

Abstract
Background Interaction of advanced glycation end products (AGE) with the receptor for advanced glycation end products (RAGE) has been implicated in the pathogenesis of atherosclerosis. Soluble receptors for advanced glycation end products (sRAGE) act as a decoy for AGE by competing with RAGE and suppressing developing atherosclerosis. Hypercholesterolemia and the oxidative stress are known factors involved in atherosclerosis. High-density lipoprotein cholesterol (HDL-C) is known to exert a protective effect against the development of atherosclerosis.
We hypothesize that hypercholesterolemia-induced atherosclerosis may be mediated through the AGE–RAGE axis.
Objectives Two objectives to be determined are: (1) if hypercholesterolemia is positively correlated with serum AGE, AGE/sRAGE, and malondialdehyde (MDA: a marker for oxidative stress) and (2) if the protective effect of HDL-C is positively associated with serum sRAGE and negatively correlated with the levels of AGE and AGE/sRAGE.
Methods Measurement of serum lipid levels from 100 patients allowed the separation into two groups (hypercholesterolemic and normocholesterolemic). Measurements of serum levels of AGE, sRAGE, and MDA were performed.
Results Serum levels of sRAGE were lower, while the levels of AGE and AGE/sRAGE were higher in hypercholesterolemic subjects as compared with normocholesterolemic subjects. sRAGE levels are positively correlated with HDL, while they are negatively correlated with low-density lipoprotein, triglycerides, total cholesterol, and MDA in hypercholesterolemic subjects.
Conclusions Hypercholesterolemia is positively correlated with serum AGE, AGE/sRAGE, and MDA. The effect of HDL-C may be due to increases in sRAGE and decreases in the levels of AGE and AGE/sRAGE. Hypercholesterolemia-induced atherosclerosis may be mediated through the AGE–RAGE axis; however, more research must be conducted.
Keywords: atherosclerosis, advanced glycation end product, hypercholesterolemia, oxidative stress, coronary artery
Reactive oxygen species (ROS) have been implicated in the initiation and the progression of hypercholesterolemia-induced atherosclerosis.1 2 3 Hypercholesterolemia increases the generation of ROS through various mechanisms.4 5 Additionally, hypercholesterolemia increases the activity of nicotinamide adenine dinucleotide phosphate-oxidase (NADPH oxidase), xanthine oxidase,6 and myeloperoxidase7 that leads to the generation of ROS. Hypercholesterolemia increases the generation of proinflammatory cytokines.8 Cytokines are known to generate ROS through mitochondria and NADPH oxidase.9 Cytokines also increase the expression of the receptor for advanced glycation end products (RAGE).10 The advanced glycation end products (AGE) receptor (RAGE) has also been implicated in the development of atherosclerosis.11 12 13
It has been demonstrated that the AGE–RAGE interaction in endothelial cells activates intracellular production of ROS that are involved in signal transduction, which in turn produce the expression of vascular cell adhesion molecule 1 (VCAM-1). Utilizing specific inhibitors (apocynin and diphenylene iodium) for NADPH oxidase and thenoyltrifluoroacetone for mitochondrial electron transport chain, the investigators found a significant decrease in AGE-induced ROS generation and VCAM-1 expression in cultured endothelial cells. The data gathered from their study lend support to studies which demonstrate that AGE interaction with RAGE induces ROS, which subsequently stimulates the redox-sensitive transcription factor NF-κB to induce the expression of response to injury genes such as VCAM-1, which has been shown to be present in early atherosclerotic lesion formation.14 15 Since it has been shown that cytokines and adhesion molecules are produced from the AGE–RAGE interaction,14 15 we have not measured these biomolecules in the present study. However, the interplay between ROS, cytokines, and AGE in hypercholesterolemic atherosclerosis is not fully understood.
Hypercholesterolemia, AGE, and sRAGE
Hypercholesterolemia has been implicated in the development of atherosclerosis.16 17 The relationship between serum levels of hypercholesterolemia and sRAGE is poorly understood. Devangelio et al have reported no significant difference in plasma sRAGE levels between normocholesterolemic control subjects and type 2 diabetic, hypercholesterolemic patients.18 However, increased levels of sRAGE in animal models treated with statin therapy have been reported.19 Specifically, the interaction of AGE with its cell-bound receptor results in the generation of ROS (i.e., hypochlorous acid, hydrogen peroxide),14 activation of NF-κB resulting in increased expression of cytokines (i.e., tumor necrosis factor-α [TNF-α], interleukin-1) adhesion molecules (i.e., VCAM-1), growth factors (i.e., vascular endothelial growth factor), tissue factor-1, and the generation of more ROS.20 21 22 In addition, the interaction of AGE with the vascular endothelial cell surface RAGE activates NADPH oxidase, resulting in the generation of ROS (superoxide anions, hydroxyl radicals).23 Hypercholesterolemic diet–induced atherosclerosis is associated with hyperlipidemia and increased oxidative stress.24 25
Based on the above background, we hypothesize that patients' with hypercholesterolemia-induced atherosclerosis will have higher levels of oxidative stress, AGE, AGE/sRAGE ratio, and lower sRAGE levels as compared with patients with normal cholesterol levels.
The main objectives are to investigate if: (1) the levels of AGE, AGE/sRAGE, and malondialdehyde (MDA: a marker for oxidative stress) are higher and that of sRAGE lower in hypercholesterolemic subjects as compared with normocholesterolemic subjects; (2) hypercholesterolemia is positively correlated with serum AGE, AGE/sRAGE, and MDA and negatively correlated with sRAGE; and (3) high-density lipoprotein cholesterol (HDL-C) is positively correlated with serum sRAGE and negatively correlated with the levels of AGE and AGE/sRAGE.
Materials and Methods
Patient Selection
One hundred consecutive, Caucasian, male and female patients diagnosed as acute coronary syndrome (ACS) non-ST-segment myocardial infarction (NSTEMI) (by the attending cardiologist) signed a written consent for participation in the study. The University of Saskatchewan Biomedical Research Ethics Board approved the study protocol. The Royal University Hospital Ethics Committee also approved the study. Blood samples from these patients were drawn prior to statin treatment. Patients were separated into two groups (hypercholesterolemic [cholesterol levels > 5.17 mmol/L] and normocholesterolemic [cholesterol levels < 5.17 mmol/L]) based upon their levels of serum cholesterol. The desirable range for total cholesterol (TC) is below 5.17 mmol/L. Therefore, we used this value as the cutoff to separate hypercholesterolemia from normal cholesterol levels.26 The clinical characteristics of the study patients were obtained through direct interviews with the patients prior to the procedure or with permission from the patients' files. The demographic and clinical characteristics are summarized in Table 1. All patients of the study met the following inclusion criteria: (1) ACS patients who are of the NSTEMI subclass and (2) age range from 40 to 70 years. The exclusion criteria for patients in the study were as follows: (1) receiving cholesterol-lowering or antioxidants supplements; (2) STEMI; (3) post–coronary artery bypass graft surgery; (4) diabetic; (5) coexisting cardiomyopathy; (6) coexisting inflammatory diseases; (7) coexisting valve disease; and (8) chronic kidney disease which is defined as either damage or a glomerular filtration of <60 mL/min/1.73 m2 for ≥3 months. Kidney damage is defined as pathologic conditions or biomarkers of damage in blood, urine, or imaging studies.27
Table 1. The demographic and clinical characteristics of normocholesterolemic and hypercholesterolemic subjects.
| Variable | Normal cholesterol n = 45 |
High cholesterol n = 55 |
p-Value |
|---|---|---|---|
| Age (mean ± SEM), y | 66 ± 7 | 64.5 ± 5 | NS |
| Gender | Male: 64% | Male: 64% | NS |
| Female: 36% | Female: 36% | NS | |
| Smoking status | Smokers: 20% | Smokers: 20% | NS |
| Nonsmokers: 80% | Nonsmokers: 80% | NS | |
| HTN | 24% | 25% | NS |
| BMI (mean ± SEM), kg/m2 | 25 ± 1.5 | 28 ± 0.7 | NS |
Abbreviations: BMI: body mass index; HTN, hypertension; NS, not significant; SEM, standard error of the mean.
Sample Collection
Blood samples were obtained after fasting and prior to any statin treatment. From each patient, 17 mL of blood was collected and transferred into 8.5-mL vacutainer serum separator tubes (EM Science, Merk KgA, Germany). Blood samples were allowed to clot and then immediately centrifuged at 1,000 RPM for 15 minutes for serum collection. Serum was aliquoted into labeled Eppendorf tubes and stored at −80°C until assay was performed. The serum was used for the measurement of sRAGE and AGE. Blood was collected into ethylenediaminetetraacetic acid tubes (1 mg/mL) for plasma and used to measure the lipid profile.
Methods for Determination of sRAGE and AGE
In the present study, only total circulating soluble RAGE was measured. The measurement of sRAGE and AGE were made using commercially available quantitative sandwich enzyme-linked immunoabsorbent assay (ELISA) kits. The measurement of sRAGE used the Quantikine ELISA kit (Human RAGE Immunoassay, R&D Systems, Minneapolis, MN), and for the measurement of AGE we used the OxiSelect Advanced Glycation End Product (AGE) Competitive ELISA Kit (Cell BioLabs, Inc., San Diego CA). Measurements were performed in duplicate and the results were averaged. The intra- and interassay coefficients of variation were <6 and < 9%, respectively.
Measurement of Serum Cholesterol
The serum TC, HDL-C, low-density lipoprotein cholesterol (LDL-C), and triglycerides (TG) were measured with a Roche Cobas 6000 analyzer (Roche Diagnostics, Indianapolis, IN).
Measurement of Malondialdehyde
MDA measurements were made by the previously described method.28 MDA in the sample was reacted with thiobarbituric acid (TBA) to generate an MDA–TBA adduct which was quantified fluorometrically at 532 nm by a computerized plate reader. MDA was measured by the colorimetric/fluorometric method (assay kit; BioVision Inc., Milpitas, CA). Measurements were performed in duplicate and the results were averaged. The intra- and interassay coefficients of variation were <5 and < 8%, respectively.
Statistical Analysis
Statistical analyses were performed utilizing the Statistical Program for the Social Sciences (SPSS) version 13.0. Sample size was calculated using a two-sided Satterthwaite t-test to have 95% power and α level of 5%. The results were expressed as mean ± standard error of the mean (SEM). In addition, the unpaired t-test was used to compare data between groups. Single linear univariate correlations (Pearson correlation coefficients) were performed to evaluate the relationships between circulating sRAGE levels and the following variables: serum TC, HDL-C, LDL-C, TG, and MDA. A value of p < 0.05 was considered significant.
Results
The Serum Levels of Total Cholesterol, Low-Density Lipoprotein Cholesterol, High-Density Lipoprotein Cholesterol, and Triglyceride
The levels of serum lipids for normocholesterolemic and hypercholesterolemic patients are summarized in Fig. 1. The serum levels of TC in normocholesterolemic subjects ranged from 3.08 to 5.19 (mean ± SE, 4.5 ± 0.05 mmol/L), while those in hypercholesterolemic patients ranged from 5.2 to 8.53 (mean ± SE, 5.83 ± 0.12 mmol/L). These levels were significantly (p = 0.0005) different from each other. The serum levels of HDL-C in normocholesterolemic subjects ranged from 0.90 to 2.3 (mean ± SE, 1.79 ± 0.10 mmol/L), while those in hypercholesterolemic patients ranged from 0.44 to 1.53 (mean ± SE, 0.86 ± 0.05 mmol/L). These levels were significantly (p > 0.001) different from each other. The serum levels of TG in normocholesterolemic subjects ranged from 0.51 to 2.3 (mean ± SE, 1.57 ± 0.10 mmol/L), compared with those in hypercholesterolemic patients, which ranged from 2.36 to 6.8 (mean ± SE, 2.76 ± 0.16 mmol/L). These levels were significantly (p = 0.027) different from each other. Serum levels of LDL-C in normocholesterolemic subjects ranged from 1.7 to 3.3 (mean ± SE, 2.45 ± 0.05 mmol/L), while the levels in hypercholesterolemic patients ranged from 3.5 to 7.8 (mean ± SE, 3.95 ± 0.15 mmol/L). These levels were significantly (p = 0.0058) different from one another.
Fig. 1.
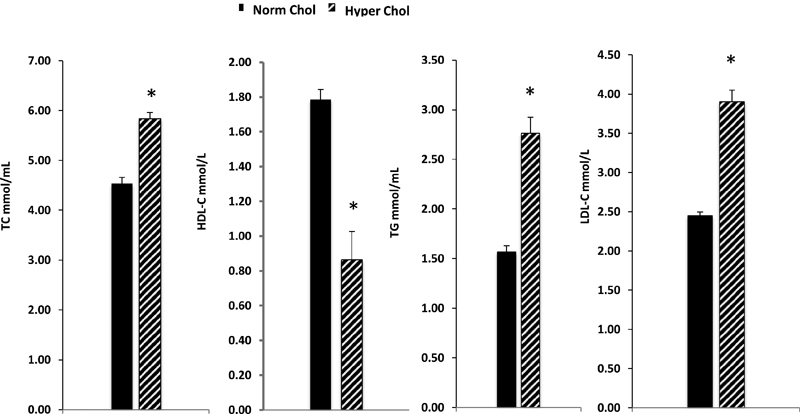
Serum levels of total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), triglyceride (TG), and low-density lipoprotein (LDL-C) in normocholesterolemic (Norm Chol.) and hypercholesterolemic (Hyper Chol.) subjects. The results are expressed as mean ± SE. *p < 0.05.
The Serum Levels of sRAGE, AGE, and AGE/sRAGE Ratio
The levels of serum sRAGE were lower (p = 0.001) in hypercholesterolemic subjects as compared with normocholesterolemic subjects (889 ± 47.6 vs. 1394.8 ± 56.5 pg/mL). The serum levels of AGE were higher (p = 0.001) in hypercholesterolemic subjects as compared with normocholesterolemic subjects (1213 ± 68.6 ng/mL vs. 642 ± 22 ng/mL). The AGE/sRAGE ratio in normocholesterolemic subjects was 0.49 ± 0.02, while the ratio was 1.71 ± 0.16 in hypercholesterolemic patients. The ratio was higher (p < 0.001) in hypercholesterolemic as compared with normocholesterolemic subjects.
The Serum Levels of MDA
The serum levels of MDA in normocholesterolemic subjects ranged from 0.22 to 1.18 (mean ± SE, 1.05 ± 0.03 nmol) as compared with hypercholesterolemic patients that ranged from 1.25 to 2.32 (mean ± SE, 1.5 ± 0.05 mmol/L). These levels were significantly (p < 0.001) different from each other.
Correlation of Serum Lipids with sRAGE in Hypercholesterolemic Patients
The correlation of serum lipid levels with sRAGE is summarized in Fig. 2. There was a negative correlation between LDL-C and sRAGE (r = − 0.59; p < 0.001), TG and sRAGE (r = − 0.43; p < 0.001), and TC and sRAGE (r = − 0.56; p < 0.001). There was a positive correlation between HDL-C and sRAGE (r = 0.49; p = 0.001).
Fig. 2.
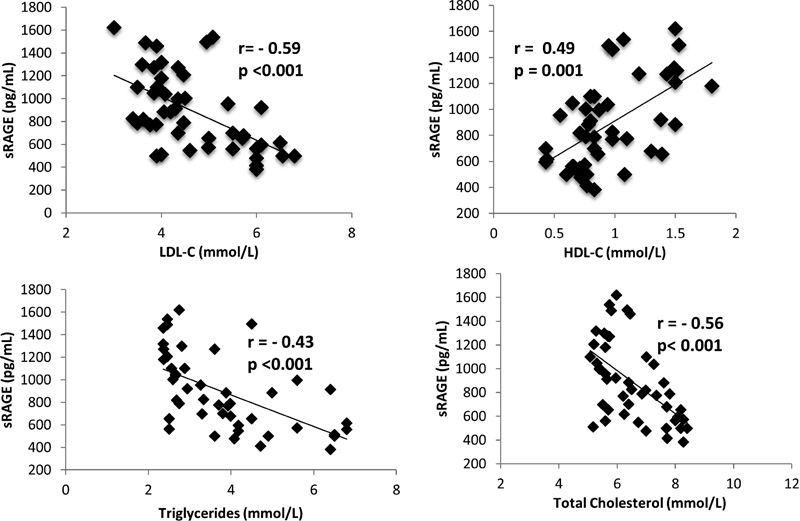
The correlation of sRAGE with LDL-C, TG, HDL-C, and TC in hypercholesterolemic subjects. HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; r, Pearson correlation coefficient; sRAGE, soluble receptor for advanced glycation end products; TC, total cholesterol; TG, triglycerides. p < 0.05 is considered significant.
Correlation of Lipids with AGE, and AGE/sRAGE Ratio in Hypercholesterolemic Patients
The correlation of serum lipids with AGE and AGE/sRAGE are summarized in Table 2. There was a positive correlation between TC and AGE (r = 0.664; p < 0.001), LDL-C and AGE (r = 0.66; p < 0.001), and TG and AGE (r = 0.741; p < 0.001). However, there was a negative correlation between HDL and AGE (r = − 0.634; p < 0.001) and between HDL and AGE/sRAGE (r = − 0.62; p < 0.001). There was a positive correlation between AGE/sRAGE and the following: TC (r = 0.73; p < 0.001); LDL-C (r = 0.74; p = 0.001); and TG (r = 0.77; p = 0.001).
Table 2. Correlation of AGE and the AGE/sRAGE ratio with total cholesterol, low-density lipoprotein cholesterol, triglycerides, and high-density lipoprotein cholesterol in hypercholesterolemic subjects.
| Parameter | AGE | AGE/sRAGE |
|---|---|---|
| Total cholesterol | p < 0.001 | p < 0.001 |
| r = 0.664 | r = 0.73 | |
| Low-density lipoprotein cholesterol | p < 0.001 | p = 0.001 |
| r = 0.66 | r = 0.74 | |
| Triglycerides | p < 0.001 | p = 0.001 |
| r = 0.741 | r = 0.77 | |
| High-density lipoprotein cholesterol | p < 0.001 | p < 0.001 |
| r = − 0.63 | r = − 0.62 |
Abbreviations: AGE, advanced glycation end product; r, Pearson correlation coefficient; sRAGE, soluble receptor for advance glycation end product ratio.
Note: p < 0.05 is considered significant.
Correlation of Serum Levels of sRAGE with MDA in Hypercholesterolemic Patients
The correlation of serum levels of sRAGE with the serum levels of MDA in hypercholesterolemic patients is summarized in Fig. 3. There was a negative correlation between sRAGE and MDA (r = − 0.46; p < 0.001).
Fig. 3.
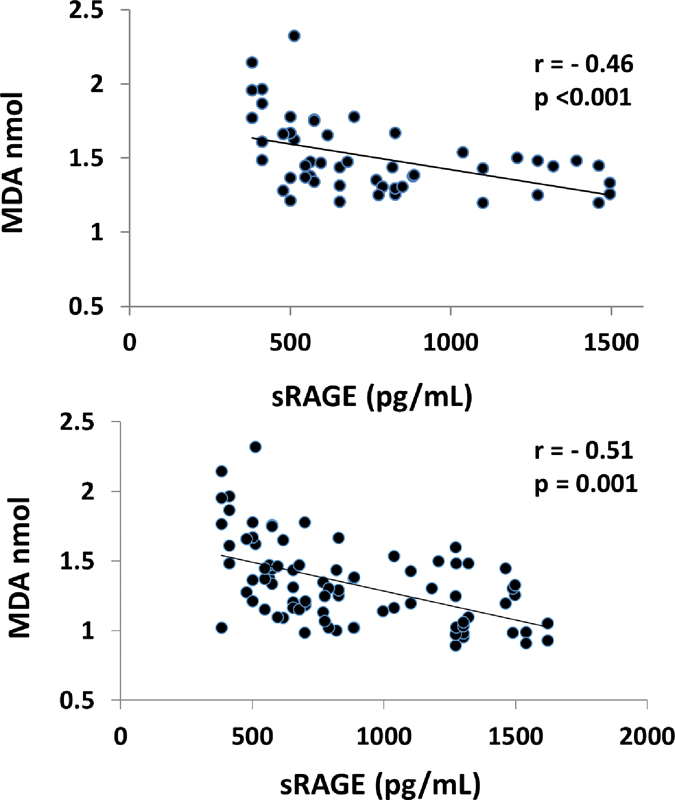
Correlation of sRAGE with MDA in hypercholesterolemic subjects (top) and hypercholesterolemic and normocholesterolemic subjects (bottom). MDA, malondialdehyde; r, Pearson correlation coefficient; sRAGE, soluble receptor for advanced glycation end products. p < 0.05 is considered significant.
Discussion
The results of the present study demonstrate that statin-free nondiabetic NSTEMI patients with hypercholesterolemia have lower serum sRAGE levels as compared with normocholesterolemic NSTEMI subjects. Devangelio et al have reported no significant difference in plasma sRAGE levels between normocholesterolemic patients and type 2 diabetic, hypercholesterolemic patients.18 However, the work of Santilli et al is consistent with our results because they have also reported that hypercholesterolemic subjects have significantly lower sRAGE levels as compared with normocholesterolemic patients.29 They also observed that hypercholesterolemic, ischemic patients on statin therapy had significantly higher plasma sRAGE levels as compared with disease-free, hypercholesterolemic patients.29 Statin therapy has been shown to reduce plasma cholesterol, and enhances the cleavage of sRAGE and its shedding into the circulation.30 Taken all together, this suggests that the higher plasma sRAGE levels in their cohort of patients29 may be due to the effect of statin therapy. The hypercholesterolemic patients in the present study had low serum levels of sRAGE and were not on statin therapy. The findings of the present study also demonstrate that there are significant correlations between serum levels of sRAGE and the components of the lipid profile (HDL-C, LDL-C, TG, and TC). Furthermore, we have demonstrated that there is a negative correlation between sRAGE and serum markers for MDA and LDL-C in hypercholesterolemic subjects. Our data are consistent with other studies that have shown that in hypercholesterolemic subjects sRAGE levels are negatively correlated with other markers for oxidative stress (8-iso-PGF2α).29 31 Our data suggest that serum lipids may affect the AGE–RAGE axis and oxidative stress. It has been reported that the treatment of diabetic apolipoprotein-E-deficient mice with sRAGE completely suppressed atherosclerotic lesion development.32 33 Our group has previously shown that the levels of serum sRAGE are lower, while the serum levels of AGE, sVCAM-1, and TNF-α are higher in subjects with NSTEMI as compared with healthy controls.34 Studies demonstrating the mechanism underlying the relationship between serum sRAGE levels and cholesterol levels are lacking. Blockade of RAGE by using sRAGE abolishes vascular complications of diabetes in animal models.35 36 Increased shedding of sRAGE during statin therapy results in high levels of sRAGE,30 suggesting a cytoprotective effect. The implication is that increased serum levels of sRAGE may offset the oxidative damage produced by hypercholesterolemia. LDL-C trapping by AGEs may augment the progression of atherosclerosis in diabetic and nondiabetic patients.37 It has been demonstrated that hypercholesterolemia produces oxidative stress in the endothelial cells of animal models.38 39 40 It is possible that vascular endothelial AGE-trapping of LDL-C is decreased in the presence of high sRAGE levels. Therefore, decreased AGE-trapping of LDL-C would attenuate the conversion of LDL-C to oxLDL-C with a resultant decline in the generation of ROS and oxidative tissue damage. Stimulation of the AGE–RAGE axis may create interplay between levels of serum cholesterol, sRAGE, and oxidative stress. The data suggest that hypercholesterolemia may lower the serum levels of sRAGE and elevate the levels of AGE and AGE/sRAGE. Increases in the levels of AGE and a reduction in the levels of sRAGE would increase the interaction of AGE with RAGE. As previously discussed, this interaction will increase the generation of ROS,22 23 cytokines, and adhesion molecules,20 21 22 leading to initiation and progression of hypercholesterolemic atherosclerosis.41 42 In addition, the data also suggest that increasing the levels of TC, TG, and LDL-C increases the levels of AGE and AGE/sRAGE and decreases the levels of sRAGE and, thereby, may increase the risk of the development of hypercholesterolemic atherosclerosis. Also, the data show that increases in the serum HDL-C increases the levels of serum sRAGE and decreases the levels of AGE and AGE/sRAGE. These changes in the AGE–RAGE axis would decrease the generation of ROS, expression of cytokines, and cell adhesion molecules, thus decreasing the risk of the development of atherosclerosis. The data show that there is an association of lipids with AGE–RAGE axis, and it suggests that the AGE–RAGE axis may be involved in hypercholesterolemic atherosclerosis. More extensive studies are needed to determine if it is an association or cause-effect.
Conclusion
Hypercholesterolemia is positively correlated with serum AGE, AGE/sRAGE, and MDA. The protective effect of HDL-C may be due to increases in serum sRAGE and decreases in the levels of AGE and AGE/sRAGE.
Limitations to This Study
The ELISA method measured total sRAGE, which could not discriminate between sRAGE that was enzymatically cleaved from the cell surface and the secretory splice variants esRAGE.43 44 45 46 47 However, the majority of circulating RAGE is derived from sRAGE. Interestingly, esRAGE comprises only 20% of the soluble RAGE level in humans.45 Another limitation to the study is that all of the patients in the study were classified as ACS NSTEMI. The results from the study may not be extrapolated to patients in other classifications without further investigations.
Significance
This study is significant because it sheds light on the relationship between the AGE–RAGE axis and hypercholesterolemia in the development and progression of atherosclerosis.
References
- 1.Prasad K, Kalra J. Oxygen free radicals and hypercholesterolemic atherosclerosis: effect of vitamin E. Am Heart J. 1993;125(4):958–973. doi: 10.1016/0002-8703(93)90102-f. [DOI] [PubMed] [Google Scholar]
- 2.Prasad K. Reduction of serum cholesterol and hypercholesterolemic atherosclerosis in rabbits by secoisolariciresinol diglucoside isolated from flaxseed. Circulation. 1999;99(10):1355–1362. doi: 10.1161/01.cir.99.10.1355. [DOI] [PubMed] [Google Scholar]
- 3.Steinberg D. Antioxidants in the prevention of human atherosclerosis. Summary of the proceedings of a National Heart, Lung, and Blood Institute Workshop: September 5-6, 1991, Bethesda, Maryland. Circulation. 1992;85(6):2337–2344. doi: 10.1161/01.cir.85.6.2337. [DOI] [PubMed] [Google Scholar]
- 4.Prasad K. New York, NY: Springer-Verlag; 2003. Pathophysiology of atherosclerosis; p. 85. [Google Scholar]
- 5.Warnholtz A, Wendt M, August M, Münzel T. Clinical aspects of reactive oxygen and nitrogen species. Biochem Soc Symp. 2004;(71):121–133. doi: 10.1042/bss0710121. [DOI] [PubMed] [Google Scholar]
- 6.White C R, Darley-Usmar V, Berrington W R. et al. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc Natl Acad Sci U S A. 1996;93(16):8745–8749. doi: 10.1073/pnas.93.16.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu H R, Tao L, Gao E. et al. Rosiglitazone inhibits hypercholesterolaemia-induced myeloperoxidase upregulation—a novel mechanism for the cardioprotective effects of PPAR agonists. Cardiovasc Res. 2009;81(2):344–352. doi: 10.1093/cvr/cvn308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu X, Lee J Y, Timmins J M. et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283(34):22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang D, Elner S G, Bian Z M, Till G O, Petty H R, Elner V M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 2007;85(4):462–472. doi: 10.1016/j.exer.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Makino H. Increased expression of receptor for advanced glycation end products by synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum. 2006;54(1):97–104. doi: 10.1002/art.21524. [DOI] [PubMed] [Google Scholar]
- 11.Basta G, Schmidt A M, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63(4):582–592. doi: 10.1016/j.cardiores.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Sakaguchi T, Yan S F, Yan S D. et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest. 2003;111(7):959–972. doi: 10.1172/JCI17115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Z, Wang K, Penn M S. et al. Receptor for AGE (RAGE) mediates neointimal formation in response to arterial injury. Circulation. 2003;107(17):2238–2243. doi: 10.1161/01.CIR.0000063577.32819.23. [DOI] [PubMed] [Google Scholar]
- 14.Yan S D, Schmidt A M, Anderson G M. et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994;269(13):9889–9897. [PubMed] [Google Scholar]
- 15.Schmidt A, Hori O, Chen J. et al. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest. 1995;96:1395–1403. doi: 10.1172/JCI118175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laitinen T T, Pahkala K, Magnussen C G. et al. Lifetime measures of ideal cardiovascular health and their association with subclinical atherosclerosis: The Cardiovascular Risk in Young Finns Study. Int J Cardiol. 2015;185:186–191. doi: 10.1016/j.ijcard.2015.03.051. [DOI] [PubMed] [Google Scholar]
- 17.Prasad K. Dietary flax seed in prevention of hypercholesterolemic atherosclerosis. Atherosclerosis. 1997;132(1):69–76. doi: 10.1016/s0021-9150(97)06110-8. [DOI] [PubMed] [Google Scholar]
- 18.Devangelio E, Santilli F, Formoso G. et al. Soluble RAGE in type 2 diabetes: association with oxidative stress. Free Radic Biol Med. 2007;43(4):511–518. doi: 10.1016/j.freeradbiomed.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 19.Quade-Lyssy P, Kanarek A M, Baiersdörfer M, Postina R, Kojro E. Statins stimulate the production of a soluble form of the receptor for advanced glycation end products. J Lipid Res. 2013;54(11):3052–3061. doi: 10.1194/jlr.M038968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffmann J A, Kafatos F C, Janeway C A, Ezekowitz R A. Phylogenetic perspectives in innate immunity. Science. 1999;284(5418):1313–1318. doi: 10.1126/science.284.5418.1313. [DOI] [PubMed] [Google Scholar]
- 21.Bierhaus A, Chevion S, Chevion M. et al. Advanced glycation end product-induced activation of NF-kappaB is suppressed by alpha-lipoic acid in cultured endothelial cells. Diabetes. 1997;46(9):1481–1490. doi: 10.2337/diab.46.9.1481. [DOI] [PubMed] [Google Scholar]
- 22.Bierhaus A, Humpert P M, Morcos M. et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl) 2005;83(11):876–886. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 23.Wautier M P, Chappey O, Corda S, Stern D M, Schmidt A M, Wautier J L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. 2001;280(5):E685–E694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 24.Prasad K, Lee P. Suppression of hypercholesterolemic atherosclerosis by pentoxifylline and its mechanism. Atherosclerosis. 2007;192(2):313–322. doi: 10.1016/j.atherosclerosis.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 25.Prasad K. A study on regression of hypercholesterolemic atherosclerosis in rabbits by flax lignan complex. J Cardiovasc Pharmacol Ther. 2007;12(4):304–313. doi: 10.1177/1074248407307853. [DOI] [PubMed] [Google Scholar]
- 26.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults . Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) JAMA. 2001;285(19):2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 27.Luo X, Jiang L, Du B. A comparison of different diagnostic criteria of acute kidney injury in critically ill patients. Crit Care Res. 2014;18:144. doi: 10.1186/cc13977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prasad K, McNair E D, Caspar-Bell G, Mabood Qureshi A. Vitamin E does not regress hypercholesterolemia-induced oxidative stress in heart. Mol Cell Biochem. 2014;391(1–2):211–216. doi: 10.1007/s11010-014-2004-8. [DOI] [PubMed] [Google Scholar]
- 29.Santilli F, Bucciarelli L, Noto D. et al. Decreased plasma soluble RAGE in patients with hypercholesterolemia: effects of statins. Free Radic Biol Med. 2007;43(9):1255–1262. doi: 10.1016/j.freeradbiomed.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 30.Cuccurullo C, Iezzi A, Fazia M L. et al. Suppression of RAGE as a basis of simvastatin-dependent plaque stabilization in type 2 diabetes. Arterioscler Thromb Vasc Biol. 2006;26(12):2716–2723. doi: 10.1161/01.ATV.0000249630.02085.12. [DOI] [PubMed] [Google Scholar]
- 31.Santilli F, Vazzana N, Bucciarelli L G, Davì G. Soluble forms of RAGE in human diseases: clinical and therapeutical implications. Curr Med Chem. 2009;16(8):940–952. doi: 10.2174/092986709787581888. [DOI] [PubMed] [Google Scholar]
- 32.Park L, Raman K G, Lee K J. et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4(9):1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 33.Basta G, Sironi A M, Lazzerini G. et al. Circulating soluble receptor for advanced glycation end products is inversely associated with glycemic control and S100A12 protein. J Clin Endocrinol Metab. 2006;91(11):4628–4634. doi: 10.1210/jc.2005-2559. [DOI] [PubMed] [Google Scholar]
- 34.McNair E D, Wells C R, Qureshi A M. et al. Low levels of soluble receptor for advanced glycation end products in non-ST elevation myocardial infarction patients. Int J Angiol. 2009;18(4):187–192. doi: 10.1055/s-0031-1278352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wautier J L, Zoukourian C, Chappey O. et al. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. J Clin Invest. 1996;97(1):238–243. doi: 10.1172/JCI118397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goova M T, Li J, Kislinger T. et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159(2):513–525. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci U S A. 1993;90(14):6434–6438. doi: 10.1073/pnas.90.14.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prasad K, McNair E D, Qureshi A M, Casper-Bell G. Vitamin E slows the progression of hypercholesterolemia-induced oxidative stress in heart, liver and kidney. Mol Cell Biochem. 2012;368(1–2):181–187. doi: 10.1007/s11010-012-1358-z. [DOI] [PubMed] [Google Scholar]
- 39.Ohara Y, Peterson T E, Harrison D G. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91(6):2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Küçükgergin C, Aydin A F, Ozdemirler-Erata G, Mehmetçik G, Koçak-Toker N, Uysal M. Effect of artichoke leaf extract on hepatic and cardiac oxidative stress in rats fed on high cholesterol diet. Biol Trace Elem Res. 2010;135(1–3):264–274. doi: 10.1007/s12011-009-8484-9. [DOI] [PubMed] [Google Scholar]
- 41.Bierhaus A, Hofmann M A, Ziegler R, Nawroth P P. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovasc Res. 1998;37(3):586–600. doi: 10.1016/s0008-6363(97)00233-2. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt A M, Yan S D, Yan S F, Stern D M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108(7):949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hudson B I, Carter A M, Harja E. et al. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008;22(5):1572–1580. doi: 10.1096/fj.07-9909com. [DOI] [PubMed] [Google Scholar]
- 44.Vazzana N, Santilli F, Cuccurullo C, Davì G. Soluble forms of RAGE in internal medicine. Intern Emerg Med. 2009;4(5):389–401. doi: 10.1007/s11739-009-0300-1. [DOI] [PubMed] [Google Scholar]
- 45.Raucci A, Cugusi S, Antonelli A. et al. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10) FASEB J. 2008;22(10):3716–3727. doi: 10.1096/fj.08-109033. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Bukulin M, Kojro E. et al. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J Biol Chem. 2008;283(51):35507–35516. doi: 10.1074/jbc.M806948200. [DOI] [PubMed] [Google Scholar]
- 47.Parkin E, Harris B. A disintegrin and metalloproteinase (ADAM)-mediated ectodomain shedding of ADAM10. J Neurochem. 2009;108(6):1464–1479. doi: 10.1111/j.1471-4159.2009.05907.x. [DOI] [PubMed] [Google Scholar]