

SUMMARY
The ability to monitor kinase activity dynamics in live cells greatly aids the study of how signaling events are spatiotemporally regulated. Here, we report on the adaptability of bimolecular Kinase Activity Reporters (bimKARs) as molecular tools to enhance the real-time visualization of kinase activity. We demonstrate that the bimKAR design is truly versatile and can be used to monitor a variety of kinases, including JNK, ERK, and AMPK. Furthermore, bimKARs can have significantly enhanced dynamic ranges over their unimolecular counterparts, allowing the elucidation of previously undetectable kinase activity dynamics. Using these newly designed bimKARs, we investigate the regulation of AMPK by PKA in the plasma membrane and demonstrate that stimulated and unstimulated (i.e., basal) PKA activity enhance and suppress stimulated AMPK activity, respectively.
Keywords: PKA, AMPK, FRET, live-cell imaging, biosensor, compartmentalized signaling
Graphical Abstract
INTRODUCTION
In live cells, signal transduction of extracellular cues requires exquisite spatial and temporal regulation of numerous molecules dispersed throughout the cellular milieu in order to elicit specific responses. For example, protein kinases aid cellular communication by altering substrate activity, cellular localization and/or association with other proteins. Cells employ a variety of mechanisms to tightly regulate protein kinase activity in space and time to ensure that signals are properly transduced. To better understand how kinase signaling functions in cells, a multitude of different methods have been employed over the years to study kinase behavior. Traditional biochemical approaches such as western blotting, immunostaining, and in vitro kinase activity assays, which have greatly fostered our general understanding of kinase signaling, only provide fixed glimpses of signal transduction, a process that is constantly in flux. Thus, various fluorescence-based kinase activity biosensors have been developed to allow real-time monitoring of signaling dynamics. For example, peptide-based biosensors incorporate organic fluorophores into peptides or protein-interaction domains such that phosphorylation yields enhanced fluorescent properties such as emission wavelength or quantum yield (Rothman et al., 2005; Sharma et al., 2008; Tarrant and Cole, 2009). These biosensors offer a large dynamic range and are able to detect kinase signaling in live cells; however, they can be unstable in cells and often require microinjection (Tarrant and Cole, 2009). Unlike peptide-based biosensors, genetically encodable fluorescence resonance energy transfer (FRET)-based biosensors circumvent cell entry issues by relying on the native cellular machinery for expression. Moreover, this method accommodates the incorporation of subcellular localization sequences and thus allows for the investigation of kinase signaling dynamics in precise spatial compartments (Mehta and Zhang, 2011).
FRET-based kinase activity reporters (KARs) serve as surrogate substrates for the kinases of interest and report their activity dynamics via changes in FRET. KARs utilize a kinase-inducible molecular switch consisting of a substrate sequence and a phosphoamino acid binding domain (PAABD) that binds to the phosphorylated substrate, sandwiched between two spectrally different fluorescent proteins (FPs) capable of undergoing FRET (Depry and Zhang, 2010). A myriad of different FRET-based activity biosensors have been created to study individual kinases, including Akt (Gao and Zhang, 2008), Src (Wang et al., 2005), InsR (Sato and Umezawa, 2004; Sato et al., 2002), EGFR (Ting et al., 2001), JNK (Fosbrink et al., 2010), and AMPK (Tsou et al., 2011), among others (Newman et al., 2011). Furthermore, the modular design of KARs has enabled the creation of many new reporters through the substitution of one kinase-specific substrate and/or PAABD for another. This tactic was employed to generate KARs for protein kinase D (D Kinase Activity Reporter, DKAR) and extracellular signal-regulated kinase (ERK Kinase Activity Reporter, EKAR), which were derived from C Kinase Activity Reporter (CKAR) and A Kinase Activity Reporter (AKAR), respectively (Harvey et al., 2008; Violin et al., 2003). Many novel insights have been made possible using the available FRET-based biosensors. For example, a recent study using EKAR revealed stochastic Erk activity pulses in cultured epithelial cells, thus illuminating the role of spontaneous Erk activity in regulating cell proliferation (Aoki et al., 2013). Another study using FRET-based reporters for FAK and Src revealed the differential activation of these two kinases in stem cells directed to adopt different cell fates (Liao et al., 2013).
KARs are invaluable tools for studying kinase activity dynamics, but their utility may be restricted by limited dynamic ranges, which can hinder the detection of subtle yet physiologically relevant changes in kinase activity (Herbst et al., 2011; Hodgson et al., 2010). For example, in insulin-secreting MIN6 β cells, PKA oscillates in sync with Ca2+ and cAMP, contributing to precise control of spatially localized or global PKA activity, despite the small amplitude of activity oscillations (Ni et al., 2011). Further building upon the modular nature of KARs in an attempt to enhance the dynamic range of these biosensors, recent advances have led to the development of bimolecular KARs (bimKARs). In bimKARs, a single polypeptide encoding the PAABD attached to one FP is expressed along with a separate polypeptide containing the kinase-specific substrate attached to the second FP of the FRET pair. The separation of the FRET pair is thought to enhance the dynamic range, likely by decreasing the occurrence of basal FRET between the FPs since they are much further apart than in a unimolecular biosensor. For example, the bimolecular A kinase activity reporter, bimAKAR, specifically detects PKA activity in cells and, when targeted to the plasma membrane, exhibits a larger dynamic range than the corresponding unimolecular AKAR (Herbst et al., 2011). Although successful at visualizing kinase signaling, only a small number of bimKARs have been created, and it remains to be tested how generalizable this design is. In this study, we show that bimKARs are highly versatile and can be used to enhance our ability to detect and monitor various kinase activities in living cells. We constructed bimKARs that incorporate kinase docking domains and utilize different PAABDs. In addition, we created bimKARs that enabled us to delve into the regulation of AMP activated kinase (AMPK) activity within discrete subcellular compartments, revealing that AMPK activity at the plasma membrane experiences both positive and negative regulation by PKA within the same cell.
RESULTS
Including a Kinase Docking Domain in the BimKAR design
A number of kinases require docking domains in order to ensure specific phosphorylation of their target substrates, including members of the mitogen-activated protein kinase (MAPK) family. MAPKs rely upon kinase-specific docking domains within scaffolds and target proteins to help mediate structural and functional organization by bringing the kinase and substrate into a multiprotein complex (Dhanasekaran et al., 2007; Sharrocks et al., 2000), thus enhancing the specificity and efficiency of substrate phosphorylation. As a subgroup of the MAPK family, the c-Jun N-terminal Kinases (JNKs) are no exception; even in the case of identical phosphoacceptor sites, JNK fails to phosphorylate substrates that specifically lack a docking domain (Katz and Aronheim, 2002). As such, the incorporation of a docking domain was essential to the successful generation of the JNK Activity Reporter (JNKAR1) (Fosbrink et al., 2010).
To determine whether a bimKAR design is compatible with the inclusion of a docking domain, we used unimolecular JNKAR1 as a model to develop bimJNKAR, making certain to keep the substrate and docking domain a part of the same fragment. BimJNKAR consists of the cyan fluorescent protein (CFP) variant Cerulean (Rizzo et al., 2004) fused to the phosphothreonine (pThr)-binding forkhead-associated 1 (FHA1) domain, and the yellow fluorescent protein (YFP) variant YPet (Nguyen and Daugherty, 2005) fused to a Jun dimerization protein 2 (JDP2) peptide linked to the JDP2 docking domain (“Substrate+DD”; Fig. 1A). Both halves of bimJNKAR are targeted to the cytoplasm via a C-terminal nuclear export signal (NES). In HeLa cells treated with anisomycin, an inhibitor of protein synthesis that robustly activates JNK, yellow fluorescence increased at the expense of cyan fluorescence, yielding a change in the yellow-to-cyan emission ratio of 36.6 ± 4.5% (n = 25) [mean ± SEM, n = number of cells]. The dynamic range of bimJNKAR is significantly larger than that of unimolecular JNKAR1-NES (20.3 ± 01.6%, n = 19, p < 0.01), most probably due to reduced levels of basal FRET in the dissociated, unstimulated state of the reporter (Fig. 1C, D). The response to anisomycin stimulation was abolished when the phosphorylation site was mutated to alanine (T/A), as well as when cells were pretreated with JNK inhibitor VIII (a JNK-specific inhibitor (Szczepankiewicz et al., 2006)) (Fig. 1B), indicating that bimJNKAR is a selective reporter of JNK activity.
Figure 1.
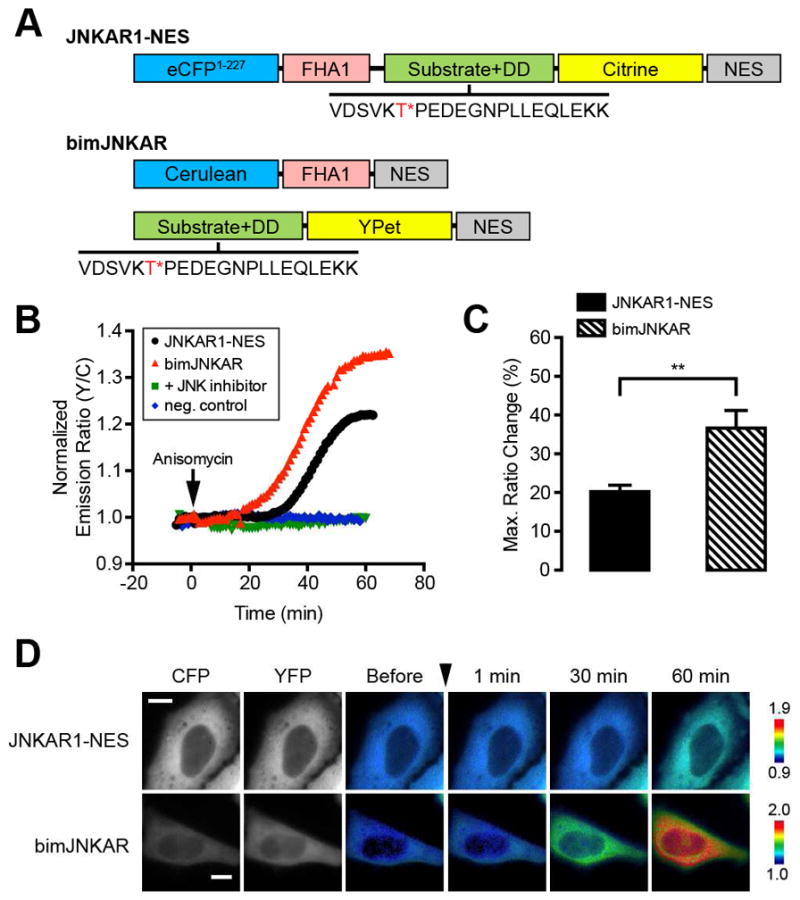
Development of a bimolecular JNK activity reporter. (A) Schematic diagram of domain structures for unimolecular JNKAR1-NES and bimolecular JNKAR (bimJNKAR). The JNK substrate/docking domain (DD) sequence is shown with the target threonine (T) residue highlighted in red and marked with an asterisk (*). (B) Representative single-cell traces of HeLa cells expressing JNKAR1-NES (black curve), bimJNKAR (red curve), or a non-phosphorylatable bimJNKAR mutant (neg. control; blue curve) treated with 5 μM anisomycin. “+ inhibitor” (green curve) indicates HeLa cells expressing bimJNKAR that were pretreated for 1 h with 20 μM JNK inhibitor VIII. (C) Summary bar graph comparing the average maximum response for JNKAR1-NES and bimJNKAR upon anisomycin treatment. Data shown represent means ± SEM. **, p < 0.01 according to unpaired Student’s t test. (D) Representative pseudocolored images showing the responses of JNKAR1-NES and bimJNKAR to 5 μM anisomycin treatment in HeLa cells. Warmer colors correspond to increasing FRET ratios. Cyan (CFP) and yellow (YFP) fluorescence images show the cellular distribution of probe fluorescence.
Substituting the PAABD in the BimKAR design
In order to further establish the adaptability of bimKARs, we evaluated the impact of changing the PAABD in addition to investigating the effect of docking domains. To date, fewer than twenty specific serine/threonine (Ser/Thr) FRET-based KARs have been generated that rely on an engineered molecular switch consisting of a kinase-specific substrate and a PAABD. Though the majority of these use FHA as the PAABD (Depry et al., 2011; Fosbrink et al., 2010; Fuller et al., 2008; Gallegos et al., 2006; Gao and Zhang, 2008; Komatsu et al., 2011; Kunkel et al., 2007; Macůrek et al., 2008; Tomida et al., 2009; Tsou et al., 2011; Wang et al., 2005), a few notable exceptions do exist. For example, EKAR utilizes the proline-directed WW domain from human Pin1 as the PAABD (Harvey et al., 2008). Unlike FHA, the WW domain recognizes both phosphoserine (pSer)- and phosphothreonine (pThr)-containing sequences. FHA1 contains eleven well-defined β sheets, whereas the much smaller WW domain consists of only two β sheets (Huang and Chang, 2011). In addition, FHA1 has a higher affinity for phosphopeptides compared to the WW domain. Dissociation constants (Kd) for FHA1 and phosphopeptides are frequently in the 100-nM range (Durocher et al., 2000; Liao et al., 2000; Yongkiettrakul et al., 2004), whereas Kds for the WW domain can range from 5 μM to 100 μM (Dalby et al., 2000; Verdecia et al., 2000; Wintjens et al., 2001). These differences in affinity pose a potential concern in the development of WW domain-based bimKARs, as the weaker affinity of the WW domain for phosphopeptides could hinder intermolecular binding to the substrate-containing fragment. To test this effect, we therefore created bimEKAR, which consists of Cerulean fused to a WW domain and Venus (YFP) fused to a Cdc25C peptide and the ERK docking site (“Substrate+DD”), with both halves tagged with an NES for cytoplasmic localization (Fig. 2A).
Figure 2.
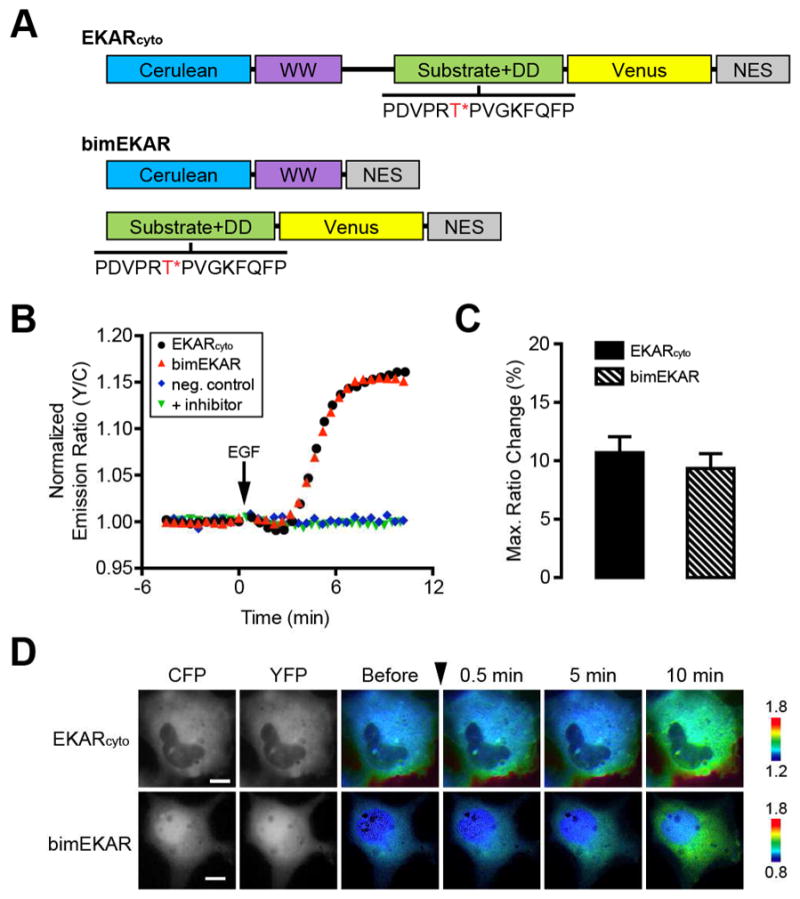
Development of a bimolecular ERK activity reporter. (A) Schematic diagram of domain structures for EKARcyto and bimEKAR. The ERK substrate/docking domain (DD) sequence is shown with the target threonine (T) residue highlighted in red and marked with an asterisk (*). (B) Representative single-cell traces of Cos7 cells expressing EKARcyto (black curve), bimEKAR (red curve), or a non-phosphorylatable bimEKAR mutant (neg. control; blue curve) treated with 100 ng/mL EGF. “+ inhibitor” (green curve) indicates Cos7 cells expressing bimEKAR that were pretreated for 1 h with 10 μM U0126. Cells were serum starved for 20 min prior to imaging. (C) Summary bar graphs comparing the average maximum responses of EKARcyto and bimEKAR upon EGF stimulation. Data shown represent means ± SEM. (D) Representative pseudocolored images showing the responses of EKARcyto and bimEKAR to 100 ng/mL EGF treatment in Cos7 cells. Warmer colors correspond to increasing FRET ratios. Cyan (CFP) and yellow (YFP) fluorescence images show the cellular distribution of probe fluorescence.
Our results suggest that the weaker binding affinity of the WW domain for phosphopeptides does not prevent it from being used in a bimolecular design. In Cos7 cells treated with epidermal growth factor (EGF), which potently stimulates ERK, bimEKAR responded with an emission ratio change of 9.3 ± 1.3% (n = 11) (Fig. 2B–D). The response to EGF stimulation was eliminated when the phosphorylation site was mutated to alanine (T/A), as well as when the cells were pretreated with the MEK inhibitor U0126 (Duncia et al., 1998) (Fig. 2B), indicating that bimEKAR selectively responds to phosphorylation by ERK. The overall dynamic range of bimEKAR is similar to that of EKARcyto (10.7 ± 1.4%, n = 7) (Fig. 2B–D); however, this may not be completely unexpected. Given that the WW domain does have a lower affinity for phosphosubstrates, it is possible that any gains in dynamic range caused by a decrease in basal FRET are being counterbalanced by this lower affinity.
Using BimKARs to Study Compartmentalized Kinase Activity
The ability to probe enzyme activities within specific subcellular compartments can provide crucial insights into how signaling pathways are organized. Given their genetic encodability, fluorescent biosensors can be targeted to different regions of the cell with relative ease by incorporating various targeting sequences (Mehta et al., 2014; Zhou et al., 2015). However, tethering a biosensor to a cellular structure such as the plasma membrane can restrict the dynamic range of the reporter, thereby making it difficult to obtain a robust signal, especially when attempting to detect subtle changes in kinase activity. For example, the previously described unimolecular reporter for AMP-dependent kinase (AMPK) activity, or AMPKAR (Tsou et al., 2011), displays a negligible response to stimulation when targeted to the plasma membrane (Fig. S1). This effect is not necessarily solvable through reporter optimization alone: we recently reported an improved AMPKAR variant, termed ABKAR (Sample et al., 2015), and while this reporter displays a substantially higher dynamic range (Fig. S1), the response from plasma membrane-tethered ABKAR is still greatly diminished (Fig. S1). Given the versatile nature of bimKARs, we therefore tested whether switching to a bimolecular design would improve the response and allow us to investigate plasma membrane compartmentalized AMPK activity.
Cytosolic bimABKAR (Fig. 3A) and plasma membrane-targeted bimABKAR-Kras (Fig. 3E) both use the same Cerulean-FHA1-NES fragment described above. For bimABKAR, the other half of the biomolecular switch contains an AMPK substrate motif identical to that found in AMPKAR and ABKAR (Sample et al., 2015; Tsou et al., 2011) fused to YPet, along with a C-terminal NES. For bimABKAR-Kras, a plasma membrane targeting motif was incorporated in place of the NES. In Cos7 cells treated with the nonhydrolyzable glucose analogue 2-deoxyglucose (2DG), which lowers ATP levels by inhibiting glycolysis, bimABKAR exhibits a FRET ratio increase of 30.6 ± 2.4% (n = 19), similar to the 36.4 ± 1.5% (n = 18) change observed using ABKAR-NES (Fig. 3B–D), while mutating the phosphorylation site within bimABKAR or pretreating cells with the AMPK-specific inhibitor compound C blocked the response to 2DG (Fig. 3B). By contrast, bimABKAR-Kras exhibited a 55.7 ± 4.1% (n = 9) ratio change, which represents a dramatic improvement over the response from ABKAR-Kras (7.0 ± 1.1%, n = 15) (Fig. 3F–H). The physical separation of the Cerulean and YPet components of bimABKAR-Kras may reduce the basal FRET and thus enhance the dynamic range of the plasma membrane-targeted bimolecular reporter. The significantly larger dynamic range of bimABKAR-Kras allows for the visualization of previously undetectable local kinase signaling and suggests that bimKAR conversion may be a useful strategy for improving the dynamic ranges of subcellularly targeted reporters, further highlighting the general utility of bimKARs.
Figure 3.

Development of a biomolecular AMKP activity reporter. (A and E) Schematic diagram of domain structures for (A) ABKAR-NES and bimABKAR or (E) ABKAR-Kras and bimABKAR-Kras. The AMPK substrate sequence is shown with the target threonine (T) residue highlighted in red and marked with an asterisk (*). (B and F) Representative single-cell traces of Cos7 cells expressing (B) ABKAR-NES (black curve), bimABKAR (red curve), or a nonphosphorylatable bimABKAR mutant (neg. control; blue curve) or (F) ABKAR-Kras (black curve) or bimABKAR-Kras (red curve) treated with 40 mM 2DG. The “+ inhibitor” (green curve) in (B) indicates Cos7 cells expressing bimABKAR that were pretreated for 1 h with 20 μM compound C. (C and G) Summary bar graphs comparing the average maximum responses of (C) ABKAR-NES and bimABKAR or (G) ABKAR-Kras and bimABKAR-Kras upon 2DG stimulation. Data shown represent means ± SEM. ***, p < 0.001; ****, p < 0.0001 according to unpaired Student’s t test. (D and H) Representative pseudocolored images showing the responses of (D) ABKAR-NES and bimABKAR or (H) ABKAR-Kras and bimABKAR-Kras to 40 mM 2DG treatment in Cos7 cells. Warmer colors correspond to increasing FRET ratios. Cyan (CFP) and yellow (YFP) fluorescence images show the cellular distribution of probe fluorescence. See also Figure S1.
Bidirectional Regulation of Plasma Membrane AMPK Activity by PKA
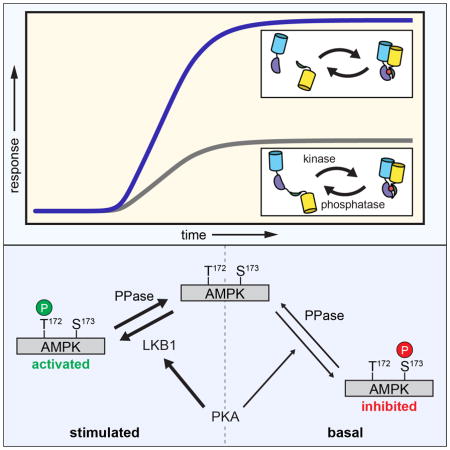
AMPK is an evolutionarily conserved master regulator of cellular and whole-body energy homeostasis. Cellular processes that deplete ATP, thus increasing AMP levels, activate AMPK, which contributes to carbohydrate, lipid, and protein metabolism, as well as cell polarity, growth, and insulin secretion (Kahn et al., 2005; Steinberg and Kemp, 2009). The latter is of particular interest, in part because metformin and thiazolidinediones, widely used drug treatments for Type 2 diabetes, have been shown to function by increasing AMPK activity (Chang et al., 2009; Puljak et al., 2008). As a “master regulator” of metabolic homeostasis, AMPK activity is modulated by at least five different kinases (Steinberg and Kemp, 2009). Prominent among these are liver kinase B1 (LKB1) and calmodulin kinase kinase (CaMKK), which both activate AMPK by phosphorylating Thr172 in the activation loop (Hawley et al., 1996; 1995; 2003; Hong et al., 2003). AMPK has also been shown to engage in a rather complex relationship with cAMP-dependent protein kinase (PKA). For instance, several studies have shown that PKA directly phosphorylates and inhibits AMPK (Cao et al., 2014; Djouder et al., 2010; Garcia-Haro et al., 2012; Hurley et al., 2006; Pulinilkunnil et al., 2011). In particular, PKA phosphorylates Ser173 within the activation loop, which occludes the activation site Thr172 (Djouder et al., 2010). However, PKA has also been shown to stimulate AMPK activity (Kimball et al., 2004; Yin et al., 2003). Notably, PKA is known to promote axon initiation by stimulating LKB1 (Shelly et al., 2007), and the elevated AMPK activity (Sample et al., 2015) and cAMP levels (Shelly et al., 2010) observed in axons may suggest a possible route for PKA to enhance AMPK activity.
Given these varying reports, we set out to further examine the dynamics of AMPK regulation by PKA in living cells. In particular, we previously demonstrated that PKA is basally active at the plasma membrane in unstimulated cells (Depry et al., 2011); therefore, we used bimABKAR-Kras to investigate how this basal PKA activity regulates AMPK activity at the plasma membrane. When Cos7 cells expressing bimABKAR-Kras were treated with H89 to inhibit basal PKA activity, we observed only a minimal change in the FRET response (1.9 ± 0.8%, n = 8) (Fig. 4A, E), although cells expressing AKAR4-Kras exhibited a 5.4 ± 0.6% (n = 9) ratio decrease, consistent with the presence of basal PKA activity (Fig. S2). However, when these cells were subsequently deprived of glucose to activate AMPK (see Experimental Procedures), we observed a dramatic emission ratio increase of 61.8 ± 8.9% (n = 8), signifying a robust plasma membrane AMPK response (Fig. 4A, E). Conversely, glucose deprivation alone resulted in a smaller ratio increase of 33.2 ± 4.2% (n = 44), which could then be increased by an additional 30.6 ± 4.0% (n = 19) upon H89 treatment (Fig. 4B, E). These results indicate that basal PKA activity is able to partially suppress the activation of plasma membrane AMPK.
Figure 4.

Bidirectional regulation of AMPK activity by PKA. Representative single-cell traces of bimABKAR-Kras-expressing Cos7 cells treated with (A) 10 μM H89 followed by glucose deprivation (−Glc), (B) glucose deprivation followed by 10 μM H89, (C) 50 μM Fsk stimulation followed by glucose deprivation, or (D) glucose deprivation followed by 50 μM Fsk. (E) Summary bar graphs showing the overall responses of Cos7 cells transfected with bimABKAR-Kras and treated with 10 μM H89 (white), glucose deprivation (−Glc, black), or 50 μM Fsk (grey). Data shown represent means ± SEM. See also Figure S2.
Interestingly, stimulating PKA activity by treating the cells with the adenylyl cyclase activator forskolin (Fsk) (Fig. S2) led to a gradual, 37.7 ± 5.6% (n = 5) increase in the bimABKAR-Kras FRET response, which could then be increased by an additional 22.4 ± 3.6% (n = 5) following glucose deprivation (Fig. 4C, E). Yet when AMPK activity was stimulated first via glucose deprivation, the resulting bimABKAR-Kras response could not be further enhanced by PKA stimulation. These results not only confirm that PKA can both negatively and positively regulate AMPK activity but also reveal that this bidirectional regulation can co-exist within the same cell.
PKA Stimulates Local AMPK Activity via LKB1 Phosphorylation
PKA is known to upregulate AMPK activity by phosphorylating Ser431 of LKB1, an upstream kinase that is required for AMPK to respond to metabolic stimuli such as glucose deprivation (Shaw et al., 2004). In keeping with these effects, we found that LKB1 phosphorylation at Ser431 was significantly increased within 5 min of Fsk treatment in Cos7 cells and blocked by H89 treatment (Fig. 5A, B). Therefore, to further explore the regulation of plasma membrane-localized AMPK activity by PKA, we repeated the above experiments in HeLa cells, which do not express LKB1 (Tiainen et al., 1999). In these cells, neither PKA activation by Fsk nor glucose deprivation were capable of eliciting a strong response from bimABKAR-Kras (Fig. 5C, D). Moreover, the AMPK response was rescued by heterologous overexpression of wild-type LKB1 but not the kinase-dead LKB1-K78I mutant (LKB1 KD) (Shaw et al., 2004) (Fig. 5C, D). In LKB1 WT-expressing cells, Fsk stimulation induced a 15.9 ± 1.7% (n = 14) FRET increase, followed by an additional 19.0 ± 2.4% (n = 14) increase upon glucose deprivation, whereas untransfected and LKB1 KD-expressing cells exhibited only 3.2 ± 0.7% (n = 14) and 4.0 ± 1.4% (n = 7) responses to Fsk and 7.5 ± 1.2% (n = 16) and 5.6 ± 0.8% (n = 12) responses to glucose deprivation (Fig. 5D), respectively. In addition, expressing the LKB1-S431A mutant (LKB1 SA), which lacks the C-terminal PKA phosphorylation site, largely abolished Fsk-stimulated AMPK activity (6.2 ± 1.1%, n = 11) in these cells while also somewhat diminishing the response to glucose deprivation (12.1 ± 1.3%, n = 11) (Fig. 5C, D). As a control, HeLa cells transfected with AKAR4-Kras displayed a clear increase and decrease in FRET following Fsk and H89 treatment, respectively, whereas glucose deprivation had no effect (Fig. S3). Interestingly, H89 treatment led to similar increases in the bimABKAR-Kras response regardless of LKB1 expression (Fig. 5C, D), which were abolished by pretreatment with the Ca2+ chelator BAPTA (Fig. S3). Like LKB1, CaMKII also serves as an upstream activating kinase for AMPK, and our results suggest the presence of basal, CaMKK-mediated AMPK activity that remains under direct negative regulation by PKA.
Figure 5.

PKA stimulates AMPK activity via LKB1 phosphorylation. (A) Representative western blot showing increase in endogenous LKB1 phosphorylation (p-LKB1S431) relative to total LKB1 levels in Cos7 cells. 0′, untreated; 5′, 50 μM Fsk for 5 min; 45′, 50 μM Fsk for 45 min; H89+Fsk; 10 μM H89 for 10 min followed by 50 μM Fsk for 45 min. (B) Quantification of western blot data from (A). Data shown represent means ± SEM (n = 3). ***, p < 0.001 vs. untreated cells (0′) according to one-way ANOVA followed by Tukey’s multiple comparisons test. (C) Representative single-cell traces showing the FRET ratio change in HeLa cells expressing bimABKAR-Kras alone (−LKB1, black curve) or cotransfected with wild-type LKB1 (LKB1 WT, red curve) kinase-dead LKB1-K781 (LKB1 KD, blue curve), or non-phosphorylatable LKB1-S431A (LKB1 SA, orange curve) and sequentially treated with 50 μM Fsk, glucose deprivation (−Glc), and 10 μM H89 at the indicated times. (D) Bar graph summarizing the maximum individual responses to each treatment shown in (C). Data shown represent means ± SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 according to unpaired Student’s t test. See also Figure S3.
DISCUSSION
Despite their overall success in providing new insights into signal transduction by visualizing real-time kinase signaling dynamics in living cells, KARs often suffer from a constrained dynamic range. Building upon the basic modular design of KARs, which consists of a FRET pair and a kinase-inducible molecular switch, we developed bimKARs to help overcome this limitation. This study cements bimKARs as versatile tools to study kinase signaling. First, we showed that bimKARs can be constructed to successfully incorporate kinase docking domains, as evidenced by bimJNKAR. Using bimEKAR, we further demonstrated that the inclusion of a lower-affinity PAABD does not hinder the function of bimKARs.
One advantage of the bimKAR system over unimolecular KARs is the potential for increased dynamic range, which allows the visualization of small, yet physiologically relevant changes in kinase signaling. For instance, the markedly enhanced dynamic range of bimABKAR-Kras allowed us to visualize stimulated AMPK activity near the plasma membrane. As stated above, the enhanced range may be explained by the inherent physical separation of the FP pair in the bimKAR system. While an increased dynamic range is desired, it is not always observed, as evidenced by bimEKAR and EKAR; we observed no difference in the dynamic range of these two biosensors. However, this may be explained by the long, 72-Gly linker found in EKAR, which already provides a considerable amount of separation between the two halves of the molecular switch, mimicking a bimolecular system. Other characteristics unique to the bimKAR system are less advantageous. For instance, the stoichiometry of the donor and the acceptor FPs is not fixed, a feature that is inherent to the unimolecular design. To ensure that signal analysis is performed correctly, special care must be taken to balance the expression levels of the two components of bimolecular reporters. Unequal amounts of the two halves result in variable ratios that will both compromise the sensitivity of the biosensors and make quantification more complex (Zhang et al., 2002). Another concern when using bimKARs is that they are more likely to interact with endogenous molecules because the individual components of the molecular switch are expressed independently (Miyawaki, 2003; Zhou et al., 2012). For instance, if the PAABD portion of the reporter were to bind endogenous substrates or to have to compete with endogenous PAABDs for the other half of the molecular switch, the performance of the reporter and/or cellular signaling machinery would be hindered. Nevertheless, this does not appear to pose a problem for the current bimKARs, given their robust responses. Moreover, as bimKARs are more sensitive to diffusion constraints, there is the potential for them to display reduced temporal sensitivity compared with their unimolecular counterparts (Zhou et al., 2012).
In addition to testing the bimKAR design, we used these reporters to investigate plasma membrane AMPK activity. We found that basal PKA activity is able to both suppress basal and dampen stimulated AMPK activity. Phosphorylation of AMPK by PKA prior to stimulation may provide an efficient means to inhibit AMPK and thus tightly control its regulation. Glucose-starvation-induced AMPK activity was previously shown to be inhibited following 30 min of PKA activation, as indicated by the reduced phosphorylation of AMPK at the activation site Thr172 (Djouder et al., 2010). Consistent with the ability of PKA to inactivate AMPK, we found that inhibiting basal PKA activity by treating cells with H89, either before or after acute glucose deprivation, enhanced plasma membrane AMPK activity. However, increasing PKA activity appeared to have no effect on glucose-starvation-induced AMPK activity (Fig. 4D), perhaps due to the different cell types used or to differences in whole-cell versus plasma membrane AMPK activity. On the other hand, cAMP-elevating stimuli have also been shown to enhance AMPK phosphorylation at Thr172 (Kimball et al., 2004; Yin et al., 2003). Indeed, we found that stimulating PKA activity in the presence of glucose actually induced AMPK activity, which was further increased by glucose deprivation.
The stimulatory effect of PKA towards AMPK is thought to be mediated by the phosphorylation of LKB1 at Ser431, though this remains controversial. For example, whereas some studies have shown PKA to be necessary for LKB1 signaling and AMPK activation (Kimball et al., 2004; Martin and St Johnston, 2003; Sapkota et al., 2001; Shelly et al., 2007), others indicate that LKB1 activity is invariant (Lizcano et al., 2004). Notably, Fogarty and colleagues found that Ser431 was not required for LKB1 to be fully active (Fogarty and Hardie, 2009). Nevertheless, our results strongly implicate PKA as a positive regulator of AMPK activity via LKB1; not only did we observe the rapid induction of LKB1 Ser431 phosphorylation in Fsk-stimulated Cos7 cells, both LKB1 catalytic activity and Ser431 were required to recapitulate PKA-induced AMPK activity in HeLa cells. In fact, the LKB1-S431A mutant was less capable of stimulating AMPK activity in response to glucose deprivation than the wild-type protein (Fig. 5C, D), in agreement with a role for PKA in promoting LKB1 function (Martin and St Johnston, 2003; Sapkota et al., 2001; Shelly et al., 2007). Meanwhile, the inhibitory effect of basal PKA activity persisted in the absence of LKB1 and appeared to correspond to the direct inhibition of AMPK seen previously (Djouder et al., 2010; Hurley et al., 2006). PKA has been observed to undergo a switch from short-term AMPK inhibition to long-term AMPK stimulation (Djouder et al., 2010); however, our results are the first to demonstrate that these opposing regulatory mechanisms can operate concurrently in the same cell. Specifically, our results are consistent with a model in which basally active PKA weakly phosphorylates AMPK (represented here by Ser173), creating a pool of inactivated AMPK. Meanwhile, stimulating PKA leads to the preferential phosphorylation of LKB1, which then rapidly and strongly activates naïve AMPK via phosphorylation at Thr172 (Fig. 6). Additional experiments are needed to refine this model, especially to clarify the specific PKA phosphorylation site(s) on AMPK.
Figure 6.

Model of AMPK regulation by PKA. In resting (i.e., fed) cells, AMPK undergoes rapid basal phosphorylation at Thr172 by activating kinases and weak basal phosphorylation by PKA at inhibitory sites (here represented by Ser173). Whereas Thr172 is rapidly dephosphorylated by cellular phosphatase activity (PPase), inhibitory phosphorylation by PKA is only weakly antagonized, leading to the accumulation of a stable pool of inactivated AMPK. At the same time, however, starvation or PKA stimulation preferentially induces the phosphorylation of LKB1, leading to the robust activation of naïve AMPK via Thr172 phosphorylation.
In conclusion, we report that bimKARs are a versatile and adaptable set of tools for the study of real-time kinase signaling dynamics in live cells, capable of accommodating docking domains, as well as various PAABDs, with the potential to offer enhanced dynamic ranges versus unimolecular KARs. Furthermore, using a plasma membrane-targeted bimKAR, we were able to visualize AMPK activity for the first time at plasma membrane and show that PKA activity can alternately serve as both a brake and accelerator in the regulation of AMPK signaling.
SIGNIFICANCE
Communication between and within cells enables our bodies to maintain proper functioning. Elucidating the mechanisms that underlie proper signaling benefits greatly from the use of real-time approaches to monitor processes whose dynamics can be lost with approaches that only offer static glimpses. Fluorescence Resonance Energy Transfer (FRET)-based biosensors allow for live-cell imaging of kinase dynamics. FRET-based biosensors are comprised of two modules: a FRET pair and a kinase-inducible molecular switch. We assessed the versatility and adaptable nature of FRET-based, bimolecular Kinase Activity Reporters (bimKARs), which have proven capable of functioning with additional or new domains. For example, a kinase-specific docking domain was added to increase phosphorylation specificity and efficiency, and an alternate phosphoamino acid-binding domain was added to demonstrate the generalizable nature of the bimKAR system. Finally, an AMP-activated protein kinase-specific bimKAR targeted to the plasma membrane revealed the complex regulation of this kinase by PKA activity. Overall, our studies highlight the many uses afforded by this system and demonstrate how this class of biosensor will aid in our understanding of signaling networks.
EXPERIMENTAL PROCEDURES
Detailed experimental procedures are provided in the Supplemental Experimental Procedures.
Plasmid Construction
All plasmids were generated using standard molecular biology techniques (see Supplemental Experimental Procedures). Details of all biosensors used in this work are summarized in Table S1.
Cell Culture and Transfection
HeLa cells were cultured in Dulbecco minimal Eagle Medium (DMEM; Gibco, Grant Island, NY) containing 1 g/L D-glucose and supplemented with 10% fetal bovine serum (FBS; Sigma, St. Louis, MO) and 1% pen/strep (Sigma-Aldrich, St. Louis, MO). Cos7 cells were cultured in DMEM containing 4.5 g/L D-glucose and supplemented with 10% FBS, 1% pen/strep, and 1 mM NaPO4. Both cell lines were maintained at 37 °C with 5% CO2. For imaging, cells were plated onto sterile 35-mm glass-bottom dishes, transfected with Lipofectamine2000 (Invitrogen) at 60–70% confluency, and then grown for 24–48 h.
Western Blotting
Cos7 cells were grown to 80% confluence in 6-cm dishes and treated with 50 μM Fsk with or without 10 μM H89 as indicated. Cells were then washed with ice-cold Dulbecco’s phosphate-buffered saline (DPBS; Gibco) and lysed in RIPA lysis buffer containing protease inhibitor cocktail (Complete, EDTA-Free; Roche), 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, and 25 nM calyculin A. Total cell lysates were incubated on ice for 30 min and then centrifuged at 13,000 rpm for 30 min at 4°C. Protein concentrations were determined using a BCA assay kit (Pierce); 20 μg of protein from each sample was separated via 7.5% SDS-PAGE and then transferred to nitrocellulose. Membranes were blocked for 1 h at room temperature in Tris-buffered saline (TBS) containing 0.1% Tween-20 (TBST) and 5% bovine serum albumin (BSA) and then incubated overnight at 4°C with primary antibodies against p-LKB1-S431 (1:1000, EMD-Millipore, Darmstadt, Germany) or human LKB1 (1:1000, Cell Signaling, Danvers, MA) in TBST plus 5% BSA. After incubation with horseradish peroxidase-conjugated goat-anti-rabbit secondary antibody (Pierce), the bands were visualized using enhanced chemiluminescent detection (Pierce). Band intensities were quantified using ImageJ software. p-LKB1 levels were first normalized to total LKB1 expression in the same sample and then divided by the untreated value to determine relative expression.
Imaging and Data Analysis
Cells were washed twice with Hank’s Balanced Salt Solution (HBSS; Gibco) supplemented with 20 mM HEPES, pH 7.4, and 2.0 g/L D-glucose, then imaged in the dark at 37 °C. Prior to imaging bimEKAR, cells were pre-incubated in HBSS for 20 min at 37 °C. Cells were treated with drugs as indicated. For glucose deprivation (−Glc), cells were initially imaged in HBSS containing 4.5 g/L (25 mM) D-glucose. The glucose-containing medium was then washed out and replaced with glucose-free HBSS. Images were acquired on an Zeiss Axiovert 200M microscope (Carl Zeiss, Thornwood, NY) equipped with a 40x/1.3 NA oil-immersion objective lens and a cooled charge-coupled device camera (Roper Scientific, Trenton, NJ) controlled by Metafluor 7.7 software (Molecular Devices, Sunnyvale, CA). Dual emission ratio imaging was performed using a 420DF20 excitation filter, a 450DRLP dichroic mirror, and two emission filters (475DF40 for CFP and 535DF25 for YFP). Filter sets were alternated using a Lambda 10-2 filter changer (Sutter Instruments, Novato, CA). Exposure times were 50–500 ms, and images were acquired every 30–60 s. Background correction of the fluorescence images was performed by subtracting the intensities of untransfected cells or regions of the imaging dish with no cells. Time-courses were normalized by setting the emission ratio before drug addition equal to one. Graphs were plotted using GraphPad Prism version 5.0f (GraphPad Software, La Jolla, CA), and statistical analyses were performed using the same software. Statistical significance was set at p < 0.05.
Supplementary Material
HIGHLIGHTS.
The versatile bimKAR design is easily adapted to monitor various kinase activities
A membrane-targeted bimKAR improves the detection of local AMPK activity
Membrane-localized AMPK activity undergoes opposing bidirectional regulation by PKA
Acknowledgments
The authors are grateful to Karel Svoboda and Lewis Cantley for generously providing constructs. We also wish to thank Vedangi Sample, Santosh Ramamurthy, and Kirill Gorshkov for their comments on the manuscript. This work was supported by the National Institutes of Health (R01 DK073368 and DP1 CA174423 to J.Z. and F31 GM087079 to C.D.)
Footnotes
AUTHOR CONTRIBUTIONS
CD and JZ conceived of the study; CD, SM, and RL generated reagents, performed experiments, and analyzed data; and CD, SM, and JZ wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoki K, Kumagai Y, Sakurai A, Komatsu N, Fujita Y, Shionyu C, Matsuda M. Stochastic ERK activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol Cell. 2013;52:529–540. doi: 10.1016/j.molcel.2013.09.015. [DOI] [PubMed] [Google Scholar]
- Cao J, Meng S, Chang E, Beckwith-Fickas K, Xiong L, Cole RN, Radovick S, Wondisford FE, He L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK) J Biol Chem. 2014;289:20435–20446. doi: 10.1074/jbc.M114.567271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TJ, Chen WP, Yang C, Lu PH, Liang YC, Su MJ, Lee SC, Chuang LM. Serine-385 phosphorylation of inwardly rectifying K+ channel subunit (Kir6.2) by AMP-dependent protein kinase plays a key role in rosiglitazone-induced closure of the K(ATP) channel and insulin secretion in rats. Diabetologia. 2009;52:1112–1121. doi: 10.1007/s00125-009-1337-4. [DOI] [PubMed] [Google Scholar]
- Dalby PA, Hoess RH, DeGrado WF. Evolution of binding affinity in a WW domain probed by phage display. Protein Sci. 2000;9:2366–2376. doi: 10.1110/ps.9.12.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depry C, Zhang J. Visualization of kinase activity with FRET-based activity biosensors. Curr Protoc Mol Biol. 2010;Chapter 18(Unit18.15) doi: 10.1002/0471142727.mb1815s91. [DOI] [PubMed] [Google Scholar]
- Depry C, Allen MD, Zhang J. Visualization of PKA activity in plasma membrane microdomains. Mol Biosyst. 2011;7:52. doi: 10.1039/c0mb00079e. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP. Scaffold proteins of MAP-kinase modules. Oncogene. 2007;26:3185–3202. doi: 10.1038/sj.onc.1210411. [DOI] [PubMed] [Google Scholar]
- Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, et al. PKA phosphorylates and inactivates AMPKα to promote efficient lipolysis. Embo J. 2010;29:469–481. doi: 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncia JV, Santella JB, Higley CA, Pitts WJ, Wityak J, Frietze WE, Rankin FW, Sun JH, Earl RA, Tabaka AC, et al. MEK inhibitors: the chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg Med Chem Lett. 1998;8:2839–2844. doi: 10.1016/s0960-894x(98)00522-8. [DOI] [PubMed] [Google Scholar]
- Durocher D, Taylor IA, Sarbassova D, Haire LF, Westcott SL, Jackson SP, Smerdon SJ, Yaffe MB. The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol Cell. 2000;6:1169–1182. doi: 10.1016/s1097-2765(00)00114-3. [DOI] [PubMed] [Google Scholar]
- Fogarty S, Hardie DG. C-terminal phosphorylation of LKB1 is not required for regulation of AMP-activated protein kinase, BRSK1, BRSK2, or cell cycle arrest. J Biol Chem. 2009;284:77–84. doi: 10.1074/jbc.M806152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosbrink M, Aye-Han NN, Cheong R, Levchenko A, Zhang J. Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc Natl Acad Sci USA. 2010;107:5459–5464. doi: 10.1073/pnas.0909671107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller BG, Lampson MA, Foley EA, Rosasco-Nitcher S, Le KV, Tobelmann P, Brautigan DL, Stukenberg PT, Kapoor TM. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature. 2008;453:1132–1136. doi: 10.1038/nature06923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos LL, Kunkel MT, Newton AC. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J Biol Chem. 2006;281:30947–30956. doi: 10.1074/jbc.M603741200. [DOI] [PubMed] [Google Scholar]
- Gao X, Zhang J. Spatiotemporal analysis of differential Akt regulation in plasma membrane microdomains. Mol Biol Cell. 2008;19:4366–4373. doi: 10.1091/mbc.E08-05-0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Haro L, Garcia-Gimeno MA, Neumann D, Beullens M, Bollen M, Sanz P. Glucose-dependent regulation of AMP-activated protein kinase in MIN6 beta cells is not affected by the protein kinase A pathway. FEBS Lett. 2012;586:4241–4247. doi: 10.1016/j.febslet.2012.10.032. [DOI] [PubMed] [Google Scholar]
- Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, Svoboda K. A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci USA. 2008;105:19264–19269. doi: 10.1073/pnas.0804598105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Selbert MA, Goldstein EG, Edelman AM, Carling D, Hardie DG. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem. 1995;270:27186–27191. doi: 10.1074/jbc.270.45.27186. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst KJ, Allen MD, Zhang J. Luminescent kinase activity biosensors based on a versatile bimolecular switch. J Am Chem Soc. 2011;133:5676–5679. doi: 10.1021/ja1117396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson L, Shen F, Hahn K. Biosensors for characterizing the dynamics of rho family GTPases in living cells. Curr Protoc Cell Biol. 2010;Chapter 14(Unit14.11.1–Unit14.11.26) doi: 10.1002/0471143030.cb1411s46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA. 2003;100:8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YMM, Chang CEA. Mechanism of PhosphoThreonine/Serine Recognition and Specificity for Modular Domains from All-atom Molecular Dynamics. BMC Biophys. 2011;4:12. doi: 10.1186/2046-1682-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley RL, Barré LK, Wood SD, Anderson KA, Kemp BE, Means AR, Witters LA. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem. 2006;281:36662–36672. doi: 10.1074/jbc.M606676200. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metabolism. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Katz S, Aronheim A. Differential targeting of the stress mitogen-activated protein kinases to the c-Jun dimerization protein 2. Biochem J. 2002;368:939–945. doi: 10.1042/BJ20021127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball SR, Siegfried BA, Jefferson LS. Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J Biol Chem. 2004;279:54103–54109. doi: 10.1074/jbc.M410755200. [DOI] [PubMed] [Google Scholar]
- Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, Matsuda M. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell. 2011;22:4647–4656. doi: 10.1091/mbc.E11-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel MT, Toker A, Tsien RY, Newton AC. Calcium-dependent regulation of protein kinase D revealed by a genetically encoded kinase activity reporter. J Biol Chem. 2007;282:6733–6742. doi: 10.1074/jbc.M608086200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H, Yuan C, Su MI, Yongkiettrakul S, Qin D, Li H, Byeon IJ, Pei D, Tsai MD. Structure of the FHA1 domain of yeast Rad53 and identification of binding sites for both FHA1 and its target protein Rad9. J Mol Biol. 2000;304:941–951. doi: 10.1006/jmbi.2000.4291. [DOI] [PubMed] [Google Scholar]
- Liao X, Lu S, Wu Y, Xu W, Zhuo Y, Peng Q, Li B, Zhang L, Wang Y. The effect of differentiation induction on FAK and Src activity in live HMSCs visualized by FRET. PLoS ONE. 2013;8:e72233. doi: 10.1371/journal.pone.0072233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455:119–123. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- Martin SG, St Johnston D. A role for Drosophila LKB1 in anterior-posterior axis formation and epithelial polarity. Nature. 2003;421:379–384. doi: 10.1038/nature01296. [DOI] [PubMed] [Google Scholar]
- Mehta S, Zhang J. Reporting from the field: genetically encoded fluorescent reporters uncover signaling dynamics in living biological systems. Annu Rev Biochem. 2011;80:375–401. doi: 10.1146/annurev-biochem-060409-093259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Aye-Han N-N, Ganesan A, Oldach L, Gorshkov K, Zhang J. Calmodulin-controlled spatial decoding of oscillatory Ca2+ signals by calcineurin. Elife. 2014:e03765. doi: 10.7554/eLife.03765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A. Visualization of the spatial and temporal dynamics of intracellular signaling. Dev Cell. 2003;4:295–305. doi: 10.1016/s1534-5807(03)00060-1. [DOI] [PubMed] [Google Scholar]
- Newman RH, Fosbrink MD, Zhang J. Genetically Encodable Fluorescent Biosensors for Tracking Signaling Dynamics in Living Cells. Chem Rev. 2011;111:3614–3666. doi: 10.1021/cr100002u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- Ni Q, Ganesan A, Aye-Han NN, Gao X, Allen MD, Levchenko A, Zhang J. Signaling diversity of PKA achieved via a Ca2+-cAMP-PKA oscillatory circuit. Nat Chem Biol. 2011;7:34–40. doi: 10.1038/nchembio.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulinilkunnil T, He H, Kong D, Asakura K, Peroni OD, Lee A, Kahn BB. Adrenergic regulation of AMP-activated protein kinase in brown adipose tissue in vivo. J Biol Chem. 2011;286:8798–8809. doi: 10.1074/jbc.M111.218719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puljak L, Parameswara V, Dolovcak S, Waldrop SL, Emmett D, Esser V, Fitz JG, Kilic G. Evidence for AMPK-dependent regulation of exocytosis of lipoproteins in a model liver cell line. Exp Cell Res. 2008;314:2100–2109. doi: 10.1016/j.yexcr.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Rothman DM, Shults MD, Imperiali B. Chemical approaches for investigating phosphorylation in signal transduction networks. Trends Cell Biol. 2005;15:502–510. doi: 10.1016/j.tcb.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Sample V, Ramamurthy S, Gorshkov K, Ronnett GV, Zhang J. Polarized Activities of AMPK and BRSK in Primary Hippocampal Neurons. Mol Biol Cell. 2015 doi: 10.1091/mbc.E14-02-0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota GP, Kieloch A, Lizcano JM, Lain S, Arthur JS, Williams MR, Morrice N, Deak M, Alessi DR. Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell growth. J Biol Chem. 2001;276:19469–19482. doi: 10.1074/jbc.M009953200. [DOI] [PubMed] [Google Scholar]
- Sato M, Umezawa Y. Imaging protein phosphorylation by fluorescence in single living cells. Methods. 2004;32:451–455. doi: 10.1016/j.ymeth.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Sato M, Ozawa T, Inukai K, Asano T, Umezawa Y. Fluorescent indicators for imaging protein phosphorylation in single living cells. Nat Biotechnol. 2002;20:287–294. doi: 10.1038/nbt0302-287. [DOI] [PubMed] [Google Scholar]
- Sharma V, Wang Q, Lawrence DS. Peptide-based fluorescent sensors of protein kinase activity: design and applications. Biochim Biophys Acta. 2008;1784:94–99. doi: 10.1016/j.bbapap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrocks AD, Yang SH, Galanis A. Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem Sci. 2000;25:448–453. doi: 10.1016/s0968-0004(00)01627-3. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565–577. doi: 10.1016/j.cell.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Shelly M, Lim BK, Cancedda L, Heilshorn SC, Gao H, Poo MM. Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science. 2010;327:547–552. doi: 10.1126/science.1179735. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz BG, Kosogof C, Nelson LTJ, Liu G, Liu B, Zhao H, Serby MD, Xin Z, Liu M, Gum RJ, et al. Aminopyridine-based c-Jun N-terminal kinase inhibitors with cellular activity and minimal cross-kinase activity. J Med Chem. 2006;49:3563–3580. doi: 10.1021/jm060199b. [DOI] [PubMed] [Google Scholar]
- Tarrant MK, Cole PA. The chemical biology of protein phosphorylation. Annu Rev Biochem. 2009;78:797–825. doi: 10.1146/annurev.biochem.78.070907.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiainen M, Ylikorkala A, Mäkelä TP. Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA. 1999;96:9248–9251. doi: 10.1073/pnas.96.16.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting AY, Kain KH, Klemke RL, Tsien RY. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc Natl Acad Sci USA. 2001;98:15003–15008. doi: 10.1073/pnas.211564598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida T, Takekawa M, O’Grady P, Saito H. Stimulus-specific distinctions in spatial and temporal dynamics of stress-activated protein kinase kinase kinases revealed by a fluorescence resonance energy transfer biosensor. Mol Cell Biol. 2009;29:6117–6127. doi: 10.1128/MCB.00571-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou P, Zheng B, Hsu CH, Sasaki AT, Cantley LC. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metabolism. 2011;13:476–486. doi: 10.1016/j.cmet.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol. 2000;7:639–643. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- Violin JD, Zhang J, Tsien RY, Newton AC. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol. 2003;161:899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, Chien S. Visualizing the mechanical activation of Src. Nature. 2005;434:1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- Wintjens R, Wieruszeski JM, Drobecq H, Rousselot-Pailley P, Buée L, Lippens G, Landrieu I. 1H NMR study on the binding of Pin1 Trp-Trp domain with phosphothreonine peptides. J Biol Chem. 2001;276:25150–25156. doi: 10.1074/jbc.M010327200. [DOI] [PubMed] [Google Scholar]
- Yin W, Mu J, Birnbaum MJ. Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis In 3T3-L1 adipocytes. J Biol Chem. 2003;278:43074–43080. doi: 10.1074/jbc.M308484200. [DOI] [PubMed] [Google Scholar]
- Yongkiettrakul S, Byeon IJL, Tsai MD. The ligand specificity of yeast Rad53 FHA domains at the +3 position is determined by nonconserved residues. Biochemistry. 2004;43:3862–3869. doi: 10.1021/bi036195f. [DOI] [PubMed] [Google Scholar]
- Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- Zhou X, Clister TL, Lowry PR, Seldin MM, Wong GW, Zhang J. Dynamic Visualization of mTORC1 Activity in Living Cells. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Herbst-Robinson KJ, Zhang J. Visualizing dynamic activities of signaling enzymes using genetically encodable FRET-based biosensors from designs to applications. Meth Enzymol. 2012;504:317–340. doi: 10.1016/B978-0-12-391857-4.00016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.