Bayat N, Ostblom E, Pan-Hammarstrom Q, Nilsson P, Hammarstrom L. Selective IgA deficiency in early life: association to infections and allergic diseases during childhood. Clin Immunol. 2009;133:78–85. doi: 10.1016/j.clim.2009.05.014. [DOI] [PubMed] [Google Scholar]
- Kalliomaki M, Salminen S, Arvilommi H, Kero P, Koskinen P, Isolauri E. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357:1076–1079. doi: 10.1016/S0140-6736(00)04259-8. [DOI] [PubMed] [Google Scholar]
- Kanok JM, Steinberg P, Cassidy JT, Petty RE, Bayne NK. Serum IgE levels in patients with selective IgA deficiency. Ann Allergy. 1978;41:22–23. [PubMed] [Google Scholar]
- Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, Tsutsui Y, Qin H, Honda K, Okada T, et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity. 2014;41:152–165. doi: 10.1016/j.immuni.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, Ley RE. Succession of microbial consortia in the developing infant gut microbiome. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Murray PR, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome research. 2012;22:850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglov AA, Grivennikov SI, Kuprash DV, Winsauer C, Prepens S, Seleznik GM, Eberl G, Littman DR, Heikenwalder M, Tumanov AV, Nedospasov SA. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science. 2013;342:1243–1246. doi: 10.1126/science.1243364. [DOI] [PubMed] [Google Scholar]
- Lack G, Fox D, Northstone K, Golding J. Factors associated with the development of peanut allergy in childhood. The New England journal of medicine. 2003;348:977–985. doi: 10.1056/NEJMoa013536. [DOI] [PubMed] [Google Scholar]
- Lai SK, Wang YY, Wirtz D, Hanes J. Micro- and macrorheology of mucus. Adv Drug Deliv Rev. 2009;61:86–100. doi: 10.1016/j.addr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh CS. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JB, Chen CY, Liu B, Mugge L, Angkasekwinai P, Facchinetti V, Dong C, Liu YJ, Rothenberg ME, Hogan SP, et al. IL-25 and CD4 T2 cells enhance type 2 innate lymphoid cell-derived IL-13 production, which promotes IgE-mediated experimental food allergy. The Journal of allergy and clinical immunology. 2015 doi: 10.1016/j.jaci.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Yu J, Ahn KM, Kim KW, Shin YH, Lee K, Hong SA, Jung YJ, Lee E, Yang SI, et al. Additive effect between IL-13 polymorphism and cesarean section delivery/prenatal antbiotic use on atopic dermatitis: A Birth Cohort Study (COCOA) PloS one. 2014;9:e96603. doi: 10.1371/journal.pone.0096603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MH, Haberman AM, Sant’Angelo DB, Hannum LG, Cancro MP, Janeway CA, Jr, Shlomchik MJ. A B-cell receptor-specific selection step governs immature to mature B cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:2743–2748. doi: 10.1073/pnas.050552997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Z, Li Z, Liu X, Cheng Y, Luo Y, Tong X, Yuan L, Wang Y, Sun J, Li L, Xiang C. Altered fecal microbiota composition associated with food allergy in infants. Applied and environmental microbiology. 2014;80:2546–2554. doi: 10.1128/AEM.00003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288:2222–2226. doi: 10.1126/science.288.5474.2222. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, Koller Y, McCoy KD. The bilateral responsiveness between intestinal microbes and IgA. Trends in immunology. 2015;36:460–470. doi: 10.1016/j.it.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, McCoy KD. Independence Day for IgA. Immunity. 2015;43:416–418. doi: 10.1016/j.immuni.2015.08.024. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, McCoy KD, Johansen FE, Brandtzaeg P. The immune geography of IgA induction and function. Mucosal immunology. 2008;1:11–22. doi: 10.1038/mi.2007.6. [DOI] [PubMed] [Google Scholar]
- Marichal T, Starkl P, Reber LL, Kalesnikoff J, Oettgen HC, Tsai M, Metz M, Galli SJ. A beneficial role for immunoglobulin E in host defense against honeybee venom. Immunity. 2013;39:963–975. doi: 10.1016/j.immuni.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59:469–478. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- McAleer MA, Irvine AD. The multifunctional role of filaggrin in allergic skin disease. The Journal of allergy and clinical immunology. 2013;131:280–291. doi: 10.1016/j.jaci.2012.12.668. [DOI] [PubMed] [Google Scholar]
- McCoy KD, Harris NL, Diener P, Hatak S, Odermatt B, Hangartner L, Senn BM, Marsland BJ, Geuking MB, Hengartner H, et al. Natural IgE production in the absence of MHC Class II cognate help. Immunity. 2006;24:329–339. doi: 10.1016/j.immuni.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Metsala J, Lundqvist A, Virta LJ, Kaila M, Gissler M, Virtanen SM. Mother’s and offspring’s use of antibiotics and infant allergy to cow’s milk. Epidemiology. 2013;24:303–309. doi: 10.1097/EDE.0b013e31827f520f. [DOI] [PubMed] [Google Scholar]
- Mondoulet L, Dioszeghy V, Puteaux E, Ligouis M, Dhelft V, Plaquet C, Dupont C, Benhamou PH. Specific epicutaneous immunotherapy prevents sensitization to new allergens in a murine model. The Journal of allergy and clinical immunology. 2015;9:01726–01726. doi: 10.1016/j.jaci.2014.11.028. [DOI] [PubMed] [Google Scholar]
- Mueller NT, Bakacs E, Combellick J, Grigoryan Z, Dominguez-Bello MG. The infant microbiome development: mom matters. Trends in molecular medicine. 2015;21:109–117. doi: 10.1016/j.molmed.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutwiri G, Watts T, Lew L, Beskorwayne T, Papp Z, Baca-Estrada ME, Griebel P. Ileal and jejunal Peyer’s patches play distinct roles in mucosal immunity of sheep. Immunology. 1999;97:455–461. doi: 10.1046/j.1365-2567.1999.00791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagler-Anderson C. Man the barrier! Strategic defences in the intestinal mucosa. Nat Rev Immunol. 2001;1:59–67. doi: 10.1038/35095573. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, Villaruz AE, Cheung GY, McGavin MJ, Travers JB, et al. Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature. 2013;503:397–401. doi: 10.1038/nature12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J, Kobayashi T, Tanaka S, Korenori Y, Tateyama A, Sakamoto N, Kiyohara C, Shirakawa T, Sonomoto K. Aberrant structures of fecal bacterial community in allergic infants profiled by 16S rRNA gene pyrosequencing. FEMS immunology and medical microbiology. 2011;63:397–406. doi: 10.1111/j.1574-695X.2011.00872.x. [DOI] [PubMed] [Google Scholar]
- Nemazee D. Receptor editing in B cells. Advances in immunology. 2000;74:89–126. doi: 10.1016/s0065-2776(08)60909-8. [DOI] [PubMed] [Google Scholar]
- Noval Rivas M, Burton OT, Wise P, Charbonnier LM, Georgiev P, Oettgen HC, Rachid R, Chatila TA. Regulatory T cell reprogramming toward a Th2-cell-like lineage impairs oral tolerance and promotes food allergy. Immunity. 2015;42:512–523. doi: 10.1016/j.immuni.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noval Rivas M, Burton OT, Wise P, Zhang YQ, Hobson SA, Garcia Lloret M, Chehoud C, Kuczynski J, DeSantis T, Warrington J, et al. A microbiota signature associated with experimental food allergy promotes allergic sensitization and anaphylaxis. The Journal of allergy and clinical immunology. 2013;131:201–212. doi: 10.1016/j.jaci.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylund L, Nermes M, Isolauri E, Salminen S, de Vos WM, Satokari R. Severity of atopic disease inversely correlates with intestinal microbiota diversity and butyrate-producing bacteria. Allergy. 2015;70:241–244. doi: 10.1111/all.12549. [DOI] [PubMed] [Google Scholar]
- Okada H, Kuhn C, Feillet H, Bach JF. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clinical and experimental immunology. 2010;160:1–9. doi: 10.1111/j.1365-2249.2010.04139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olle B. Medicines from microbiota. Nature biotechnology. 2013;31:309–315. doi: 10.1038/nbt.2548. [DOI] [PubMed] [Google Scholar]
- Olmsted SS, Padgett JL, Yudin AI, Whaley KJ, Moench TR, Cone RA. Diffusion of macromolecules and virus-like particles in human cervical mucus. Biophys J. 2001;81:1930–1937. doi: 10.1016/S0006-3495(01)75844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, Blumberg RS. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst O. New concepts in the generation and functions of IgA. Nat Rev Immunol. 2012;12:821–832. doi: 10.1038/nri3322. [DOI] [PubMed] [Google Scholar]
- Pabst O, Mowat AM. Oral tolerance to food protein. Mucosal immunology. 2012;5:232–239. doi: 10.1038/mi.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158:1000–1010. doi: 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, Rosenstein RK, Medzhitov R. Allergic host defences. Nature. 2012;484:465–472. doi: 10.1038/nature11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, Rosenstein RK, Yu S, Schenten DD, Florsheim E, Medzhitov R. Bee venom phospholipase A2 induces a primary type 2 response that is dependent on the receptor ST2 and confers protective immunity. Immunity. 2013;39:976–985. doi: 10.1016/j.immuni.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, Goudie DR, Sandilands A, Campbell LE, Smith FJ, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nature genetics. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- Palmer CN, Ismail T, Lee SP, Terron-Kwiatkowski A, Zhao Y, Liao H, Smith FJ, McLean WH, Mukhopadhyay S. Filaggrin null mutations are associated with increased asthma severity in children and young adults. The Journal of allergy and clinical immunology. 2007;120:64–68. doi: 10.1016/j.jaci.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Pantoja-Feliciano IG, Clemente JC, Costello EK, Perez ME, Blaser MJ, Knight R, Dominguez-Bello MG. Biphasic assembly of the murine intestinal microbiota during early development. The ISME journal. 2013;7:1112–1115. doi: 10.1038/ismej.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul WE. Fundamental Immunology. Seventh. Philadelphia, New York: Lippincott-Raven; 2013. [Google Scholar]
- Penders J, Gerhold K, Stobberingh EE, Thijs C, Zimmermann K, Lau S, Hamelmann E. Establishment of the intestinal microbiota and its role for atopic dermatitis in early childhood. The Journal of allergy and clinical immunology. 2013;132:601–607. doi: 10.1016/j.jaci.2013.05.043. [DOI] [PubMed] [Google Scholar]
- Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe. 2007;2:328–339. doi: 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- Plebani A, Monafo V, Ugazio AG, Monti C, Avanzini MA, Massimi P, Burgio GR. Comparison of the frequency of atopic diseases in children with severe and partial IgA deficiency. Int Arch Allergy Appl Immunol. 1987;82:485–486. doi: 10.1159/000234261. [DOI] [PubMed] [Google Scholar]
- Prausnitz C, Kustner H. Studien uber die Ueberempfindlichkeit. Zentralblatt fur Bakteriologie. 1921;86:160–169. [Google Scholar]
- Prescott S, Allen KJ. Food allergy: riding the second wave of the allergy epidemic. Pediatric allergy and immunology: official publication of the European Society of Pediatric Allergy and Immunology. 2011;22:155–160. doi: 10.1111/j.1399-3038.2011.01145.x. [DOI] [PubMed] [Google Scholar]
- Prescott SL, Pawankar R, Allen KJ, Campbell DE, Sinn J, Fiocchi A, Ebisawa M, Sampson HA, Beyer K, Lee BW. A global survey of changing patterns of food allergy burden in children. The World Allergy Organization journal. 2013;6:21. doi: 10.1186/1939-4551-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prioult G, Nagler-Anderson C. Mucosal immunity and allergic responses: lack of regulation and/or lack of microbial stimulation? Immunol Rev. 2005;206:204–218. doi: 10.1111/j.0105-2896.2005.00277.x. [DOI] [PubMed] [Google Scholar]
- Profet M. The function of allergy: immunological defense against toxins. The Quarterly review of biology. 1991;66:23–62. doi: 10.1086/417049. [DOI] [PubMed] [Google Scholar]
- Pulendran B, Artis D. New paradigms in type 2 immunity. Science. 2012;337:431–435. doi: 10.1126/science.1221064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee KJ, Jasper PJ, Sethupathi P, Shanmugam M, Lanning D, Knight KL. Positive selection of the peripheral B cell repertoire in gut-associated lymphoid tissues. The Journal of experimental medicine. 2005;201:55–62. doi: 10.1084/jem.20041849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogier EW, Frantz AL, Bruno ME, Wedlund L, Cohen DA, Stromberg AJ, Kaetzel CS. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:3074–3079. doi: 10.1073/pnas.1315792111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SL, Gold MJ, Hartmann M, Willing BP, Thorson L, Wlodarska M, Gill N, Blanchet MR, Mohn WW, McNagny KM, Finlay BB. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012;13:440–447. doi: 10.1038/embor.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nature reviews Drug discovery. 2014;13:21–38. doi: 10.1038/nrd4176. [DOI] [PubMed] [Google Scholar]
- Savage DC. Microbial ecology of the gastrointestinal tract. Annual review of microbiology. 1977;31:107–133. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- Shek LP, Bardina L, Castro R, Sampson HA, Beyer K. Humoral and cellular responses to cow milk proteins in patients with milk-induced IgE-mediated and non-IgE-mediated disorders. Allergy. 2005;60:912–919. doi: 10.1111/j.1398-9995.2005.00705.x. [DOI] [PubMed] [Google Scholar]
- Shroff KE, Meslin K, Cebra JJ. Commensal enteric bacteria engender a self-limiting humoral mucosal immune response while permanently colonizing the gut. Infect Immun. 1995;63:3904–3913. doi: 10.1128/iai.63.10.3904-3913.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicherer SH, Sampson HA. Food allergy: Epidemiology, pathogenesis, diagnosis, and treatment. The Journal of allergy and clinical immunology. 2014;133:291–307. doi: 10.1016/j.jaci.2013.11.020. quiz 308. [DOI] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefka AT, Feehley T, Tripathi P, Qiu J, McCoy K, Mazmanian SK, Tjota MY, Seo GY, Cao S, Theriault BR, et al. Commensal bacteria protect against food allergen sensitization. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:13145–13150. doi: 10.1073/pnas.1412008111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Voehringer D, Grogan JL, Xu M, Reinhardt RL, Scheu S, Kelly BL, Locksley RM. Th2 cells: orchestrating barrier immunity. Advances in immunology. 2004;83:163–189. doi: 10.1016/S0065-2776(04)83005-0. [DOI] [PubMed] [Google Scholar]
- Strid J, Sobolev O, Zafirova B, Polic B, Hayday A. The intraepithelial T cell response to NKG2D-ligands links lymphoid stress surveillance to atopy. Science. 2011;334:1293–1297. doi: 10.1126/science.1211250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Meek B, Doi Y, Muramatsu M, Chiba T, Honjo T, Fagarasan S. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1981–1986. doi: 10.1073/pnas.0307317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson-Chagoyan OC, Fallani M, Maldonado J, Vieites JM, Khanna S, Edwards C, Dore J, Gil A. Faecal microbiota and short-chain fatty acid levels in faeces from infants with cow’s milk protein allergy. International archives of allergy and immunology. 2011;156:325–332. doi: 10.1159/000323893. [DOI] [PubMed] [Google Scholar]
- Thompson-Chagoyan OC, Vieites JM, Maldonado J, Edwards C, Gil A. Changes in faecal microbiota of infants with cow’s milk protein allergy–a Spanish prospective case-control 6-month follow-up study. Pediatric allergy and immunology: official publication of the European Society of Pediatric Allergy and Immunology. 2010;21:e394–400. doi: 10.1111/j.1399-3038.2009.00961.x. [DOI] [PubMed] [Google Scholar]
- Thorburn AN, Macia L, Mackay CR. Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity. 2014;40:833–842. doi: 10.1016/j.immuni.2014.05.014. [DOI] [PubMed] [Google Scholar]
- Tong P, Wesemann DR. Molecular Mechanisms of IgE Class Switch Recombination. Curr Top Microbiol Immunol. 2015;388:21–37. doi: 10.1007/978-3-319-13725-4_2. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, Fagarasan S. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science. 2009;323:1488–1492. doi: 10.1126/science.1169152. [DOI] [PubMed] [Google Scholar]
- Valenta R, Hochwallner H, Linhart B, Pahr S. Food allergies: the basics. Gastroenterology. 2015;148:1120–1131. doi: 10.1053/j.gastro.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Shinkura R, Doi Y, Maruya M, Fagarasan S, Honjo T. Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nature immunology. 2011;12:264–270. doi: 10.1038/ni.1991. [DOI] [PubMed] [Google Scholar]
- Weidinger S, Illig T, Baurecht H, Irvine AD, Rodriguez E, Diaz-Lacava A, Klopp N, Wagenpfeil S, Zhao Y, Liao H, et al. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. The Journal of allergy and clinical immunology. 2006;118:214–219. doi: 10.1016/j.jaci.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Weill JC, Reynaud CA. The chicken B cell compartment. Science. 1987;238:1094–1098. doi: 10.1126/science.3317827. [DOI] [PubMed] [Google Scholar]
- Wesemann DR, Portuguese AJ, Meyers RM, Gallagher MP, Cluff-Jones K, Magee JM, Panchakshari RA, Rodig SJ, Kepler TB, Alt FW. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature. 2013;501:112–115. doi: 10.1038/nature12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood RA, Sampson HA. Oral immunotherapy for the treatment of peanut allergy: is it ready for prime time? The Journal of allergy and clinical immunology. 2014;2:97–98. doi: 10.1016/j.jaip.2013.11.010. In practice. [DOI] [PubMed] [Google Scholar]
- Yurasov S, Nussenzweig MC. Regulation of autoreactive antibodies. Curr Opin Rheumatol. 2007;19:421–426. doi: 10.1097/BOR.0b013e328277ef3b. [DOI] [PubMed] [Google Scholar]