

Abstract
This article presents systematic investigations on the relationship between the catalytic property and the surface ligand density/core size of thiolate ligand-capped Pd nanoparticles (PdNPs). The systematic variations in the two-phase synthesis of PdNPs generated from sodium S-dodecylthiosulfate were performed. The resulting PdNPs were characterized by transmission electron microscopy (TEM), thermogravimetric analysis (TGA), and 1H NMR and UV–vis spectroscopy. The decrease in the molar equivalent of sodium S-dodecylthiosulfate (Bunte salts) resulted in the formation of nanoparticles with lower surface ligand density and larger particle core size. A decrease in the molar equivalent of tetra-n-octylammonium bromide or an increase in reaction temperature generated nanoparticles with higher surface ligand density and smaller particle core size. As the molar equivalent of NaBH4 decreased, the particle core size increased. The catalysis studies on various PdNPs with different surface ligand density and average core size showed a strong correlation between the PdNP composition and the turnover frequency (TOF) of the isomerization of allyl alcohol. Optimized “good” PdNPs with lower surface ligand coverage and larger core size catalyzed the isomerization of various allyl alcohols to carbonyl analogues with high activity and selectivity.

Metallic nanoparticles (NPs) display highly reactive surfaces due to the surface atom arrangement which displays a low number of atomic neighbors along with a high surface atom to volume ratio.1 Controllable syntheses of noble metal NPs with desired shape and size has created considerable interests from both academia and industry and have been in demand for uses in the fields of catalysis,2-6 electronics,7,8 optics,9,10 biosensing,11 drug delivery,12-15 and biology.16,17 A particular challenge with the synthesis of metal NP is irreversible agglomeration in which smaller particles grow uncontrollably and ripen into larger particles.18-20 Solid supports and particle encapsulations have been employed to alleviate this issue.21-26 Additionally, introducing organic ligands prior to nanoparticle formation has also served well in preventing NPs from excessive growth while adding stability and in some cases increasing solubility in particular solvents.27-32
Since the performance of a catalyst, in addition to stability and solubility, can also be influenced by the presence of surface adsorbent, various surface ligand stabilizers including polymers,23,24 dendrimers,25,26 and small organic ligands such as thiols,27-29 thioethers,30 isocyanides,31 and amines32 have been used. Among these systems, alkanethiolate-capped NPs are considered as a popular and well-defined system. However, they are generally not recognized as efficient catalytic materials for organic reactions besides a few examples such as C–C coupling reactions (Suzuki and Suzuki–Miyaura) and simple hydrogenation reactions due to their high ligand surface coverage.33-36 Understanding and controlling the effects of alkanethiolate ligands on the catalytic properties of nanoparticles is important due to the tremendous potential of a well-defined system to provide a spatial control in reactivity and selectivity for various applications.
In our previous studies, palladium nanoparticles (PdNPs) were synthesized by a variation of the Brust-Schiffin method, in which sodium S-alkanethiosulfate ligands were employed rather than alkanethiolate ligands.37,38 Characterization results for PdNPs generated from sodium S-alkanethiosulfate suggested a lower ligand surface coverage for these NPs compared to the PdNPs synthesized with alkanethiols. The cleavage of a sulfite moiety from the surface of the PdNPs generated from S-alkylthiosulfate during the nucleation-passivation was believed to be the reason for the formation of PdNPs with lower ligand surface coverage.37,39 These PdNPs were shown to selectively catalyze the isomerization of allyl alcohol to propanal in the presence of hydrogen gas; however, the isomerization of substituted allyl alcohols was shown to be kinetically slower and/or thermodynamically less favorable.37,38
In this study, systematic variations of the two-phase synthesis of PdNPs using sodium S-dodecylthiosulfate were attempted in order to produce stable PdNPs with different surface ligand densities and particle core sizes. It is important to stress that the milder reactivity of S-alkanethiosulfates40 compared to that of alkanethiols allow variations in surface ligand density not previously possible in alkanethiolate-capped PdNPs. Controlling the ligand density on metal nanoparticle surfaces was not previously observed for the Brust-Schiffrin method since alkanethiols tend to generate well-packed monolayer ligands around nanoparticle core even with low alkanethiol concentrations.41,42 The effect of the variation in each synthetic parameter was analyzed by both characterizing the composition of PdNPs and testing their catalytic activity for the isomerization of allyl alcohol. The optimized PdNPs that exhibited the highest activity and selectivity for the isomerization of allyl alcohol were tested against the isomerization of a variety of substituted allyl alcohols.
RESULTS AND DISCUSSION
General Synthesis of Pd Nanoparticles
The synthesis employed in this study using sodium S-alkylthiosulfate (Bunte salts) is based on the two-phase Brust-Schiffrin method that has been popularly used for the synthesis of thiol-protected gold nanoparticles.42 The Brust-Schiffrin method has been extensively documented and studied due to its ease and popularity still eminent after nearly two decades after the first publication.41,42 To investigate the precursor species present in solution and the chemical interactions throughout the nanoparticle synthesis (Scheme 1), the reduction of tetrachloropalladate(II) with NaBH4 in the presence of S-alkylthiosulfate in a two phase system was performed in deuterated solvents and the reaction progress was monitored using 1H NMR and UV–vis spectroscopy.
Scheme 1. Reaction Scheme for Synthesis of Palladium Nanoparticles Using Sodium S-Dodecylthiosulfate.

Initially, the mixture of aqueous K2PdCl4 solution (D2O) and nonpolar solution (CDCl3) containing TOAB led to a phase transfer of the PdX PdX42−, with a mixture of both Br− and Cl− ions as ligands, to the organic phase. Upon addition of the ligand, the 1H NMR spectra of the organic layer (CDCl3) revealed a weak triplet at δ 3.04 for the CH2S2O3− and other peaks corresponding to the –CH2– and –CH3 of S-dodecylth-iosulfate and tetra-n-octylammonium (TOA) surfactant (Figure 1a).37 The NMR analysis of the aqueous layer (D2O) showed an absence of any relevant peaks other than H2O and CH3OH as trace impurities. These results suggested the TOA was able to fully transfer the S-dodecylthiosulfate into the nonpolar organic layer in spite of the low solubility of alkanethiosulfate in CDCl3.
Figure 1.

1H NMR spectra of TOAB + (TOA)2PdCl4 in CDCl3 with the addition of (a) 2 and (b) 3 equiv. of sodium S-dodecylthiosulfate; (c) the CDCl3 layer and (d) the D2O layer after the addition of NaBH4; (e) the purified PdNPs.
Previous studies on the synthesis of alkanethiolate-capped AuNPs were able to provide the evidence of the reduction of the Au3+ complex, [TOA][AuBr4], to the Au1+ complex, [TOA][AuBr2], along with the oxidation of thiols to disulfides.43,44 The present study in two phase system using thiosulfates was unable to show any chemical change of that magnitude (e.g., thiosulfate to disulfide or thiolate). Even when an increased amount of sodium S-dodecylthiosulfate was used, no chemical change was observed in the NMR spectra (Figure 1b) which implied no chemical reactions between PdX42− and alkanethiosulfate in the reaction mixture.
UV–vis spectra showed an intense band centered at 430 nm in the aqueous phase before the addition of TOAB and thiosulfates (Figure 2). This band was shifted to 445 nm in the organic phase after the phase transfer by TOAB implying the formation of [TOA]2[PdX4] complex.47 UV–vis spectra of the aqueous phase after the addition of TOAB again showed no peaks of interest. The results clearly indicated that TOA in the nonpolar solvent could completely transfer PdX42− in the aqueous phase into the organic phase. It should be noted that the phase transfer of both PdX42− and S-dodecylthiosulfate can be fully achieved with at least 5 equivalents of TOAB.
Figure 2.

UV–vis spectra of PdCl42− in H2O and dodecanethiolate (C12S)-capped Pd nanoparticles in CH2Cl2.
The recent study from Ferguson’s group suggested that the capping of Au surfaces by S-alkylthiosulfate took place by the adsorption of alkanethiolate that is generated from thiosulfate by the initial hydrolysis of thiosulfate groups in aqueous solution.45 Based on the 1H NMR spectra, which did not show the presence of αH from free dialkyldisulfide or alkylthiolate in the organic phase (Figure 1b), the possibility of alkanethiolate formation before the adsorption of alkylthiosulfates on PdNP surfaces can be eliminated. In addition, another possibility of S-dodecylthiosulfate being reduced to dodecylthiolate was also examined by reacting S-dodecylthiosulfate with NaBH4 in the absence of Pd2+ complex. However, the 1H NMR spectra of the organic phase which contained both S-dodecylthiosulfate and NaBH4 showed no S–S bond reduction (Figure 1c). Therefore, the SO32− moiety from the S-dodecylthiosulfate is only expunged from the monolayer/metallic interface upon the interaction with PdNP surface which is clearly in accordance with our previous conclusions.37,39
The presence of −BH4, the reducing agent, was monitored during the reaction in both organic and aqueous phases by 1H NMR. In the NMR spectra of D2O layer, a quartet centered at δ −0.03 was observed due to the presence of unreacted NaBH4 (Figure 1d).46 However, this quartet was absent in the 1H NMR spectra of CDCl3 layer since the −BH4 was consumed in the reduction of PdX42−. In comparison, the NMR spectra of the prior solution lacking PdX42− showed a strong quartet at δ −0.03 in the NMR spectra of CDCl3 layer (Figure 1c). The results confirmed the reduction of Pd2+ rapidly consumes −BH4 in the CDCl3 layer, while the NaBH4 in the aqueous layer reacts mildly with H2O to form H2 gas. Eventually after 24 h, the 1H NMR spectra still showed a weak quartet at δ −0.03 in the CDCl3 layer, but an absence of this peak in the D2O layer, indicating the relatively high stability of −BH4 in organic phase and the complete hydrolysis of NaBH4 in aqueous environment.
1H NMR spectra of the isolated PdNPs were identical to those previously reported spectra of alkanethiolate-capped metal nanoparticles displaying broad peaks at δ 1.20 for the CH2 and δ 0.87 for the CH3 without the presence of αCH2 and βCH2 from the surface bound thiolate functional group (Figure 1e).37,41 UV–vis spectra showed no interesting features other than an exponential rise to lower energy since PdNPs have no surface plasmon bands (Figure 2).47 The absence of Pd(II) bands in the UV–vis spectra of PdNPs generated from alkylthiosulfate should be noted, particularly due to the residual unreacted thiol ligands are known to oxidize the Pd core causing instability in PdNPs.47 The spectroscopic results confirmed that the synthesis of PdNPs from sodium S-dodecylthiosulfate leads to the generation of dodecanethiolate-capped NPs as illustrated in the previous reports.37
Surface Ligand Density/Core Size vs Catalytic Property of Pd Nanoparticles
The systematic variations presented in Table 1 were performed during the NP synthesis to control the factors (NP core size and ligand surface coverage) that might affect the activity and selectivity of these nanocatalysts.
Table 1. Systematic Variations Applied for the Synthesis of PdNPsa.
The numbers (equivalents) in the first three columns are against 1 equiv of K2PdCl4.
When less than 1 equiv of sodium S-dodecylthiosulfate is used, the irreversible aggregation of PdNPs took place after the addition of NaBH4.
A minimum of 5 equiv of tetra-n-octylammonium bromide was needed for the complete phase transfer of ligand precursors and reducing agents to the organic phase.
A minimum of 5 equiv of sodium borohydride was necessary for the complete reduction of PdCL42−.
Each synthesized PdNP was characterized by the analytical techniques described in the Experimental Section. TEM images and histograms shown in Figure 3 provided the average core diameters and dispersity of PdNPs. These results were used to mathematically determine the total theoretical number of Pd atoms present in the particle surface and in the core by using a truncated octahedron model.41 Furthermore, the results from thermogravimetric analysis (TGA), which provides the palladium and organic weight percentages in the NP, were used to obtain the average number of surface thiolate ligands surrounding each PdNP. Finally, the ligand surface coverage was obtained by dividing the number of average ligands on a PdNP by the theoretical amount of surface palladium atoms in the nanoparticle model.41 These calculated values could involve some errors, because the calculation is based on the assumed average molecular weight of somewhat polydisperse nanoparticles. Therefore, the obtained results must be viewed as rough estimates and just as guidelines for comparison of various PdNPs.
Figure 3.

TEM images (a–e) and the size distribution histograms (i–v) of dodecanethiolate-capped PdNPs generated from sodium S-dodecylthiosulfates: (a/i) PdNPs generated using the standard condition, (b/ii) PdNPs generated at 60 °C, (c/iii) PdNPs generated with one-half tetra-n-octylammonium bromide, (d/iv) PdNPs generated with one-half sodium S-dodecylthiosulfate, (e/v) PdNPs generated with one-quarter sodium borohydride from the condition (iv). Scale bars are 20 nm.
The increment of core size using Bunte salts method has been previously observed from the synthesis of water-soluble AuNPs at different temperatures.48 In this study, the reaction was performed by heating the two-phase mixture at 60 °C prior to NaBH4 reduction and maintaining the constant temperature for additional 3 h. The results in Table 2 clearly indicated increasing the reaction temperature to 60 °C (ii) from 22 °C (i) led to a significant decrease in Pd content and a large increase in organic fraction. TEM results along with histograms showed a decrease in average core size, which was close to 1.5 nm (Figure 3). The calculated ligand surface coverage was in accordance with the formation of PdNPs with higher ligand density. From these results, it is believed that the escalation in reaction temperature increases the activity of sodium S-dodecylthiosulfate stabilizers and makes the passivation of the nanoparticle nucleation–growth faster than the reactions performed at the lower temperature. The catalytic ability of PdNPs synthesized at high temperature (ii) toward the isomerization of allyl alcohol proved to be quite poor, displaying less than 20% of the catalytic conversion of allyl alcohol (Table 3). In comparison, PdNPs synthesized at the standard condition (i) converted over 90% of allyl alcohol to either isomerization (81%) or hydrogenation (12%) products. The results showed that the selectivity toward isomerization of PdNPs prepared at high temperature (ii) was also significantly lower (63% for ii vs 87% for i).
Table 2. Characterization Results and Theoretical Models of Each PdNP Variant.
| TGA (% Pd) |
TEM (dia, nm)a |
approximate average molecular formulab |
ligand surf. coverage (ligands/surface atoms)c |
|
|---|---|---|---|---|
| i | 68.6 | 2.59 ± 1.15 | ~Pd586L142 | 0.52 |
| ii | 51.0 | 1.51 ± 0.46 | ~Pd116L59 | 0.75 |
| iii | 55.7 | 1.66 ± 0.62 | ~Pd140L59 | 0.61 |
| iv | 77.0 | 2.76 ± 1.15 | ~Pd807L127 | 0.37 |
| v | 80.6 | 3.38 ± 0.95 | ~Pd1289L164 | 0.34 |
Average core size was calculated from histogram analysis of TEM images.
Calculations were based on theoretical model of a truncoctahedron.41
Ligand surface coverage was calculated by dividing the total ligands by surface Pd atoms of PdNP.
Table 3. Catalysis Results with 5 mol % Pd and 12 mmol H2 Gas.
| catalysis yields (%) |
|||||
|---|---|---|---|---|---|
| reaction time (h) | allyl alcohol | 1-propanol | 1-propanal | TOFa | |
| i | 4 | 7 | 12 | 81 | 407 |
| ii | 4 | 81 | 7 | 12 | 61 |
| iii | 4 | 38 | 47 | 15 | 75 |
| iv | 4 | 0 | 15 | 85 | 426 |
| v | 4 | 0 | 10 | 90 | 449 |
Turnover frequenices (initial TOFs) of fresh PdNPs (5 mol % in Pd atoms) are based on the mole isomerized per mol Pd atoms per hour.
In our synthetic method, the main purpose of TOAB is to phase transfer both metal precursor (e.g., tetrachloropalladate-(II)) and the stabilizing ligands (e.g., S-dodecylthiosulfate) into the organic layer. As described earlier, a large excess of TOAB (≥5 equiv) is required in order to guarantee the complete transfer of both reactants to the organic layer. A variation in the amount of TOAB was performed in order to investigate the effects of the phase transfer reagent concentration on the nanoparticle composition. Typically, the Brust-Schiffrin reaction is not influenced by this deviation due to the high reactivity of alkanethiols.41 Should a decreased amount of TOAB be employed, the presence of a hydrophilic thiosulfate functional group makes the partitioning of Bunte salts into the aqueous layer more plausible. Would this is the case, a fraction of Bunte salts would still be in the aqueous layer, while the Pd(II) is reduced in the organic layer. Therefore, the decrease in the amount of TOAB was anticipated to lower the concentration of S-dodecylthiosulfate in the organic phase and decrease the passivation activity of ligands. The decrease in the amount of TOAB by one-half (iii in Table 1) from the standard condition (i in Table 1), however, resulted in the formation of PdNPs with a smaller average core size and a higher ligand surface coverage (iii in Table 2). Hence, the nanoparticle composition shown in Table 2 for the variation (iii) suggested an increase in passivation activity of S-dodecylthiosulfate with the decreased amount of TOAB. This interesting result supports the idea that the excess TOAB in the mixture actually hinders the access of S-dodecylthiosulfate to the Pd surface during the nanoparticle formation. The intrinsic lower reactivity of alkanethiosulfates compared to that of thiols is likely the cause for such kinetic phenomena during the ligand passivation process. The catalysis results using PdNPs synthesized from the method using the decreased amount of TOAB (iii) showed a slightly better catalytic reactivity than PdNPs generated from the method using a higher reaction temperature (ii) but a lower activity than those generated from the standard condition (i) (Table 3). The catalytic results correspond well with the trend observed for the combination of the surface ligand density and average core size of PdNPs. It was also found that a further increase in the amount of TOAB from 10 to 20 equilibriums was mostly ineffective in terms of both PdNP composition and the catalytic activity.
Table 2 shows that when the amount of sodium S-dodecylthiosulfate was decreased by one-half (iv), it led to an increase in average core size and a decrease in organic ligand fraction. Less sodium S-dodecylthiosulfate ligands clearly hindered the passivation during the nucleation–growth process of PdNPs. The approximate molecular formula and the calculated number of surface Pd atoms shown in Table 2 indicated a decrease in ligand surface coverage to 0.37 ligands per surface atoms compared to 0.52 from the standard condition (i). These results suggested that a decrease in ligand surface coverage increases the overall reactivity of PdNPs, which was indicated from the increased turnover frequency for isomerization of allyl alcohol (Table 3). Moreover, it is important to note that reducing the L/Pd ratio past a 1:1 led to an increased formation of agglomerated product resulting in the loss of solubility in organic solvents.
The studies by Dasog et al. have recently shown that the synthesis of AuNPs could be further controlled with the variation of borohydride salts.36 In order to study the effects of NaBH4 in the Bunte salts synthesis, the variation (v) in Table 1 was performed by decreasing the sodium borohydride concentration to one-quarter from the condition (iv) which has so far produced the best catalytic results. The reduction of PdCl42− with a decreased amount of NaBH4 exhibited a slow color change from dark orange to brown and was unlike other reactions, in which displayed an aggressive effervescence during the Pd reduction. The results in Table 2 show that a decrease in the concentration of reducing agent led to a large increase in Pd core diameter and a small decrease in surface ligand coverage. The results confirm that, due to the low reactivity of S-dodecylthiosulfate, the role of NaBH4 during the nucleation–growth–passivation becomes more important for the synthesis of nanoparticles with alkanethiosulfates. Decreasing the amount of reducing agents likely produces Pd seed clusters in a lower concentration. The slower passivation by S-dodecylthiosulfate along with the lower concentration of Pd seed clusters causes the PdNPs to grow into larger particles. As for the catalytic properties toward the isomerization of allyl alcohol (Table 3), the PdNPs generated in this condition using the decreased amount of NaBH4 (v) were more catalytically active with a full conversion and slightly more selective toward isomerization.
The overall analysis between the systematic variations in the synthetic method and the catalysis results revealed several interesting trends. First, there is a clear correspondence between the ligand surface coverage and the turnover frequencies (TOFs). Our systematic variation studies support the original hypothesis which states “the primary factor in allowing highly successful isomerization reactions results from a lower surface ligand density of alkanethiolate ligands on PdNPs synthesized from sodium S-dodecylthiosulfate.”37,38 As the ligand surface coverage of each nanoparticle decreases, the isomerization reactions are more successful. As the ligand surface coverage of each variation increases, the isomerization of allyl alcohol is practically inhibited and the overall catalytic activity of PdNPs decreases. Second, the core size of PdNPs is also an important contributor for catalytic reactivity and selectivity. In the case of the variation (v), a larger increase in core size was accompanied by only a small change in surface ligand density, which still led to a notable enhancement in the catalytic selectivity for isomerization of allyl alcohol. These results suggest that the nanoparticle core size itself is also an important parameter for controlling catalytic activity and selectivity for the isomerization of allyl alcohol. The recent discovery by Cothey et al. regarding the presence of a submonolayer of sulfide species on PdNPs generated from alkanethiols is interesting and could be important for our studies as well.49 We are currently investigating whether or not PdNPs generated from alkylthiosulfate contain a PdSx layer and will report the results in a future manuscript.
PdNPs generated from the variation (iv) and (v) showing the effect of NaBH4 were further tested in catalytic reactions using less than one-half of hydrogen gas (~5 mmol, Table 4) compared to the standard catalytic reaction condition (12 mmol, Table 3). Hypothetically, the amount of H2 gas should be an important factor in determining the selectivity between isomerization and hydrogenation. Hydrogen deficient PdNPs are less likely to provide a second hydrogen to complete hydrogenation and far more likely to accept the allylic proton, which is the final and critical step for the isomerization. The results shown in Table 4, entry 1, can be compared with those shown in Table 3 for the catalytic results for PdNPs synthesized using the variations (iv) and (v). These results suggest the selectivity toward isomerization for both PdNPs increases when the decreased amount of H2 gas is used during the reaction.
Table 4. Catalysis Results with 5 mol % Pd and 5 mmol H2 Gas.
| Entry | PdNP catalyst |
Time | Allyl alcohol |
Hydrogenation Yielda |
Isomerization Yielda |
|---|---|---|---|---|---|
| 1 | iv | 4 h |
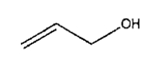
|
2% | 98% |
| v | 4 h |
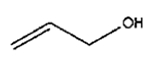
|
0% | 100% | |
| 2 | iv | 8 h |
|
4% | 51% |
| v | 8 h |
|
3% | 93% |
Yields were obtained by 1H NMR.
Previous studies of TiO2 supported Pd catalysts documenting the competition between hydrogenation and isomerization of allyl alcohols have offered interesting mechanistic insights into the overall reaction.50,51 In particular, the studies performed by Musolino et al. explained the different selectivity toward saturated alcohols and carbonyl analogues on the basis of interactions between σ-alkyl palladium bonded intermediates.50 Their proposed scheme implies the hydrogen addition to the terminal C-sp2 (γ from OH) would yield a carbonyl analogue via an enol intermediate and the hydrogen addition to the internal C-sp2 (β from OH) would yield a saturated alcohol. Their conclusion indicated the selectivity toward carbonyl analogues decrease with the presence of larger C-sp2 substituents. In fact, our previous report showed that the catalytic reaction of crotyl alcohol resulted in the isomerization product in less than 10%.38 In comparison, the isomerization yields for PdNPs generated from the variations (iv) and (v) were clearly superior with 51% and 93%, respectively (Table 4, entry 2).
As discussed in previous studies, our PdNPs could only isomerize a small variety of allyl alcohols with low to mild yields.39 Considering the high reactivity and selectivity toward the isomerization of allyl alcohol and crotyl alcohol, the catalyst synthesized using the method (v) was selected as an optimized “good” PdNP catalyst and further tested against additional substituted allyl alcohols as summarized in Table 5.
Table 5. Isomerization of Various Substituted Allyl Alcohols Using “Good” Pd Nanoparticle (5% mol Pd and 5 mmol H2 gas).
| Entry | Allyl alcohol |
Time | Hydrogenation product |
Yielda | Isomerization product |
Yielda |
|---|---|---|---|---|---|---|
| 3 |
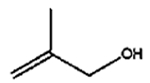
|
8 h |
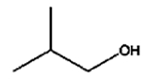
|
0% |
|
100% |
| 4 |
|
8 h |
|
0% |
|
100% |
| 5 |
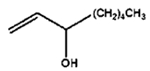
|
8 h |
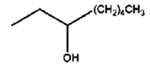
|
0% |
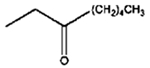
|
100% |
| 6 |
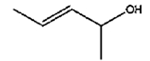
|
8 h |
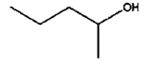
|
0% |
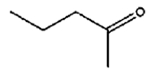
|
57% |
| 7 |
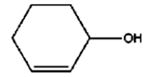
|
8 h |
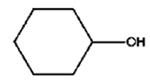
|
6% |
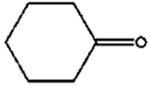
|
33% |
| 8 |
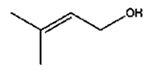
|
8 h |
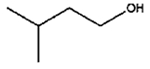
|
3% |
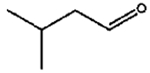
|
3% |
Yields were obtained by 1H NMR.
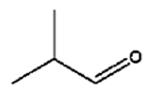
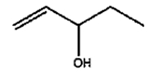
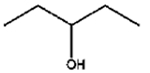
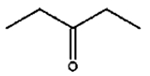
The isomerization of 2-methylprop-2-en-1-ol (entry 3) resulted in a very high selectivity and produced purely an isomerized product. Moreover, the complete isomerization of pent-1-en-3-ol (entry 4) and oct-1-en-3-ol (entry 5) confirms that the effects of an alkyl substituent at the R1 position (Scheme 2) have little to no effect in the reaction selectivity and the isomerization yields. The isomerization was a bit more difficult for crotyl alcohol (entry 2 in Table 4) since this compound required the formation of a less stable enol intermediate. As for the isomerization of both trans-pent-3-en-2-ol (entry 6) and cylcohex-2-enol (entry 7), the isomerization of these reactants was drastically decreased likely due to the increased steric hindrance. The isomerization of prenol (entry 8) was ineffective due to the presence of alkyl substituents affording a trisubstituted alkene which is too stable to undergo the reaction. In comparison with the previous results, the optimization of PdNPs by controlling surface ligand density and particle core size was successfully achieved.
Scheme 2. Catalytic Reaction Scheme for the Isomerization of Allyl Alcohols Using “Good” PdNPs.

CONCLUSION
A systematic investigation in the two-phase synthesis of palladium nanoparticles generated from sodium S-dodecylthiosulfate suggested that more catalytically active and selective Pd nanoparticles could be synthesized by using lower equivalents of sodium S-dodecylthiosulfate and sodium borohydride and higher concentrations of tetra-n-octylammonium bromide. Correlations between ligand surface coverage and core diameter against the TOF of allyl alcohol suggested that there is a strong correlation between the catalytic property and the surface ligand density/particle core size of palladium nanoparticles. The results demonstrate that controlling the surface density of well-defined alkanethiolate ligands and the particle core size bring about the development of highly selective and efficient catalytic materials. The utilization of a well-known strategy associated with the assembly of various thiolate monolayers on metal nanoparticles will likely enable better controls in the activity and selectivity of metal nanoparticle catalysts in the future. Moreover, this study describes new insights regarding the nucleation–growth-passivation of PdNPs in the presence of Bunte salts and its effect on the role of other common reagents (tetraoctylammonium bromide and sodium borohydride) used for the nanoparticle synthesis.
EXPERIMENTAL SECTION
Materials
The following materials were purchased from the indicated suppliers and used as received: Potassium tetrachloropalladate (K2PdCl4), tetra-n-octylammonium bromide (TOAB), 1-bromododecane, prop-2-en-1-ol (allyl alcohol), trans-pent-3-en-2-ol, 2-cyclohexen-1-ol, and sodium borohydride (NaBH4) were purchased from ACROS. 1-Bromododecane, pent-1-en-3-ol, 2-methylprop-2-en-1-ol, oct-1-en-3-ol, but-2-en-1-ol (crotyl alcohol), and 3-methylbut-2-en-1-ol (prenol) were purchased from Sigma-Aldrich. Sodium thiosulfate (Na2S2O3·5H2O), toluene, acetone, acetonitrile, hexane, methanol, and ethyl alcohol were obtained from Fisher Scientific. Chloroform-d, methyl alcohol-d4, and deuterium oxide were purchased from Chembridge Isotope Laboratories. Sodium S-dodecylthiosulfate was synthesized using a previously published method.37 Water was purified by using a Barnstead NANOpure Diamond ion exchange resins purification unit.
Synthesis of Pd Nanoparticles
The following synthesis is a standard procedure for all PdNP syntheses. Reaction conditions which were systematically varied were (1) the mole ratio of TOAB; (2) the mole ratio of sodium S-dodecylthiosulfate; (3) the mole ratio of NaBH4; (4) the reaction temperature prior to reduction by NaBH4. In addition to the systematic variations, the delivery method for NaBH4 was changed to an instantaneous addition from a slower addition (≥10 s) used in the previous studies.37,38 This modification was implemented to accommodate the same delivery condition for each synthetic variation.
Potassium tetrachloropalladate (K2PdCl4; 0.4 mmol) was dissolved in 12 mL of nanopure water. TOAB (2.0 mmol) was dissolved in 25 mL of toluene. Both solutions were mixed and continuously stirred until the organic layer turned dark orange and the aqueous layer cleared, indicating the completion of the phase transfer of PdCl42−. The aqueous layer was discarded and the organic layer was placed in a 250 mL round-bottom flask. Sodium S-dodecylthiosulfate (0.8 mmol) dissolved in 10 mL of 25% methanol was added to the organic layer. Additionally, TOAB (2.0 mmol) was added to the reaction flask. The reaction mixture was continuously stirred for 15 min. Afterward, sodium borohydride (NaBH4; 8.0 mmol) fully dissolved in 7 mL of nanopure water was rapidly delivered to the vigorously stirred reaction mixture. Consequently, a rapid color change (black) was observed indicating the formation of nanoparticles. Upon the completion of 3 h of continuous stirring, the aqueous layer was removed by using a separatory funnel and the toluene was removed by vacuum. The resulting crude nanoparticles were suspended by using 25 mL of ethanol and poured down on a coarse funnel frit (F). The Pd nanoparticles were then further washed with ethanol, acetonitrile, and acetone. The resulting nanoparticles were dried in vacuum overnight at a pressure of 25 Psi.
Characterization of Pd Nanoparticles
Proton NMR spectra were recorded on a Bruker AC400 FT-NMR spectrometer operating at 400 MHz in CDCl3 solutions and internally referenced to δ 7.26 ppm. UV–visible spectra varying from wavelengths of 800 to 290 nm were obtained using a Shimadzu UV-2450 UV-spectrometer. Transmission electron microscope (TEM) images were obtained with a JEOL 1200 EX II electron microscope operating a 90 keV. Samples were prepared by placing 25 μL of a Pd nanoparticle THF solution (~1 mg/mL) on a 200 mesh copper grid with formvar film. Size distribution analysis of Pd nanoparticle core microscope images was executed with Scion Image Beta Release 2TM. Background subtraction was done by Rolling Ball at a set radius of 25. Measurement options were done by Ellipse Major Axis. Thermogravimetric analysis (TGA) was conducted using a TA Instruments SDT Q600 with a flow rate of 100 mL/min of N2 with heating from room temperature to 600 °C.
Catalytic Isomerization Reactions
Catalysis experiments were performed by placing 3 mL of CDCl3 along with 5 mol % Pd nanoparticle catalyst in a glass round-bottom flask equipped with a rubber stopper. This solution was purged with H2 gas for 10 min. After the influx of H2 gas was removed, 50 μL of allyl alcohol was injected into the sealed flask. The reaction was continuously stirred at room temperature. An aliquot of the solution was quickly transferred to a NMR tube to obtain 1H NMR spectra.
ACKNOWLEDGMENTS
This research was supported in part by a grant from the ACS-PRF (PRF49407-UR7) and CSULB (MGSS award).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Klabunde KJ, Richards RM. Nanoscale Materials in Chemistry. 2nd ed. John Wiley & Sons, Inc.; Hoboken, NJ: 2009. [Google Scholar]
- (2).Cong H, Porco JA. ACS Catal. 2012;2:65–70. doi: 10.1021/cs200495s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Scholten JD, Leal BC, Dupont J. ACS Catal. 2012;2:184–200. [Google Scholar]
- (4).Moreno-Mañas M, Pleixats R. Acc. Chem. Res. 2003;36:638–643. doi: 10.1021/ar020267y. [DOI] [PubMed] [Google Scholar]
- (5).Stratakis M, Garcia H. Chem. Rev. 2012;112:4469–4506. doi: 10.1021/cr3000785. [DOI] [PubMed] [Google Scholar]
- (6).Roucoux A, Schulz J, Patin H. Chem. Rev. 2002;102:3757–3778. doi: 10.1021/cr010350j. [DOI] [PubMed] [Google Scholar]
- (7).McConnell WP, Novak JP, Brousseau LC, Fuierer RR, Tenent RC, Feldheim DL. J. Phys. Chem. B. 2000;104:8925–8930. [Google Scholar]
- (8).Reimers JR, Hush NS. J. Phys. Chem. B. 2001;105:8979–8988. [Google Scholar]
- (9).Haynes CL, Van Duyne RP. J. Phys. Chem. B. 2001;105:5599–5611. [Google Scholar]
- (10).Zhao W-W, Tian C-Y, Xu J-J, Chen H-Y. Chem. Commun. 2012;48:895–897. doi: 10.1039/c1cc16775h. [DOI] [PubMed] [Google Scholar]
- (11).Shon Y-S, Aquino M, Pham TV, Rave D, Ramirez M, Lin K, Vaccarello P, Lopez G, Gredig T, Kwon C. J. Phys. Chem. C. 2011;115:10597–10605. doi: 10.1021/jp110531x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Namiki Y, Fuchigami T, Tada N, Kawamura R, Matsunama S, Kitamoto Y, Nakagawa M. Acc. Chem. Res. 2011;44:1080–1093. doi: 10.1021/ar200011r. [DOI] [PubMed] [Google Scholar]
- (13).Yang H-W, Hua M-Y, Liu H-L, Tsai R-Y, Chuang C-K, Chu P-C, Wu P-Y, Chang Y-H, Chang H-C, Yu K-J, Pang S-T. ACS Nano. 2012;6:1795–1805. doi: 10.1021/nn2048526. [DOI] [PubMed] [Google Scholar]
- (14).Zhang X-Q, Xu X, Lam R, Giljohann D, Ho D, Mirkin CA. ACS Nano. 2011;5:6962–6970. doi: 10.1021/nn201446c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Roa W, Zhang X, Guo L, Shaw A, Hu X, Xiong Y, Gulavita S, Patel S, Sun X, Chen J, Moore R, Xing JZ. Nanotechnology. 2009;20:375101. doi: 10.1088/0957-4484/20/37/375101. [DOI] [PubMed] [Google Scholar]
- (16).Murphy CJ, Gole AM, Stone JW, Sisco PN, Alkilany AM, Goldsmith EC, Baxter SC. Acc. Chem. Res. 2008;41:1721–1730. doi: 10.1021/ar800035u. [DOI] [PubMed] [Google Scholar]
- (17).Wang H-H, Lin C-AJ, Lee C-H, Lin Y-C, Tseng Y-M, Hsieh C-L, Chen C-H, Tsai C-H, Hsieh C-T, Shen J-L, Chan W-H, Chang W-H, Yeh H-I. ACS Nano. 2011;5:4337–4344. doi: 10.1021/nn102752a. [DOI] [PubMed] [Google Scholar]
- (18).Hornstein BJ, Finke RG. Chem. Mater. 2004;16:139–150. [Google Scholar]
- (19).Ott LS, Finke RG. Chem. Mater. 2008;20:2592–2601. [Google Scholar]
- (20).Liu HH, Surawanvijit S, Rallo R, Orkoulas G, Cohen Y. Environ. Sci. Technol. 2011;45:9284–9292. doi: 10.1021/es202134p. [DOI] [PubMed] [Google Scholar]
- (21).Marshall ST, O’Brien M, Oetter B, Corpuz A, Richards RM, Schwartz DK, Medlin JW. Nat. Mater. 2010;9:853–858. doi: 10.1038/nmat2849. [DOI] [PubMed] [Google Scholar]
- (22).Lopez-Sanchez JA, Dimitratos N, Hammond C, Brett GL, Kesavan L, White S, Miedziak P, Tiruvalam R, Jenkins RL, Carley AF, Knight D, Kiely CJ, Hutchings GJ. Nature Chem. 2011;3:551–556. doi: 10.1038/nchem.1066. [DOI] [PubMed] [Google Scholar]
- (23).Bhattacharjee S, Dotzauer DM, Bruening ML. J. Am. Chem. Soc. 2009;131:3601–3610. doi: 10.1021/ja807415k. [DOI] [PubMed] [Google Scholar]
- (24).Hu J, Liu Y. Langmuir. 2005;21:2121–2123. doi: 10.1021/la0471902. [DOI] [PubMed] [Google Scholar]
- (25).Wilson OM, Knecht MR, Garcia-Martinez JC, Crooks RM. J. Am. Chem. Soc. 2006;128:4510–4511. doi: 10.1021/ja058217m. [DOI] [PubMed] [Google Scholar]
- (26).Oh S-K, Niu Y, Crooks RM. Langmuir. 2005;21:10209–10213. doi: 10.1021/la050524i. [DOI] [PubMed] [Google Scholar]
- (27).Castro EG, Salvatierra RV, Schreiner WH, Oliveira MM, Zarbin AJG. Chem. Mater. 2010;22:360–370. [Google Scholar]
- (28).Eklund SE, Cliffel DE. Langmuir. 2004;20:6012–6018. doi: 10.1021/la049787n. [DOI] [PubMed] [Google Scholar]
- (29).Alvarez J, Liu J, Roman E, Kaifer AE. Chem. Commun. 2000:1151–1152. [Google Scholar]
- (30).Ganesan M, Freemantle RG, Obare SO. Chem. Mater. 2007;19:3464–3471. [Google Scholar]
- (31).Angelici RJ, Lazar M. Inorg. Chem. 2008;47:9155–9165. doi: 10.1021/ic800513t. [DOI] [PubMed] [Google Scholar]
- (32).Li Z, Gao J, Xing X, Wu S, Shuang S, Dong C, Paau MC, Choi MMF. J. Phys. Chem. C. 2010;114:723–733. [Google Scholar]
- (33).Cargnello M, Wieder NL, Canton P, Montini T, Giambastiani G, Benedetti A, Gorte RJ, Fornasiero P. Chem. Mater. 2011;23:3961–3969. [Google Scholar]
- (34).Lu F, Ruiz J, Astruc D. Tetrahedron Lett. 2004;45:9443–9445. [Google Scholar]
- (35).Zhu Y, Qian H, Drake BA, Jin R. Angew. Chem., Int. Ed. 2010;49:1295–1298. doi: 10.1002/anie.200906249. [DOI] [PubMed] [Google Scholar]
- (36).Dasog M, Wenbo H, Scott RWJ. Chem. Commun. 2011;47:8569–8571. doi: 10.1039/c1cc11813g. [DOI] [PubMed] [Google Scholar]
- (37).Sadeghmoghaddam E, Lam C, Choi D, Shon Y-S. J. Mater. Chem. 2011;21:307–312. [Google Scholar]
- (38).Sadeghmoghaddam E, Gaïeb K, Shon Y,-S. Appl. Catal., A. 2011;405:137–141. [Google Scholar]
- (39).Shon Y-S, Gross SM, Dawson B, Porter M, Murray RW. Langmuir. 2000;16:6555–6561. [Google Scholar]
- (40).Lukkari J, Meretoja M, Kartio I, Laajalehto K, Rajamäki M, Lindström M, Kankare J. Langmuir. 1999;15:3529–3537. [Google Scholar]
- (41).Hostetler MJ, Wingate JE, Zhong C-J, Harris JE, Vachet RW, Clark MR, Londono JD, Green SJ, Stokes JJ, Wignall GD, Glish GL, Porter MD, Evans ND, Murray RW. Langmuir. 1998;14:17–30. [Google Scholar]
- (42).Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R. J. Chem. Soc., Chem. Commun. 1994:801–802. [Google Scholar]
- (43).Goulet PJG, Lennox RB. J. Am. Chem. Soc. 2010;132:9582–9584. doi: 10.1021/ja104011b. [DOI] [PubMed] [Google Scholar]
- (44).Li Y, Zaluzhna O, Tong YS. Langmuir. 2011;27:7366–7370. doi: 10.1021/la201158v. [DOI] [PubMed] [Google Scholar]
- (45).Fealy RJ, Ackerman SR, Ferguson GS. Langmuir. 2011;27:5371–5376. doi: 10.1021/la200143b. [DOI] [PubMed] [Google Scholar]
- (46).The coupling of 1H with 11B gives a quartet with same intensities. The presence of small septets with low intensities is due to the coupling of 1H with 10B.
- (47).Zamborini FP, Gross SM, Murray RW. Langmuir. 2001;17:481–488. [Google Scholar]
- (48).Lohse SE, Dahl JA, Hutchison JE. Langmuir. 2010;26:7504–7511. doi: 10.1021/la904306a. [DOI] [PubMed] [Google Scholar]
- (49).Corthey G, Rubert AA, Picone AL, Casillas G, Giovanetti LJ, Ramallo-López JM, Zelaya E, Benitez GA, Requejo FG, Josó-Yacamán M, Salvarezza RC, Fonticelli MH. J. Phys. Chem. C. 2012;116:9830–9837. [Google Scholar]
- (50).Musolino MG, Maio PD, Donato A, Pietropaolo R. J. Mol. Catal. A: Chem. 2004;208:219–224. [Google Scholar]
- (51).Uma R, Crévisy C, Grée R. Chem. Rev. 2003;103:27–51. doi: 10.1021/cr0103165. [DOI] [PubMed] [Google Scholar]