

Abstract
Many of the widely used anticancer drugs induce dose-limiting peripheral neuropathies that undermine their therapeutic efficacy. Animal models of chemotherapy-induced painful peripheral neuropathy (CIPN) evoked by a variety of drug classes, including taxanes, vinca alkaloids, platinum-complexes, and proteasome-inhibitors, suggest that the common underlying mechanism in the development of these neuropathies is mitotoxicity in primary nerve sensory axons (PNSAs) arising from reduced mitochondrial bioenergetics [eg adenosine triphosphate (ATP) production deficits due to compromised respiratory complex I and II activity]. The causative mechanisms of this mitotoxicity remain poorly defined. However, peroxynitrite, an important pro-nociceptive agent, has been linked to mitotoxicity in several disease states and may also drive the mitotoxicity associated with CIPN. Our findings reveal that the development of mechano-hypersensitivity induced by paclitaxel, oxaliplatin, and bortezomib was prevented by administration of the peroxynitrite decomposition catalyst Mn(III) 5,10,15,20-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+) without interfering with their anti-tumor effects. Peak CIPN was associated with the nitration and inactivation of superoxide dismutase in the mitochondria, but not in the cytosol, as well as a significant decrease in ATP production within the PNSAs; all of these events were attenuated by MnTE-2-PyP5+. Our results provide continued support for the role of mitotoxicity in the development of CIPN across chemotherapeutic drug classes, and identify peroxynitrite as a key mediator in these processes, thereby providing the rationale towards development of “peroxynitrite-targeted” therapeutics for CIPN.
Keywords: Peroxynitrite, Chemotherapy-induced painful peripheral, neuropathy, Neuropathic pain, Paclitaxel, Oxaliplatin, Bortezomib, Mitochondrial dysfunction, Peripheral nerve sensory axons, Superoxide dismutase, ATP depletion
1. Introduction
Chemotherapy-induced peripheral neuropathy (CIPN) is a common dose-limiting complication of cancer chemotherapy and a frequent cause of discontinuation of what is otherwise successful therapy [49]. This chronic neuropathy is characterized by bilaterally symmetrical sensory symptoms (eg numbness, tingling, and pain) appearing in the feet, or in both the feet and hands [49], and occurs with chemotherapeutics across drug classes with distinctly different anti-tumor mechanisms, such as taxanes (eg paclitaxel), platinum-complexes (eg oxaliplatin), and proteasome-inhibitors (eg bortezomib). Recent work in rats indicates a common pathophysiology for CIPN from these agents due to a long-lasting dysfunction in mitochondria of peripheral nerve sensory axons (PNSAs) [51,53,55,56] that can be blocked by mitoprotective agents, such as acetyl-L-carnitine [53,56] and olesoxime [52], and exacerbated by mitochondrial poisons [51]. These, and other, observations have led to a peripheral mitotoxicity hypothesis for CIPN, which posits that mitotoxicity within the PNSAs gives rise to persistent energy deficits leading to dysfunction of the sodium–potassium pump and a subsequent increase in abnormal spontaneous discharge in both A- and C-fibers, as well as degeneration of the sensory afferents’ terminal arbors (intra-epidermal nerve fibers, the neuronal compartment with the highest energy requirement) [14,51,53–56]. Increased spontaneous discharge of PNSAs may then initiate pathways ultimately leading to central sensitization mechanisms, and the promotion of allodynia and hyperalgesia [12].
The triggering mechanisms of this mitotoxicity are unknown, but the potent nitroxidative species peroxynitrite (PN) may play a critical role. We have recently reported that PN contributes to the development of paclitaxel-induced neuropathic pain and that administration of a peroxynitrite decomposition catalyst (PNDC) can prevent this mechano-hypersensitivity [12]. Protein nitration by PN, the product of superoxide (SO) and nitric oxide (NO) [4], in pathological settings can lead to gain or loss of protein function [17]. Mitochondria constitute a primary locus for the formation and reactions of PN [35,46]; in excess, protein nitration by PN within the mitochondria is detrimental to normal cellular bioenergetics and has been linked to mitochondrial dysfunction in several diseases [26]. Increased levels of PN are further sustained by the PN-mediated nitration of manganese superoxide dismutase (MnSOD) at Tyr-34 via a Mn-catalyzed process that leads to its inactivation [26,27]. MnSOD is the most important mitochondrial antioxidant enzyme keeping SO [28], and thus PN, in check. As a result, nitration and inactivation of MnSOD favors a “feed-forward” mechanism that sustains elevated levels of SO/PN in mitochondria and further nitration of various mitochondrial proteins critical to sustained bioenergetics. Thus, the beneficial effects of removing PN with a PNDC in the prevention of CIPN [12] may be to protect against PNSA mitochondrial protein nitration and therefore preserve cellular bioenergetics.
The objectives of this study were to establish whether (1) PN is a common mediator of bioenergetic deficits and mitochondrial dysfunction in PNSAs during paclitaxel-, oxaliplatin-, and bortezomib-induced CIPN and, if so, whether (2) removing PN with PNDCs is mitoprotective and a beneficial approach to preventing CIPN.
2. Materials and methods
2.1. Experimental animals
Male Sprague Dawley rats (200–220 g starting weight) from Harlan Laboratories (Indianapolis, IN; Frederick, MD, breeding colony) were housed 3–4 per cage in a controlled environment (12-hour light/dark cycle) with food and water available ad libitum. All experiments were performed in accordance with the International Association for the Study of Pain and the National Institutes of Health guidelines on laboratory animal welfare, and the recommendations by Saint Louis University Institutional Animal Care and Use Committee. All experiments were conducted with the experimenters blinded to treatment conditions.
2.2. Chemotherapy-induced neuropathic pain
Paclitaxel (Parenta Pharmaceuticals, Yardley, PA) or its vehicle (Cremophor EL and 95% dehydrated ethanol in 1:1 ratio; Sigma, St. Louis, MO) was injected intraperitoneally (i.p.) in rats on 4 alternate days (D): D0, 2, 4, and 6 with a final cumulative dose of 8 mg/kg [34]. Oxaliplatin (Oncology Supply, Dothan, AL) or its vehicle (5% dextrose) was injected i.p. in rats on 5 consecutive days (D0–D4) for a final cumulative dose of 10 mg/kg. This low-dose paradigm does not cause kidney injury, as reported by Xiao et al. [53]. Bortezomib (Selleck Chemicals, Houston, TX) or its vehicle (5% Tween80, 5% ethanol in saline) was injected i.p. in rats on 5 consecutive days (D0–D4; 0.2 mg/kg) for a final cumulative dose of 1 mg/kg [56].
2.2.1. Test compounds
Mn(III) 5,10,15,20-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+) was synthesized as described previously and kindly provided by Dr. Batinic-Haberle (Duke University School of Medicine) [2]. MnTE-2-PyP5+ was always given 30 minutes before the chemotherapeutic or its vehicle. Rats were treated with paclitaxel, oxaliplatin, or bortezomib and injected subcutaneously (0.2 ml) with MnTE-2-PyP5+ or its vehicle (saline) from D0 to D15/17, with behavioral testing performed on D0, D11/D12, D16/D17, and D25.
2.3. Behavior testing
2.3.1. Mechano-allodynia
Rats were placed in elevated Plexiglass chambers (28 × 40 × 35 cm) on a wire mesh floor and allowed to acclimate for 15 minutes before measuring the mechanical paw withdrawal thresholds in grams [PWT, (g)] using calibrated von Frey (VF) filaments [Stoelting, ranging from 3.61 (0.407 g) to 5.46 (26 g) bending force] according to the “up-and-down” method [10] for paclitaxel, or with an electronic version of the VF test (dynamic plantar aesthesiometer, model 37450; Ugo Basile, Italy) with a cut-off set at 50 g for oxaliplatin and bortezomib. The development of mechano-allodynia is evidenced by a significant (P < .05) reduction in mechanical mean absolute PWT (g) at forces that failed to elicit withdrawal responses before chemotherapy treatment (D0).
2.3.2. Mechanical hyperalgesia
PWTs (g) were measured by the Randall and Sellitto paw pressure test [36] using a Ugo-Basile analgesiometer (Comerio, Italy, model 37215), which applies a linearly increasing mechanical force to the dorsum of the rat’s hind paw. The nociceptive threshold was defined as the force (g) at which the rat withdrew its paw (cut-off set at 250 g). If testing coincided with a day when rats received test substance, behavioral measurements were always taken before the injection of the test substance. Chemotherapeutic treatment results in bilateral allodynia and hyperalgesia with no differences between left and right hind PWT (g) at any time point in any group; thus, the values from both paws were averaged. Animals receiving chemotherapeutic agents in the presence or absence of the experimental test substances tested did not display signs of any toxicities, that is they exhibited normal posture, grooming, and locomotor behavior; hair coat was normal; there were no signs of piloerection or ocular porphyrin discharge; and they gained body weight normally and comparably to vehicle-treated rats.
2.4. Anti-tumor activity
The effects of MnTE-2-PyP5+ on the anti-tumor activity of oxaliplatin on human colon carcinoma cells (SW480) [9] and bortezomib on human multiple myeloma cells [Roswell Park Memorial Institute (RPMI) medium 8226] [30] were assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay adapted from a previously described assay [21,40]. Cells were cultured and assayed in their recommended media [L15 for SW480 (Sigma) or RPMI 1640 for RPMI 8226 (Sigma)] supplemented with heat-denatured 10% fetal bovine serum (HyClone; Thermo Fisher Scientific, Waltham, MA) and penicillin/streptomycin (Invitrogen, Grand Island, NY) at 37°C, 0% (SW480), or 5% carbon dioxide (RPMI 8226), and 95% humidity. Cells (3.13 × 104 cells/cm2) were cultured overnight in 24-well plates (SW480; BD Biosciences, Franklin Lakes, NJ) or 12 mm × 7.5 mm culture tube (RPMI 8226; BD Biosciences, Franklin Lakes, NJ) in complete media. This regimen yielded 60% confluency for testing. After equilibrating in fresh media (5 h), cells were treated for 48 h with MnTE-2-PyP5+ (60 μM) or its vehicle (PBS) and chemotherapeutics: oxaliplatin (0–30 μM) or its vehicle (0.01% dimethyl sulfoxide, final concentration), or bortezomib (0–100 nM) or its vehicle (0.1% dextrose, final concentration). Two naïve control wells (media only) were included as a control for the anti-tumor effects of MnTE-2-PyP5+ alone. Cell survival was assessed by incubating cells with MTT (500 μg/ml) for 75 minutes, removing media, and dissolving the resulting tetrazolium crystals in isopropanol. The tetrazolium absorption (A560–570 nm) was measured using a Glomax Multi-Detection system (Promega, Madison, WI). The anti-tumor effects of MnTE-2-PyP5+ alone were determined as % Survivability = (A560–570 nm of test well)/(mean A560–570 nm of the vehicle control well) × 100. The LC50 of each chemotherapeutic agent + MnTE-2-PyP5+ or its vehicle was determined by three-parameter non-linear analysis of % Survivability where the top and bottom plateaus were constrained using GraphPad Prism (version 5.03).
2.5. Assessment of mitochondrial abnormalities by electron microscopy
On D25, saphenous nerves were harvested and immersed in primary fixative solution within seconds of being removed from rats. Primary fixation was with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.25 containing 2% sucrose and 2 mM calcium chloride. Secondary fixation was with 1% osmium tetroxide in the same buffer. Nerves were then processed into EmBed resin following standard Electron Microscopy (EM) protocols. C-fibers and myelinated A-fibers were identified in post-stained thin sections in a JEOL 1200 transmission electron microscope and mitochondrial morphology evaluated at 20,000×. The criteria for abnormal mitochondrial ultrastructure were swelling, matrix extraction, membrane inflation (“ballooning”), and membrane rupture [14]. The scoring of normal vs abnormal mitochondria was always done blind (the grids are coded so that the scorers did not know if a given grid of sections was control or experimental), in 250 axon profiles per rat. As mitochondria can be subject to artefactual swelling and vacuolization due to poor fixation [3] we designed the primary and secondary fixative solutions to minimize such problems (the sucrose is an osmotic balancer and the calcium stabilizes membranes).
2.6. ATP assay
On the final day of treatment, the saphenous nerve was excised and placed in ice-cold mitochondrial preservation media and then prepared and tested using a technique adapted for the saphenous nerve as fully described in Zheng et al. [55]. Briefly, after the saphenous tissue was prepped in ice-cold preservation medium, the preservation medium was removed and replaced with room temperature (RT) respiration medium; the sample was incubated with substrates for complex I and II (5.0 mM glutamate and 2.5 mM malate, 5.0 mM succinate) after which a sample was taken of the media (100 μl). The sample was then stimulated with adenosine diphosphate (ADP) (1.0 mM) and another sample of the medium was taken. Samples were stored at −80°C until they could be assessed with a flash luciferin–luciferase assay (Promega Enliten ATP Assay; Promega) according to the manufacturer’s protocol. To normalize adenosine triphosphate (ATP) concentrations, the saphenous tissue preparation was homogenized and processed, and a citrate synthase (mitochondrion-specific enzyme) enzyme assay kit (Sigma) was utilized to assess the levels of citrate synthase activity within the supernatant.
2.7. Immunoprecipitation
Saphenous nerves were obtained and stored immediately at −80°C. Immunoprecipitation and Western blot analyses were performed as described [29,48]. Proteins were resolved with 4–20% (MnSOD) sodium dodecyl sulfate-polyacrylamide gel electrophoresis prior to electrophoretic transfer. Membranes were blocked for 1 h at RT in 1% bovine serum albumin in TBS-T (50 mM Tris·hydrochloric acid, pH 7.4), 150 mM sodium chloride, 0.01% Tween-20, and 0.1% thimerosal, and then probed with rabbit polyclonal anti-MnSOD (1:1000). Membranes were washed with TBS-T and visualized with horseradish peroxidase-conjugated secondary antibodies for 1 hour at RT and enhanced chemiluminescence. The relative density of the protein bands of interest were determined using the Gel Logic Pro 2200 system (Carestream Molecular Imaging, Rochester, NY) in conjunction with GL2200 PRO Acquire Software module (version 5.0.7.24). The blots were stripped and probed with a murine monoclonal anti-β-actin antibody (1:2000), and nitrated MnSOD bands were normalized to β-actin.
2.8. Measurement of mitochondrial MnSOD activities
Saphenous nerves were homogenized with 10 mM phosphate buffered saline (PBS) (pH 7.4), sonicated on ice for 1 min (20 seconds, three times), and centrifuged at 1100×g for 10 min. The superoxide dismutase (SOD) activity in the supernatants was measured, as described previously [48], by the ability to competitively inhibit xanthine–xanthine oxidase-derived superoxide reduction of nitroblue tetrazolium (NBT) to blue tetrazolium salt. Copper, zinc SOD (CuZnSOD) activity was blocked in this assay by the addition of 2 mM sodium cyanide after pre-incubation for 30 minutes. The rate of NBT reduction was monitored spectrophotometrically at 560 nm. The amount of protein required to inhibit the rate of NBT reduction by 50% was defined as one unit of enzyme activity. Enzymatic activity was expressed in units per mg of protein [48].
2.9. Determining ED50 values
The ED50 and the corresponding 95% confidence interval (95% CI) were determined by curve-fitting the % Prevention of mechano-allodynia or mechano-hyperalgesia using the least sum of squares method by a normalized three-parameter non-linear analysis using GraphPad Prism (version 5.03).
where PWT is the paw withdrawal threshold, drug is MnTE-2-PyP5+, chemo is paclitaxel, oxaliplatin, or bortezomib, and Veh is the vehicle.
2.10. Statistical analysis
Data are expressed as mean ± SD for n animals. Differences between data from the full-time course studies and in the ATP assay were analyzed by two-way repeated measures analysis of variance (ANOVA) with Bonferroni-corrected comparisons. All other biochemical data collected on D25 were analyzed by one-way ANOVA with Dunnett’s-corrected comparisons. Significant differences were defined as P < .05. All statistical analyses were performed using GraphPad Prism (version 5.03).
3. Results
3.1. Targeting PN blocks the development of chemotherapy-induced neuropathic pain
We have recently reported that PN plays a key role in the development of paclitaxel-induced neuropathic pain [12] and its removal with MnTE-2-PyP5+, a well-characterized potent mangano porphyrin-based PNDC [1,37], blocked the development of mechano-allodynia and mechano-hyperalgesia without interfering with paclitaxel’s anti-tumor effects [12]. We now extend these beneficial effects of MnTE-2-PyP5+ to CIPN that develops with two other chemotherapeutic agents, oxaliplatin and bortezomib. In agreement with previous studies [53,56], oxaliplatin- (Fig. 1A, B) and bortezomib-induced (Fig. 2A, B) mechano-allodynia and mechano-hyperalgesia developed by D11/12 (onset) and reached its peak by D16/17 at a level that was maintained throughout our observation period (D25; times relative to first chemotherapy injection). Although chemotherapy dosing is completed within a few days, we continued administration of MnTE-2-PyP5+ until peak hypersensitivity to account for the delay to symptom onset (also noted in patients) that introduces uncertainty about the timeline of relevant pathological process. Subcutaneous injections of MnTE-2-PyP5+ attenuated the development of oxaliplatin-induced neuropathic pain in a dose-dependent manner (0.3–3 mg/kg/d, n = 5–7) with ED50s on D25 of 0.6 mg/kg/d (95% CI = 0.4–0.8 mg/kg/d) and of 0.4 mg/kg/d (95% CI = 0.3–0.5 mg/kg/d) for mechano-allodynia and mechano-hyperalgesia, respectively (Fig. 1A, B). MnTE-2-PyP5+, when tested at the highest dose, was found to block paclitaxel- (Fig. 3A, B), oxaliplatin- (Fig. 1), and bortezomib-induced mechano-allodynia and hyperalgesia (Fig. 2A, B). We have previously reported that when given alone, and at the highest dose tested (10 mg/kg/d), MnTE-2-PyP5+ does not affect baseline withdrawal thresholds [12]; therefore, Vehicle + MnTE-2-PyP5+ groups were unnecessary. None of the drugs tested affected body weight, and all animals gained weight to the same extent over the course of the experiment (data not shown).
Fig. 1.
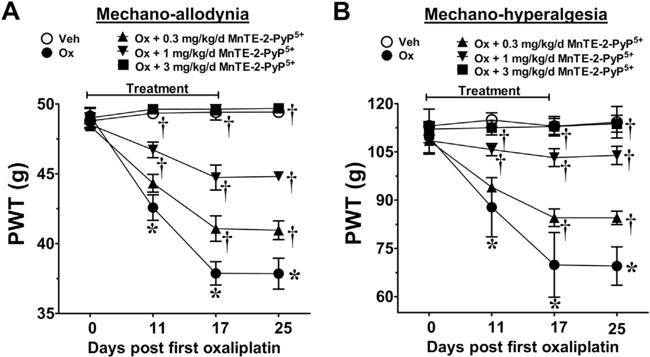
Mn(III) 5,10,15,20-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+) blocks oxaliplatin (Ox)-induced neuropathic pain in a dose-dependent manner. When compared with baseline [day (D)0], treatment with oxaliplatin (●), but not with its vehicle (Veh) (○), led to the time-dependent development of mechano-allodynia (A) and mechano-hyperalgesia (B). Daily (D0–17) injections of MnTE-2-PYP5+ (0.3 mg/kg/d, ▲; 1 mg/kg/d, ▼; 3 mg/kg/d, ■) significantly attenuated the development of oxaliplatin-induced mechano-hypersensitivity in a dose-dependent manner (A, B). Results are expressed as mean ± SD, n = 5–7 and analyzed by two-way analysis of variance with Bonferroni’s post-hoc comparisons. *P < .001 (tday vs tday 0); †P < .001 (Ox + MnTE-2-PyP5+ vs Ox). PWT = paw withdrawal threshold.
Fig. 2.
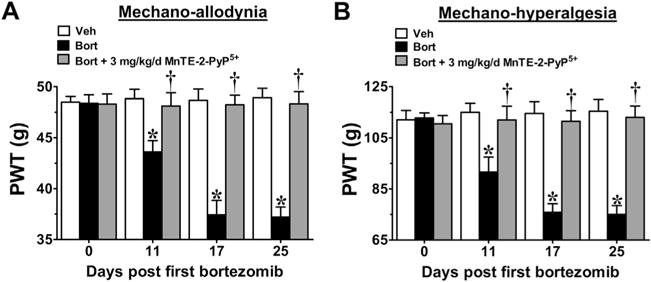
Mn(III) 5,10,15,20-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+) prevents the development of bortezomib (Bort)-induced neuropathic pain. Compared with baseline [day (D)0], administration of bortezomib (black bars), but not its vehicle (Veh) (open bars), led to the development of mechano-allodynia (A) and mechano-hyperalgesia (B). Prophylactic daily injections (D0–17) of MnTE-2-PyP5+ (3 mg/kg/d; gray bars) blocked the development of both bortezomib-induced mechano-allodynia (A) and mechano-hyperalgesia (B). Hypersensitivity did not appear after drug termination up until the end of testing on D25 (A, B). Results are expressed as mean ± SD, n = 5–6, and analyzed by two-way analysis of variance with Bonferroni’s post-hoc comparisons. *P < .001 (tday vs tday 0); †P < .001 (Bort + MnTE-2-PyP5+ vs Bort). PWT = paw withdrawal threshold.
Fig. 3.
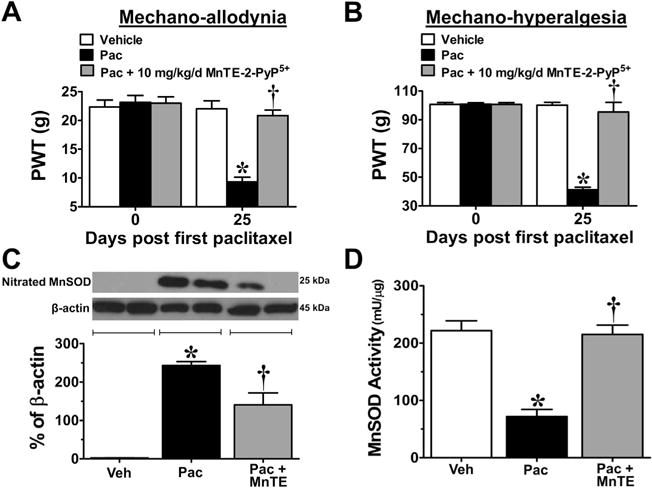
Increased nitration and inactivation of manganese superoxide (MnSOD) in peripheral nerve sensory axons (PNSAs) with paclitaxel-treatment; prevention with Mn(III) 5,10,15,20-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+). Treatment with paclitaxel (Pac) (black bars), but not vehicle (Veh) (open bars), resulted in the development of mechano-allodynia (A) and mechano-hyperalgesia (B) on day (D)25 compared with D0 and was prevented by daily (D0–16) injections of 10 mg/kg/d MnTE-2-PyP5+ (gray bars) (A,B). At peak hypersensitivity, a significant increase in the nitration of MnSOD (C) and an associated decrease in its activity (D) were observed in the PNSAs of paclitaxel-treated animals compared with vehicle; treatment with MnTE-2-PyP5+ prevented this (C,D). Representative blots are shown above the quantitative bar graph corresponding to mean ± SD for an n = 4/group (C). Results are expressed as mean ± SD, n = 4–6, and analyzed by one-way analysis of variance (ANOVA) with Dunnett’s post-hoc comparisons or two-way ANOVA with Bonferroni’s post-hoc comparisons. *P < .001 (tday vs tday 0); †P < .001 (Pac + MnTE vs Pac). PWT = paw withdrawal threshold.
3.2. MnTE-2-PyP5+ does not compromise the in vitro anti-tumor activity of oxaliplatin and bortezomib
Pharmacokinetic studies demonstrated plasma MnTE-2-PyP5+ levels in male rats when given systemically at a dose providing near-to-maximal inhibition of neuropathic pain (10 mg/kg) were approximately 17 μM at Cmax [42]. MnTE-2-PyP5+ (60 μM, a concentration three times higher than Cmax) did not diminish the anti-tumor effects of oxaliplatin in human colon cancer cells (SW480) [9] or bortezomib in human multiple myeloma cells (RPMI 8226) [30] (Table 1). Higher doses of MnTE-2-PyP5+ were not used because these doses had direct anti-tumor effects on all three cell lines tested. The presence of direct anti-tumor effects suggests the possibility that PNDCs might not only prevent CIPN, but may also enhance anti-tumor therapy.
Table 1.
Mn(III) 5,10,15,20-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+) does not interfere with the anti-tumor activity of oxaliplatin or bortezomib in vitro. Treatment of SW480 cells with oxaliplatin (Ox) or RPMI8226 cells with bortezomib (Bort) resulted in decreased cell survival in a dose-dependent manner. Co-administration of MnTE-2-PyP5+ with either oxaliplatin or bortezomib did not reduce their anti-tumor activity.
| Treatment | LC50 | 95% CI | n |
|---|---|---|---|
| Colon carcinoma cells (SW480) | |||
| Veh + Ox | 5.4 μM | 1.9–15 μM | 4 |
| Ox + MnTE-2-PyP5+ | 6.7 μM | 2.4–18 μM | 4 |
| Multiple myeloma cells (RPMI8226) | |||
| Veh + Bort | 33.2 nM | 27.2–40.5 nM | 5 |
| Bort + MnTE-2-PyP5+ | 32.9 nM | 27–40.2 nM | 5 |
Veh = vehicle; 95% CI = 95% confidence interval.
3.3. PN post-translationally nitrates mitochondrial MnSOD compromising mitochondrial bioenergetics and leading to dysfunction in PNSA
When compared with baseline (D0), development of paclitaxel-induced pain at peak plateau phase (D25; Fig. 3A, B) is associated with significant nitration (Fig. 3C) and reduced SOD activity of mitochondrial MnSOD (Fig. 3D). The activity of cytosolic CuZnSOD was not affected (Fig. 4A). Furthermore, in the saphenous nerves of paclitaxel-treated animals there is an increased appearance of abnormal and swollen/vacuolated mitochondria in both A- and C-fibers, as shown by electron microscopy (Fig. 5A–F) and impaired ATP production due to complex I and II deficits in PNSAs (Fig. 5G). Mitochondria in Schwann cells were normal confirming previous studies (data not shown) [53,54,56]. Removal of PN through catalytic decomposition with MnTE-2-PyP5+ (10 mg/kg/d) blocked the paclitaxel-induced nitration and inactivation of MnSOD (Fig. 3C, D), and mitochondrial dysfunction, that is MnTE-2-PyP5+ reduced the increased prevalence of abnormal mitochondria (Fig. 5E, F) and attenuated the loss of ATP production (Fig. 5G), implicating a critical role of PN in such mitochondrial dysfunction. Furthermore, similar nitration (Figs. 6A and 7A) and inactivation (Figs. 6B and 7B) of MnSOD, but not cytosolic CuZnSOD (Fig. 4B, C), and loss of ATP production from complex I and II deficits (Figs. 6C and 7C), were associated with oxaliplatin- (Fig. 6) and bortezomib- (Fig. 7) induced neuropathic pain at peak (D25). Of note, MnTE-2-PyP5+ blocked nitration and enzymatic inactivation of MnSOD, and restored ATP production (Figs. 6 and 7). These findings underscore the commonality and importance of mitochondrial PN-mediated nitroxidative stress in PNSAs to the development of neuropathic pain evoked by diverse chemotherapeutic agents.
Fig. 4.
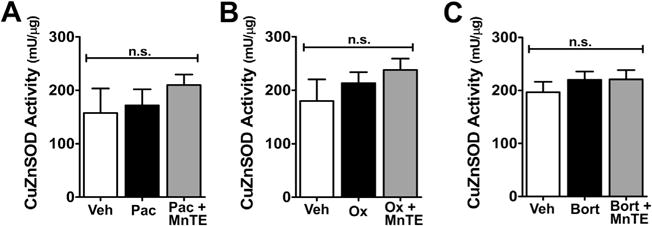
Copper zinc superoxide dismutase (CuZnSOD) activity was not altered by treatment with chemotherapeutics [paclitaxel (Pac), oxaliplatin (Ox), and bortezomib (Bort)] or Mn(III) 5,10,15,20-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+). Treatment with the chemotherapeutics paclitaxel (A), oxaliplatin (B), or bortezomib (C) either alone (black bars) or in combination with MnTE-2-PyP5+ (gray bars) had no effect on the activity of CuZnSOD compared with vehicle treatment (open bars). Results are expressed as mean ± SD, n = 6 and analyzed by one-way analysis of variance with Dunnett’s post-hoc comparisons. n.s. = not significant.
Fig. 5.
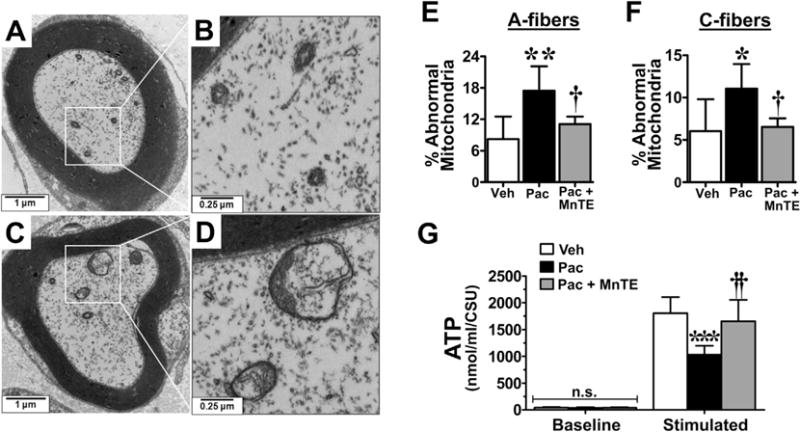
Mn(III) 5,10,15,20-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+) prevents mitochondrial abnormalities and dysfunction in the peripheral nerve sensory axons (PNSAs) associated with paclitaxel (Pac)-treatment. Representative electron micrographs of the myelinated A-fiber axons of the saphenous nerve from vehicle- (A, B) and paclitaxel- (C, D) treated animals showing swollen and vacuolated mitochondria within the nerves of the paclitaxel-treated group (C, D). Paclitaxel treatment (black bars) led to an increased appearance of abnormal mitochondria (swollen and vacuolated) in both the A- (E) and C-fibers (F) compared with treatment with vehicle (Veh) (open bars). Paclitaxel-treatment also resulted in a significant impairment of adenosine triphosphate (ATP) production after stimulation in the PNSAs compared with vehicle treatment (G) on day 25. MnTE-2-PyP5+ prevented both the paclitaxel-associated increase in mitochondrial abnormalities (E, F) and dysfunction (G). Results are expressed as mean ± SD, n = 5–6, and analyzed by one-way analysis of variance (ANOVA) with Dunnett’s post-hoc comparisons (E, F) or two-way ANOVA with Bonferroni’s post-hoc comparisons (G). *P < .05, **P < .01, ***P < .001 (Pac vs Veh); †P < .05, ††P < .001 (Pac + MnTE vs Pac). CSU = Citrate Synthase Units.
Fig. 6.
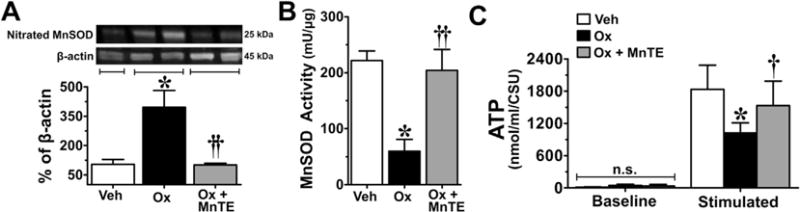
The oxaliplatin (Ox)-associated nitration/inactivation of manganese superoxide dismutase (MnSOD) and impaired adenosine triphosphate (ATP) production in peripheral nerve sensory axons (PNSAs) is attenuated with Mn(III) 5,10,15,20-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+). On day 25, PNSAs collected from oxaliplatin-treated animals (black bars) displayed increased levels of nitrated MnSOD (A), decreased MnSOD activity (B), and impaired ATP production (C) compared with tissues from vehicle(Veh)-treated animals (open bars). These alterations were prevented by administration of MnTE-2-PyP5+ (gray bars). Representative blots are shown above the quantitative bar graph corresponding to mean ± SD for an n = 6/group (A). Results are expressed as mean ± SD, n = 6 and analyzed by one-way analysis of variance (ANOVA) with Dunnett’s post-hoc comparisons (A, B) or two-way ANOVA with Bonferroni’s post-hoc comparisons (C). *P < .001 (Ox vs Veh); †P < .01, ††P < .001 (Ox + MnTE vs Ox). CSU = XXX.
Fig. 7.
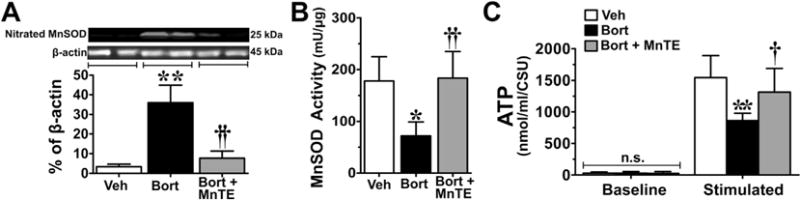
Mn(III) 5,10,15,20-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+) blocks manganese superoxide dismutase (MnSOD) nitration/inactivation and reduced adenosine triphosphate (ATP) production in the peripheral nerve sensory axons (PNSAs) of bortezomib (Bort)-treated animals. On day 25 of treatment, increased levels of nitrated MnSOD (A), decreased MnSOD activity (B), and impaired ATP production (C) were observed in the tissue of the group that received bortezomib (black bars) compared with the vehicle (Veh) group (open bars), and was prevented by administration of MnTE-2-PyP5+ (gray bars). Representative blots are shown above the quantitative bar graph corresponding to mean ± SD for an n = 6/group (A). Results are expressed as mean ± SD, n = 5–6 and analyzed by one-way analysis of variance (ANOVA) with Dunnett’s post-hoc comparisons (A, B) or two-way ANOVA with Bonferroni’s post-hoc comparisons (C). *P < .01, **P < .001 (Bort vs Veh); †P < .01, ††P < .001 (Bort + MnTE vs Bort). CSU = XXX.
4. Discussion
Chronic neuropathic pain accompanying CIPN, which generally resolves within weeks to months, but can last for years in some patients [32], greatly reduces the success of widely used chemotherapeutic agents by limiting dose and by decreasing the patient’s quality of life via psychological distress, fatigue, sleep disorders, cognitive deficit, and diminished activity [13,19]. Patients receiving aggressive therapy with doses exceeding the recommended levels or patients with pre-existing symptomatic or asymptomatic nerve damage (eg diabetes or prior chemotherapy) are at an increased risk [23,49]. CIPN largely affects sensory function leading to the development of central sensitization, although effects on motor function have been noted, especially acutely [25]; these are absent or rare with chronic dosing. The clinical management of these patients is difficult as current pain-management drugs are only marginally effective and display unacceptable side effects [13]. As the estimated incidence of CIPN can be as high as 30–90%, advances in understanding the underlying pathology are essential in order to identify effective therapeutic strategies to address this growing problem [13].
Here we confirm our recent findings that PN is a critical determinant of paclitaxel-induced neuropathic pain [12] and now extend to CIPN observed with two other distinct chemotherapeutic agents: oxaliplatin, which is used for metastatic colon cancer and other gastrointestinal tumors, and bortezomib, which is used for multiple myelomas [13]. The finding that neuropathic pain caused by these agents did not emerge for several days after discontinuing the PNDC treatment suggests that PNDCs could be used to prevent underlying causative mechanisms necessary for neuropathic pain development, rather than simply providing a transient decrease in enhanced nociceptive processing, as would occur with other analgesics (eg gabapentin, opiates). These results broaden the importance of PN in CIPN and suggest that there may be a common PN-dependent pathway independent of the chemotherapeutics’ anti-tumor mechanisms of action. One common denominator in the development of paclitaxel-, oxaliplatin-, and bortezomib-induced neuropathic pain is mitochondrial dysfunction in PNSAs leading to a persistent deficit in axonal energy supply [14,51,55,56]. Our results confirm these previous studies and extend them to implicate, for the first time, PN as one of the key players in the development of mitochondrial dysfunction in all three settings.
ATP production was assayed in saphenous nerve samples composed of axons, Schwann cells, and vascular endothelial cells. The axons of the rat’s saphenous nerve are all, or very nearly all, sensory [44,45] and we can thus discount any contribution from mitochondria in motor axons. If there is any contribution from mitochondria in endothelial cells it is certain to be quite small as these are very few in number compared with the number of axons. It seems unlikely that Schwann cell mitochondria make any contribution to the chemotherapy-induced change—first, because the number of mitochondria in Schwann cell cytoplasm is low compared with that in axoplasm [53,55,56], and, second, and more importantly, because no chemotherapy-induced change in mitochondrial structure is seen [53,55,56], suggesting that they are functionally normal. It thus seems most likely that chemotherapy-induced damage is to the mitochondria within PNSAs and that these are the targets of the PNDC’s actions.
Consistent with the mitotoxicity hypothesis, the results of our study clearly demonstrate that peak neuropathic pain was associated with significant nitration and inactivation of mitochondrial MnSOD and impairment of ATP production arising from complex I and II deficits. Moreover, the removal of PN with MnTE-2-PyP5+ prevented MnSOD nitration and inactivation, as well as the bioenergetic deficits, ultimately preventing the development of CIPN hypersensitivity. More importantly, this occurs without interfering with anti-tumor effects. The mitotoxicity hypothesis in CIPN is further supported by the fact that cytosolic CuZnSOD activity was unaffected, which is consistent with the fact that PN does not affect the catalytic activity of CuZnSOD [41]. These results therefore suggest that overt production of PN nitrates and inactivates MnSOD, which enables a feed-forward mechanism that sustains PN and PN-mediated nitroxidative stress leading to bioenergetic deficits by damage to various mitochondrial proteins, which probably includes the proteins that form respiratory complexes I and II.
While the initial source of PN responsible for the nitration and inactivation of MnSOD is unknown, we believe the intramitochondrial compartment to be the likely source as PN generated at extramitochondrial sites generally bears little effect on mitochondrial function [35,46]. In support of this, the enzymatic sources leading to the production of PN, nitric oxide synthase (NOS) and nicotinamide adenine dinucleotide phosphate oxidase, which provide the precursors for the biosynthesis of PN, namely NO and SO [4], have been identified within the mitochondria. These enzymes are either present in [16,33] or docked to [5,15] mitochondria and are activated in response to stress, such as inflammation [18,20]. Furthermore, in vitro studies have demonstrated that in isolated mitochondria, treatment with paclitaxel or cisplatin increases the formation of reactive oxygen species [47] and upregulates NOS [47], respectively. Regardless of the source, PN formed near the site of SO generation has a half-life of about 10 milliseconds, and can easily diffuse through membranes [46] where it undergoes fast, direct, and free radical-dependent molecular reactions with proteins in all mitochondrial compartments: the matrix, inner and outer membranes, and intermembrane space. These PN-mediated reactions can, ultimately, lead to mitochondrial dysfunction [35]. For example, nitrated mitochondrial electron transport chain (mETC) proteins (complex I–IV), ATP synthase, ANT1, and VDAC1 have been found in association with mitochondrial dysfunction in other pathological states [8,43]. Nitration of mETC proteins can disrupt ATP production through the loss of activity in complexes I, II, and, to a lesser extent, complex IV [6,35] and/or the inability of these proteins to physically interact with and transfer electrons to other complexes (eg nitration of complex II reduces protein–protein interactions with, and electron transfer to, complex III) [7]. Tyrosine nitration of ATP synthase reduces its activity [35] by interfering with the proton flow necessary for its function [11]. ATP production and mitochondrial integrity can also be compromised by the nitration of ANT1, an ADP–ATP exchanger across the inner membrane [57], and/or VDAC1, which imports anions and ADP, and exports ATP across the outer membrane [24]. Impairment of their activities can lead to reduced availability of mETC substrates [24] and, at least in the case of ANT, could also contribute to the assembly/activation of the mitochondrial permeability transition pore and consequential loss of mitochondrial membrane potential [57].
A question remains as to what drives PN formation in PNSAs. Many possibilities exist, but one strong contender is pro-inflammatory cytokines, which are known to be increased in blood [50] or can be released locally by activated macrophages or Schwann cells in response to chemotherapy [31]. Pro-inflammatory cytokines, such as tumor necrosis factor-α and interleukin-1β, are potent stimulating mediators in PN production [22], and, reciprocally, PN can drive the production of pro-inflammatory cytokines. Nitration/inactivation of MnSOD promotes the sustained presence of PN contributing to signal transduction changes known to be important in modulating neuronal excitability, such as activation of mitogen-activated protein kinases and redox-sensitive transcription factors, including nuclear factor kappa B and extracellular regulated kinase, and release of various inflammatory cytokines (reviewed in [38]) thereby creating a potent amplification loop. This reciprocal regulation between cytokines and PN has been defined in spinal cord during the development of pain of several etiologies [38], including paclitaxel-induced neuropathic pain [12] and in the development of opioid-induced hypersensitivity and antinociceptive tolerance [39]. Therefore, it may be that cytokines are involved in favoring mitochondrial dysfunction in the PNSAs during CIPN by stimulating the production of PN thereby sustaining such dysfunction via a feedback loop.
As mitochondrial dysfunction underlies CIPN associated with several chemotherapeutics, understanding the interactions between PN and mitochondria in PNSAs, dorsal root ganglia, and spinal cord are an important focus for future studies aimed at furthering our understanding of the pathogenesis of CIPN.
Acknowledgments
This study was funded by grants from the Leukemia & Lymphoma Society Translational Research Program and the Saint Louis University President Research Fund with additional support from the Saint Louis Cancer Center.
Footnotes
The authors have no conflict of interest.
References
- 1.Batinic-Haberle I, Reboucas JS, Spasojevich I. Superoxide dismutase mimics: chemistry, pharmacology and therapeutic potential. Antioxid Redox Signal. 2010;15:877–918. doi: 10.1089/ars.2009.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batinić-Haberle I, Spasojević I, Stevens RD, Hambright P, Fridovich I. Manganese(III) meso-tetrakis(ortho-N-alkylpyridyl)porphyrins. Synthesis, characterization, and catalysis of dismutation. Dalton Trans. 2002;13:2689–96. [Google Scholar]
- 3.Bazzola JJ, Russell LD. Electron microscopy: principles and techniques for biologists. 2nd. Boston MA: Jones and Bartlett Publishers; 1999. pp. 459–90. [Google Scholar]
- 4.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–4. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A. 2009;106:14385–90. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–16. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- 7.Chen CL, Chen J, Rawale S, Varadharaj S, Kaumaya PP, Zweier JL, Chen YR. Protein tyrosine nitration of the flavin subunit is associated with oxidative modification of mitochondrial complex II in the post-ischemic myocardium. J Biol Chem. 2008;283:27991–8003. doi: 10.1074/jbc.M802691200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chinta SJ, Andersen JK. Nitrosylation and nitration of mitochondrial complex I in Parkinson’s disease. Free Radic Res. 2011;45:53–8. doi: 10.3109/10715762.2010.509398. [DOI] [PubMed] [Google Scholar]
- 9.Dahan L, Sadok A, Formento JL, Seitz JF, Kovacic H. Modulation of cellular redox state underlies antagonism between oxaliplatin and cetuximab in human colorectal cancer cell lines. Br J Pharmacol. 2009;158:610–20. doi: 10.1111/j.1476-5381.2009.00341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–62. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 11.Dorgan LJ, Schuster SM. The effect of nitration and D2O on the kinetics of beef heart mitochondrial adenosine triphosphatase. J Biol Chem. 1981;256:3910–6. [PubMed] [Google Scholar]
- 12.Doyle T, Chen Z, Muscoli C, Bryant L, Esposito E, Cuzzocrea S, Dagostino C, Ryerse J, Rausaria S, Kamadulski A, Neumann WL, Salvemini D. Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J Neurosci. 2012;32:6149–60. doi: 10.1523/JNEUROSCI.6343-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farquhar-Smith P. Chemotherapy-induced neuropathic pain. Curr Opin Support Palliat Care. 2011;5:1–7. doi: 10.1097/SPC.0b013e328342f9cc. [DOI] [PubMed] [Google Scholar]
- 14.Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. PAIN®. 2006;122:245–57. doi: 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao S, Chen J, Brodsky SV, Huang H, Adler S, Lee JH, Dhadwal N, Cohen-Gould L, Gross SS, Goligorsky MS. Docking of endothelial nitric oxide synthase (eNOS) to the mitochondrial outer membrane: a pentabasic amino acid sequence in the autoinhibitory domain of eNOS targets a proteinase K-cleavable peptide on the cytoplasmic face of mitochondria. J Biol Chem. 2004;279:15968–74. doi: 10.1074/jbc.M308504200. [DOI] [PubMed] [Google Scholar]
- 16.Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci. 2005;26:190–5. doi: 10.1016/j.tips.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 17.Ischiropoulos H. Protein tyrosine nitration—an update. Arch Biochem Biophys. 2009;484:117–21. doi: 10.1016/j.abb.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 18.Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63:218–42. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 19.Kannarkat G, Lasher EE, Schiff D. Neurologic complications of chemotherapy agents. Curr Opin Neurol. 2007;20:719–25. doi: 10.1097/WCO.0b013e3282f1a06e. [DOI] [PubMed] [Google Scholar]
- 20.Korhonen R, Lahti A, Kankaanranta H, Moilanen E. Nitric oxide production and signaling in inflammation. Curr Drug Targets Inflamm Allergy. 2005;4:471–9. doi: 10.2174/1568010054526359. [DOI] [PubMed] [Google Scholar]
- 21.Kriedt CL, Baldassare J, Shah M, Klein C. Zinc functions as a cytotoxic agent for prostate cancer cells independent of culture and growth conditions. J Exp Ther Oncol. 2010;8:287–95. [PubMed] [Google Scholar]
- 22.Lakey JR, Suarez-Pinzon WL, Strynadka K, Korbutt GS, Rajotte RV, Mabley JG, Szabo C, Rabinovitch A. Peroxynitrite is a mediator of cytokine-induced destruction of human pancreatic islet beta cells. Lab Invest. 2001;81:1683–92. doi: 10.1038/labinvest.3780381. [DOI] [PubMed] [Google Scholar]
- 23.Lanzani F, Mattavelli L, Frigeni B, Rossini F, Cammarota S, Petro D, Jann S, Cavaletti G. Role of a pre-existing neuropathy on the course of bortezomib-induced peripheral neurotoxicity. J Peripher Nerv Syst. 2008;13:267–74. doi: 10.1111/j.1529-8027.2008.00192.x. [DOI] [PubMed] [Google Scholar]
- 24.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator – thinking outside the box. Biochim Biophys Acta. 2006;1762:181–90. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Loprinzi CL, Reeves BN, Dakhil SR, Sloan JA, Wolf SL, Burger KN, Kamal A, Le-Lindqwister NA, Soori GS, Jaslowski AJ, Novotny PJ, Lachance DH. Natural history of paclitaxel-associated acute pain syndrome: prospective cohort study NCCTG N08C1. J Clin Oncol. 2011;29:1472–8. doi: 10.1200/JCO.2010.33.0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macmillan-Crow LA, Cruthirds DL. Invited review: manganese superoxide dismutase in disease. Free Radic Res. 2001;34:325–36. doi: 10.1080/10715760100300281. [DOI] [PubMed] [Google Scholar]
- 27.MacMillan-Crow LA, Thompson JA. Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34F) by peroxynitrite. Arch Biochem Biophys. 1999;366:82–8. doi: 10.1006/abbi.1999.1202. [DOI] [PubMed] [Google Scholar]
- 28.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–55. [PubMed] [Google Scholar]
- 29.Muscoli C, Cuzzocrea S, Ndengele MM, Mollace V, Porreca F, Fabrizi F, Esposito E, Masini E, Matuschak GM, Salvemini D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest. 2007;117:3530–9. doi: 10.1172/JCI32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pellat-Deceunynk C, Amiot M, Bataille R, Van Riet I, Van Camp B, Omede P, Boccadoro M. Human myeloma cell lines as a tool for studying the biology of multiple myeloma: a reappraisal 18 years after [letter] Blood. 1995;86:4001–2. [PubMed] [Google Scholar]
- 31.Peters CM, Jimenez-Andrade JM, Jonas BM, Sevcik MA, Koewler NJ, Ghilardi JR, Wong GY, Mantyh PW. Intravenous paclitaxel administration in the rat induces a peripheral sensory neuropathy characterized by macrophage infiltration and injury to sensory neurons and their supporting cells. Exp Neurol. 2007;203:42–54. doi: 10.1016/j.expneurol.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 32.Pietrangeli A, Leandri M, Terzoli E, Jandolo B, Garufi C. Persistence of high-dose oxaliplatin-induced neuropathy at long-term follow-up. Eur Neurol. 2006;56:13–6. doi: 10.1159/000094376. [DOI] [PubMed] [Google Scholar]
- 33.Poderoso JJ, Carreras MC, Lisdero C, Riobo N, Schopfer F, Boveris A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys. 1996;328:85–92. doi: 10.1006/abbi.1996.0146. [DOI] [PubMed] [Google Scholar]
- 34.Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. PAIN®. 2001;94:293–304. doi: 10.1016/S0304-3959(01)00363-3. [DOI] [PubMed] [Google Scholar]
- 35.Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med. 2002;33:1451–64. doi: 10.1016/s0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- 36.Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111:409–19. [PubMed] [Google Scholar]
- 37.Salvemini D, Jensen MP, Riley DP, Misko TP. Therapeutic manipulations of peroxynitrite. Drug News Perspect. 1998;11:204–14. [PubMed] [Google Scholar]
- 38.Salvemini D, Little JW, Doyle T, Neumann WL. Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med. 2011;51:951–66. doi: 10.1016/j.freeradbiomed.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salvemini D, Neumann WL. Peroxynitrite: a strategic linchpin of opioid analgesic tolerance. Trends Pharmacol Sci. 2009;30:194–202. doi: 10.1016/j.tips.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 40.Shah MR, Kriedt CL, Lents NH, Hoyer MK, Jamaluddin N, Klein C, Baldassare J. Direct intra-tumoral injection of zinc-acetate halts tumor growth in a xenograft model of prostate cancer. J Exp Clin Cancer Res. 2009;28:84. doi: 10.1186/1756-9966-28-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith CD, Carson M, van der Woerd M, Chen J, Ischiropoulos H, Beckman JS. Crystal structure of peroxynitrite-modified bovine Cu, Zn superoxide dismutase. Arch Biochem Biophys. 1992;299:350–5. doi: 10.1016/0003-9861(92)90286-6. [DOI] [PubMed] [Google Scholar]
- 42.Spasojevic I, Chen Y, Noel TJ, Fan P, Zhang L, Reboucas JS, St Clair DK, Batinic-Haberle I. Pharmacokinetics of the potent redox-modulating manganese porphyrin, MnTE-2-PyP(5+), in plasma and major organs of B6C3F1 mice. Free Radic Biol Med. 2008;45:943–9. doi: 10.1016/j.freeradbiomed.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Swett JE, Torigoe Y, Elie VR, Bourassa CM, Miller PG. Sensory neurons of the rat sciatic nerve. Exp Neurol. 1991;114:82–103. doi: 10.1016/0014-4886(91)90087-s. [DOI] [PubMed] [Google Scholar]
- 45.Swett JE, Wikholm RP, Blanks RH, Swett AL, Conley LC. Motoneurons of the rat sciatic nerve. Exp Neurol. 1986;93:227–52. doi: 10.1016/0014-4886(86)90161-5. [DOI] [PubMed] [Google Scholar]
- 46.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–80. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 47.Varbiro G, Veres B, Gallyas F, Jr, Sumegi B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic Biol Med. 2001;31:548–58. doi: 10.1016/s0891-5849(01)00616-5. [DOI] [PubMed] [Google Scholar]
- 48.Wang ZQ, Porreca F, Cuzzocrea S, Galen K, Lightfoot R, Masini E, Muscoli C, Mollace V, Ndengele M, Ischiropoulos H, Salvemini D. A newly identified role for superoxide in inflammatory pain. J Pharmacol Exp Ther. 2004;309:869–78. doi: 10.1124/jpet.103.064154. [DOI] [PubMed] [Google Scholar]
- 49.Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13:27–46. doi: 10.1111/j.1529-8027.2008.00156.x. [DOI] [PubMed] [Google Scholar]
- 50.Wood LJ, Nail LM, Gilster A, Winters KA, Elsea CR. Cancer chemotherapy-related symptoms: evidence to suggest a role for proinflammatory cytokines. Oncol Nurs Forum. 2006;33:535–42. doi: 10.1188/06.ONF.535-542. [DOI] [PubMed] [Google Scholar]
- 51.Xiao WH, Bennett GJ. Effects of mitochondrial poisons on the neuropathic pain produced by the chemotherapeutic agents, paclitaxel and oxaliplatin. PAIN®. 2012;153:704–9. doi: 10.1016/j.pain.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao WH, Zheng FY, Bennett GJ, Bordet T, Pruss RM. Olesoxime (cholest-4-en-3-one, oxime): analgesic and neuroprotective effects in a rat model of painful peripheral neuropathy produced by the chemotherapeutic agent, paclitaxel. PAIN®. 2009;147:202–9. doi: 10.1016/j.pain.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiao WH, Zheng H, Bennett GJ. Characterization of oxaliplatin-induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience. 2012;203:194–206. doi: 10.1016/j.neuroscience.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao WH, Zheng H, Zheng FY, Nuydens R, Meert TF, Bennett GJ. Mitochondrial abnormality in sensory, but not motor, axons in paclitaxel-evoked painful peripheral neuropathy in the rat. Neuroscience. 2011;199:461–9. doi: 10.1016/j.neuroscience.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng H, Xiao WH, Bennett GJ. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp Neurol. 2011;232:154–61. doi: 10.1016/j.expneurol.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng H, Xiao WH, Bennett GJ. Mitotoxicity and bortezomib-induced chronic painful peripheral neuropathy. Exp Neurol. 2012;238:225–34. doi: 10.1016/j.expneurol.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 57.Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res. 2009;83:213–25. doi: 10.1093/cvr/cvp151. [DOI] [PMC free article] [PubMed] [Google Scholar]