

Abstract
AIM: To assess the effects of ischemic preconditioning (IPC, 10-min ischemia/10-min reperfusion) on steatotic liver mitochondrial function after normothermic ischemia-reperfusion injury (IRI).
METHODS: Sixty male Sprague-Dawley rats were fed 8-wk with either control chow or high-fat/high-sucrose diet inducing > 60% mixed steatosis. Three groups (n = 10/group) for each dietary state were tested: (1) the IRI group underwent 60 min partial hepatic ischemia and 4 h reperfusion; (2) the IPC group underwent IPC prior to same standard IRI; and (3) sham underwent the same surgery without IRI or IPC. Hepatic mitochondrial function was analyzed by oxygraphs. Mitochondrial Complex-I, Complex-II enzyme activity, serum alanine aminotransferase (ALT), and histological injury were measured.
RESULTS: Steatotic-IRI livers had a greater increase in ALT (2476 ± 166 vs 1457 ± 103 IU/L, P < 0.01) and histological injury following IRI compared to the lean liver group. Steatotic-IRI demonstrated lower Complex-I activity at baseline [78.4 ± 2.5 vs 116.4 ± 6.0 nmol/(min.mg protein), P < 0.001] and following IRI [28.0 ± 6.2 vs 104.3 ± 12.6 nmol/(min.mg protein), P < 0.001]. Steatotic-IRI also demonstrated impaired Complex-I function post-IRI compared to the lean liver IRI group. Complex-II activity was unaffected by hepatic steatosis or IRI. Lean liver mitochondrial function was unchanged following IRI. IPC normalized ALT and histological injury in steatotic livers but had no effect on overall steatotic liver mitochondrial function or individual mitochondrial complex enzyme activities.
CONCLUSION: Warm IRI impairs steatotic liver Complex-I activity and function. The protective effects of IPC in steatotic livers may not be mediated through mitochondria.
Keywords: Mitochondrial respiration, Fatty liver, Liver ischemia, Oxidative phosphorylation, Liver injury, Hepatic steatosis, Ischemic preconditioning
Core tip: We report a detailed mitochondrial function analysis of dietary-induced hepatic steatosis, which was not choline-deficient, during warm ischemia and after ischemia-reperfusion injury. We evaluated mitochondrial complex I and II activities as well as the impact of ischemic preconditioning on mitochondrial function. This study demonstrates that steatotic livers have decreased Complex-I activity at baseline and that Complex-I function is further impaired after warm ischemia-reperfusion injury. Ischemic preconditioning was unable to attenuate the harmful effect of ischemia-reperfusion on mitochondrial function.
INTRODUCTION
Hepatic steatosis is the most common liver disease found in clinical liver biopsies[1], and autopsy-based studies estimate that the prevalence of hepatic steatosis is 15%-30% in the Western world[1]. Consequently, the number of patients with hepatic steatosis encountered during liver surgery is increasing. Hepatic steatosis has been associated with a 2-3 fold increase in post-operative complication rates following liver resection[2,3]. It has been proposed that steatotic livers are more susceptible to ischemia-reperfusion injury (IRI), which impairs liver regeneration and is a major cause of liver damage, leading to worse outcomes[3].
The exact mechanism for the increased susceptibility of steatotic livers to IRI is not fully understood. Steatotic livers have been shown to have decreased recovery of adenosine triphosphate (ATP) concentrations following IRI[4]. One of the proposed underlying mechanisms behind the decreased ATP recovery and increased steatotic liver susceptibility to IRI is mitochondrial dysfunction[5]. Mitochondria are responsible for producing the bulk of cellular ATP and are, therefore, fundamental for cellular viability[6]. Impaired mitochondrial function (MF) disrupts normal cellular bioenergetics, which leads to cell death[7].
To attenuate the deleterious effect of IRI, ischemic preconditioning (IPC) of the liver has been used[8]. IPC involves a brief period of ischemia followed by reperfusion (generally, 10 min ischemia and 10 min reperfusion) prior to a period of sustained ischemic insult[9,10]. IPC has been reported to improve post-IRI liver injury in experimental[9] and clinical[10] steatotic livers. IPC has also been reported to improve ATP levels in steatotic livers[11]. While the mechanism by which IPC improves hepatic outcome following IRI is unknown, it has been postulated that IPC modulates and somehow preserves MF[9]. The aim of this study was to evaluate the impact of IPC on the MF of rat livers with steatosis after being subjected to warm (normothermic) IRI. Mitochondrial bioenergetics and liver injury markers were evaluated.
MATERIALS AND METHODS
Animals
All reagents were purchased from Sigma-Aldrich (St. Louis, MO, United States) unless otherwise specified. All experiments were performed in 11-wk old male Sprague-Dawley rats. The animal protocol was designed to minimize discomfort to the animals. The animals were enrolled when 3-wk old and removed from the mother at this age. They were then randomized to receive standard chow (lean animals; Teklad TB 2018; Harlan, Madison, WI, United States; 18% kcal fat, 58% kcal carbohydrate) or a high-fat/high-sucrose diet (steatotic animals; Rodent Diet D03021303; Research Diets, Inc., New Brunswick, NJ, United States; 45 kcal% fat, 25% kcal sucrose; Table 1)[12]. The animals were kept under a 12-h light/dark cycle (50%-70% humidity, 22 ± 2 °C) with ad libitum access to food and water. Bodyweight and blood glucose were measured weekly. Rats were fasted for 6 h prior to surgery to mimic pre-operative fasting. All surgical procedures were started between 12:00-1:00 pm. All experiments were approved by the University of Auckland Animal Ethics Committee (R965).
Table 1.
Content of high-fat/high-sucrose diet (D03021303; Research Diets, Inc., NJ, United States)
| Gram% | Kcal% | |
| Protein | 23.7 | 20 |
| Carbohydrate | 41.4 | 35 |
| Fat | 23.6 | 45 |
| Total | 100 | |
| Kcal/g | 4.73 | |
| Ingredient | ||
| Casein, 80 Mesh | 200 | 800 |
| L-Cystine | 3 | 12 |
| Corn Starch | 50 | 200 |
| Maltodextrin 10 | 45.6 | 182 |
| Sucrose | 250 | 1000 |
| Cellulose, BW200 | 50 | 0 |
| Soybean Oil | 25 | 225 |
| Lard | 177.5 | 1598 |
| Mineral Mix S10026 | 10 | 0 |
| DiCalcium Phosphate | 13 | 0 |
| Calcium Carbonate | 5.5 | 0 |
| Potassium Citrate. 1 H20 | 16.5 | 0 |
| Vitamin Mix V10001 | 10 | 40 |
| Choline Bitartrate | 2 | 0 |
| FD and C Blue Dye #1 | 0.05 | 0 |
| Total | 858.15 | 4057 |
Experimental design and surgical procedures
Sixty animals were randomized into one of six groups (n = 10 each): (1) Lean + Sham (Lean-Sham); (2) Lean + IRI (Lean-IRI); (3) Lean + IRI + IPC (Lean-IPC); (4) Steatotic + Sham (Steatotic-Sham); (5) Steatotic+IRI (Steatotic-IRI); and (6) Steatotic + IRI + IPC (Steatotic-IPC).
A model of partial (70%) hepatic ischemia was used that prevented mesenteric venous congestion by permitting portal decompression through the right and caudate lobes[9]. Rats were anesthetized with isofluorane inhalation. Following tracheostomy, anesthesia was maintained (1%-2% isofluorane) through a pressure-controlled ventilator (Kent Scientific Corporation, Torrington, CT, United States). Core body temperature was maintained (37-38 °C) by a thermostatically-controlled warming plate. Fluid administration via the right femoral vein and mean arterial pressure monitoring via the right carotid artery were undertaken with a radio-opaque 22G catheter and 2F solid-state pressure transducer (SPR-320 pressure catheter; Millar Instruments Inc., Houston, TX, United States), respectively.
Following a transverse laparotomy, the hepatic artery and portal vein to the left and median lobes were occluded for 60 min with a microvascular clip. Removal of the clip initiated the 240 min of reperfusion. Rats receiving IPC received 10 min of ischemia and 10 min of reperfusion prior to 60 min of ischemia. In the sham group, the rats were anesthetized, and a laparotomy was performed for 5.5 h without induction of ischemia.
The placement of a tracheostomy (reflecting endotracheal intubation) and prolonged anesthesia mimic clinical liver resection whereby patients are anesthetized continuously. We did not perform short intervals of anesthesia and repeated mini-laparotomies for our protocol, as it does not reflect clinical practice.
Tissue collections
Liver samples were obtained by “cheese-wire” ligating the liver with 4-0 silk tie. This technique caused minimal bleeding from the cut surface of the liver and allowed repeated sampling from each rat. Liver samples were obtained at various time-points: “A”: baseline (immediately following laparotomy), “B”: 10 min (after 10 min ischemia), “C”: 20 min (end of IPC), “D”: 80 min (after 60 min ischemia), and “E”: 320 min (end of 240 min reperfusion phase). Liver samples were obtained for histology, MF, and enzymatic analysis. The amount of liver tissue removed did not exceed 20% of total hepatic mass. At the end of the procedure, serum (5 mL) was collected from the inferior vena cava.
Histology
Histology was performed on time-points A and E to assess severity of the hepatic steatosis and IRI, respectively. Formalin-fixed and paraffin-embedded liver samples were stained with hematoxylin and eosin (HE). A consultant specialist histopathologist (BD) blinded to the group assessed the severity of steatosis with a published clinical grading system[13]. Severity of the IRI was assessed using a 4-point grading system previously described[14].
Assessment of hepatocyte injury
The severity of hepatic injury was assessed by serum levels of alanine aminotransferase (ALT) and was analyzed using a Roche Cobas 8000 modular analyzer (c702 module, Basel, Switzerland).
Homogenized tissue preparation for mitochondrial function analysis
Liver samples were immediately placed in ice-cold (about 4 °C) mitochondrial respiration media (Table 2). The samples were then removed from the media, blotted, weighed (20-30 mg), and placed in a 2-mL flat-bottom scintillation vial with 500 μL of mitochondrial respiration media. The sample was homogenized for 5 s with an Omni TH homogenizer (Omni International, Kennesaw, GA, United States) before analysis. Homogenates were utilized as they are more physiological and decrease the risk of any organelle selection bias inherent to the process of mitochondrial isolation, allowing for the preferential selection of more healthy organelles during isolation[15]. Our protocol also permits shorter processing time and rapid measurement of the function of the entire mitochondrial population within the tissue to provide an immediate measure of mass specific flux.
Table 2.
Mitochondrial respiration media
| Chemical | Final concentration (mmol/L) |
| EGTA | 0.5 |
| MgCl2 | 3 |
| K-Lactobionate | 60 |
| Taurine | 20 |
| KH2PO4 | 10 |
| Sucrose | 110 |
| Bovine serum albumin | 1 mg/mL |
| HEPES | 20 (pH 7.0 at 37 °C) |
Mitochondrial respiration assays
Mitochondrial respiration of liver homogenate was measured at each time-point. Respiration was measured in 2-mL chambers using an OROBOROS Oxygraph 2K (Anton Paar, Graz, Austria) at 37 °C in mitochondrial respiration media, with a calculated saturated oxygen concentration of 190 nmol O2 per milliliter at 100 kPa barometric pressure, and oxygen flux calculated using the DatLab 5 analysis software. Liver homogenate (50 μL) was added to each chamber, and the remainder was stored at -80 °C for later analysis. To account for potential variations in mitochondrial mass, citrate synthase (CS)-normalized[16] oxygen flux (pmol O2.s-1.U CS-1) was calculated.
A multiple substrate-inhibitor titration protocol was used to explore relative contributions of complex I (CI), complex II (CII), and combined CI+CII in the electron transport system (ETS). Respiration states were defined according to Gnaiger[17], where leak respiration and oxidative phosphorylation (OXPHOS) were the flux measured before and after addition of adenosine diphosphate (ADP), respectively. The assay protocol steps are described in Table 3. The integrity of tissue preparations and comparison of coupling efficiencies were made from the respiratory control ratio (RCR).
Table 3.
Mitochondrial respiration assay protocol
| Reagents added | Final concentration in oxygraph chamber (mmol/L) | Action of reagent | Measurement output |
| Step 1 | |||
| Glutamate | 10 | CI substrates | CI leak respiration |
| Malate | 5 | (CILeak) | |
| Pyruvate | 10 | ||
| Step 2 | |||
| ADP | 1.25 | Substrate for ATP generation | CI oxidative phosphorylation |
| Step 3 | |||
| Succinate | 10 | CII substrate | CI+CII oxidative phosphorylation1 |
| Step 4 | |||
| Rotenone | 0.001 | CI inhibitor | Isolate flux to CII [CII(rot)] |
| Step 5 | |||
| Oligomycin | 0.0025 | ATP-Synthase inhibitor | CI+CII leak respiration (CI,IILeak) |
| Step 6 | |||
| FCCP | 0.0015 | Mitochondrial uncoupler | ETS capacity |
| Step 7 | |||
| Antimycin A | 0.0050 | CIII inhibitor | Residual oxidase consumption |
The individual contribution of CII to oxidative phosphorylation (CII-OXPHOS) can also be derived (CI+II-OXPHOS minus CI-OXPHOS). ADP: Adenosine diphosphate; ATP: Adenosine triphosphate; CI: Complex I; CI-OXPHOS: Complex I oxidative phosphorylation; CI+II-OXPHOS: Complex I + Complex II oxidative phosphorylation; CI,IILeak: Complex I + Complex II leak respiration; CILeak: Complex I leak respiration; CII: Complex II; CIII: Complex III; ETS: Electron transfer system; FCCP: Carbonylcyanide p-trifluoromethoxy-phenylhydrazone; Rot: Rotenone.
Citrate synthase and total protein measurement
Liver homogenate, as used in mitochondrial respiration assay, was analysed for CS activity and total protein content. CS activity was measured as a surrogate for mitochondrial mass[16]. Frozen (-80 °C) liver homogenate was thawed, and CS activities were determined following the method of Srere and modified to microtitre-plate[18]. Protein content was determined by the Biuret test with bovine serum albumin as standard.
Complex I and Complex II activities
Liver homogenate from time-points A, D, and E were used to measure individual CI and CII enzyme activities using the NADH-oxidation and dichlorophenolindophenol-oxidation method, respectively[19]. Frozen liver homogenate was thawed and centrifuged for 10 min at 600 g (4 °C), and the supernatant used for analysis. Assays were conducted using a 96-well format, and results were normalized to total protein content, as determined by the Biuret method. In this report, “CI function” refers to assessments undertaken in the oxygraph, and “CI activity” refers to the isolated activity studies just described.
Statistical and data analysis
All study data were recorded in an EXCEL database. Statistical analyses were performed using GraphPad Prism version 5.0 (GraphPad Software, La Jolla, CA, United States) and SAS version 9.2 (SAS institute, Cary, NC, United States). Statistical tests were set at a 5% significance level (two-sided). Student’s t-tests were conducted on body weight and random blood glucose level to compare obese and lean rats. Difference in outcome measure between the groups of interest was tested using the analysis of covariance regression, adjusting for baseline value, bodyweight, and blood glucose measured before the procedures as appropriate. Repeated measures mixed model was used to evaluate the treatment differences at different time-points, controlling for the correlation of data collected on the same animal. The results are presented as mean ± SE of mean, with associated P-value.
RESULTS
Rats fed high-fat/high-sucrose diet were obese
Sprague-Dawley rats fed high-fat/high-sucrose diet for 8 weeks showed increased body weight (507 ± 10 vs 437 ± 6 g, P < 0.0001; Figure 1A) and increased random blood glucose level (6.1 ± 0.1 vs 4.6 ± 0.1 mmol/L, P < 0.0001; Figure 1B) relative to age-matched lean rats.
Figure 1.
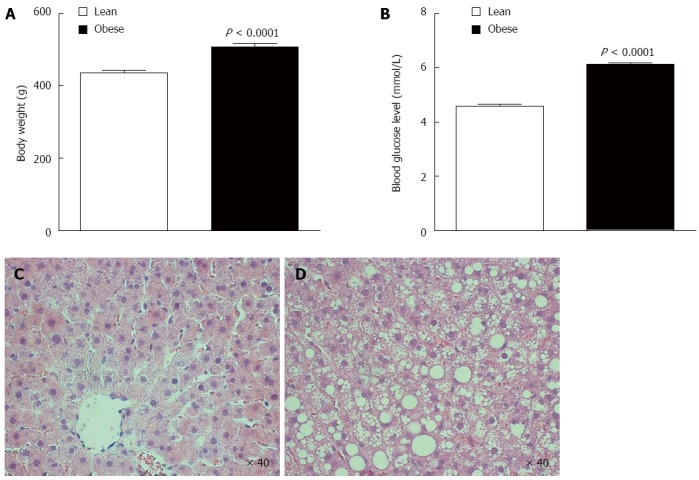
Bodyweight, random blood glucose, and baseline histology of high-fat/high-sucrose-fed (obese) and lean Sprague-Dawley rats. Obese Sprague-Dawley rats were significantly heavier (A) with higher random blood glucose (B) than age-matched lean rats. Baseline liver tissue sections were stained for hemotoxylin and eosin (x 40 magnification). Representative slides are displayed, and obese rats (D) showed severe mixed hepatic steatosis while lean rat livers (C) showed mild microvesicular steatosis. Data are shown as mean ± SE (n = 30 rat/group). Statistical analyses were performed using Students t-tests for body weight and random blood glucose.
Obese rat livers had severe mixed steatosis
Obese rat livers had gross macroscopic fat accumulation. All lean rat livers had normal baseline underlying tissue architecture with only some mild (8% ± 1%) microvesicular steatosis when evaluated by H&E staining (Figure 1C). Obese rat livers had severe (65% ± 3%) baseline mixed steatosis with prominent macrovesicular steatosis features evident (Figure 1D). There were no signs of fibrosis or inflammation in any of the groups consistent with hepatic steatosis.
Steatotic livers had increased liver injury following ischemia-reperfusion
Effect of steatosis, IRI, and IPC on liver injury biomarkers, tissue injury scores, and histology are shown in Figure 2 and 3, respectively. Both Lean-Sham and Steatotic-Sham livers did not have any biochemical (Figure 2A) or histological evidence of injury induced by the sham surgery (Figures 2B, 3A and 3B). Conversely, IRI was associated with increased serum ALT (Figure 2A) and worse liver histology injury scores in both Steatotic-IRI and Lean-IRI rats compared to Steatotic-Sham and Lean-Sham rats, respectively (Figure 2B). These same injury markers were also found to be significantly higher in Steatotic-IRI rats compared to Lean-IRI rats (Figure 2). IPC led to improvement in the serum ALT in Steatotic-IPC and Lean-IPC rats compared to Steatotic-IRI and Lean-IRI rats, respectively (Figure 2A). IPC also decreased injury score in Steatotic-IPC rats compared to Steatotic-IRI rats (Figure 2B). These results indicate that IPC was able to attenuate liver injury in steatotic livers.
Figure 2.

Serum alanine aminotransferase levels and histology injury score following ischemia-reperfusion. Serum alanine aminotransferase (ALT) (A) and histology injury score (B) following reperfusion were significantly higher in rats subjected to IRI compared to sham rats. Both injury markers were significantly higher in obese rats compared to lean rats. Compared to corresponding IRI groups, IPC decreased ALT levels in both lean and obese rats and decreased injury score in obese rats. Data are shown as mean ± SEM (n = 10 rat/group; lean rats, open bar; obese rats, closed bar). IRI: Ischemia-reperfusion injury; IPC: Ischemic preconditioning.
Baseline mitochondrial function in steatotic livers were similar to lean livers
The baseline mitochondrial functions were similar between steatotic and lean rat livers. Baseline (T = 0) samples from steatotic rat livers (n = 30) had similar MF to lean rat livers (n = 30) (Figure 4A-F). These results indicate that steatotic liver mitochondria were initially functioning adequately in vivo.
Figure 4.
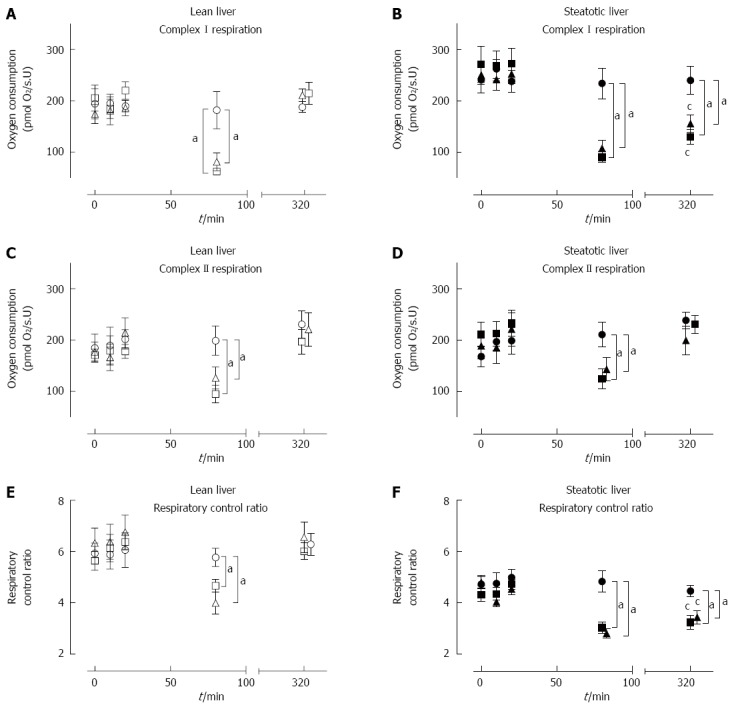
Mitochondrial function of lean and steatotic livers subjected to sham or ischemia-reperfusion injury with or without ischemic preconditioning. Baseline MF was similar between lean and steatotic livers in all outcome measures. Lean-Sham and Steatotic-Sham had stable CI-OXPHOS (A, B), CII-OXPHOS (C, D), and RCR (E, F) throughout the procedure. CI-OXPHOS (A, B), CII-OXPHOS (C, D), and RCR (E, F) were significantly lower following 60 min of ischemia in Lean-IRI, Lean-IPC, Steatotic- IRI, and Steatotic-IPC livers compared to the corresponding sham group. Following reperfusion, CI-OXPHOS (B) and RCR (F) were significantly lower in Steatotic-IRI and Steatotic-IPC livers compared to Steatotic-Sham or lean livers, while CII-OXPHOS (D) returned to pre-ischemic levels comparable to Steatotic-Sham or lean livers (D). Data are shown as mean ± SE (n = 10 rat/group; Lean-Sham, open circle; Lean-IRI, open square; Lean-IPC, open triangle; Steatotic-Sham, closed circle; Steatotic-IRI, closed square; Steatotic-IPC, closed triangle). aP < 0.05 vs Lean-IRI; cP < 0.05 vs Lean-IPC (end of reperfusion). IRI: Ischemia-reperfusion injury; IPC: Ischemic preconditioning.
Sham-operated rat liver mitochondrial function remained stable
There were no changes in MF in both Lean-Sham and Steatotic-Sham livers with a stable CI-OXPHOS, CII-OXPHOS, and RCR throughout all time-points (Figure 4A-F). These data indicate that the act of repeated liver sampling from each rat did not in itself significantly influence the underlying MF.
Prolonged ischemia led to impaired mitochondrial function
At the end of 60 min of ischemia, both lean and steatotic livers demonstrated impaired MF with significantly lower CI-OXPHOS (about 30%-40%), CII-OXPHOS (45%-60%), and RCR (about 60%-80%) compared to pre-ischemic levels or corresponding Sham livers (Figure 4A-F). There was no observable difference in MF between Lean-IRI and Steatotic-IRI livers at the end of ischemia. These findings indicate impaired MF occurs to the same extent in all groups immediately following 60 min of ischemic insult.
Reperfusion injury led to decreased Complex I mediated respiration in steatotic livers
After 60 min of ischemia and 240 min of reperfusion, MF in Lean-IRI returned to pre-ischemic levels and was comparable to Lean-Sham livers (Figure 4A, C, E). In Steatotic-IRI livers, CI-OXPHOS flux and RCR were significantly lower compared to baseline levels or Steatotic-Sham livers (Figure 4B, F) whereas CII-OXPHOS flux rates returned to pre-ischemic levels and was comparable to Steatotic-Sham livers (Figure 4D). CI-OXPHOS flux rates and RCR in Steatotic-IRI livers were observed to be 57% and 54% relative to Lean-IRI livers post-reperfusion (P < 0.01). These results indicate that, unlike lean livers, steatotic liver CI function was impaired by IRI, leading to decreased RCR.
Ischemic preconditioning had no significant effect on hepatic MF
MF in both types of livers subjected to IPC was similar to livers subjected to IRI only (Figure 4A-F). Lean-IPC and Steatotic-IPC livers demonstrated a similar pattern of CI-OXPHOS, CII-OXPHOS, and RCR at each of the sampling time-points as Lean-IRI and Steatotic-IRI livers, respectively (Figure 4A-F). Post-reperfusion, CI-OXPHOS, and RCR in Steatotic-IPC livers remained impaired, while CII-OXPHOS was comparable to pre-ischemic levels. These results indicate that despite the improvement in injury markers (ALT and histology) in IPC livers (Figures 2 and 3), IPC did not influence underlying MF over this timeframe.
Figure 3.
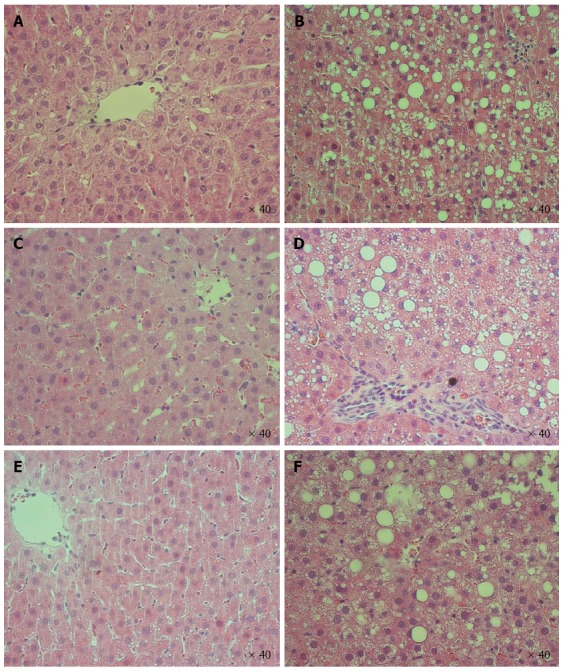
Liver histology following ischemia-reperfusion. No evidence of injury was observed in Lean-Sham (A) and Steatotic-Sham (B) livers. Lean-IRI livers (C) had mild injury while Steatotic-IRI livers (D) had moderate-severe injury. Both Lean-IPC (E) and Steatotic-IPC livers (F) were observed to have mild injury following reperfusion. Representative slides are shown (n = 10 rat/group). IRI: Ischemia-reperfusion injury; IPC: Ischemic preconditioning.
Citrate synthase activity was unaffected by ischemia-reperfusion injury
Citrate synthase (CS) activity in lean and steatotic livers was stable throughout the experiment and was not affected by IRI or IPC (Figure 5A and B). There was also no difference in CS activity between lean and steatotic livers at all time-points measured.
Figure 5.
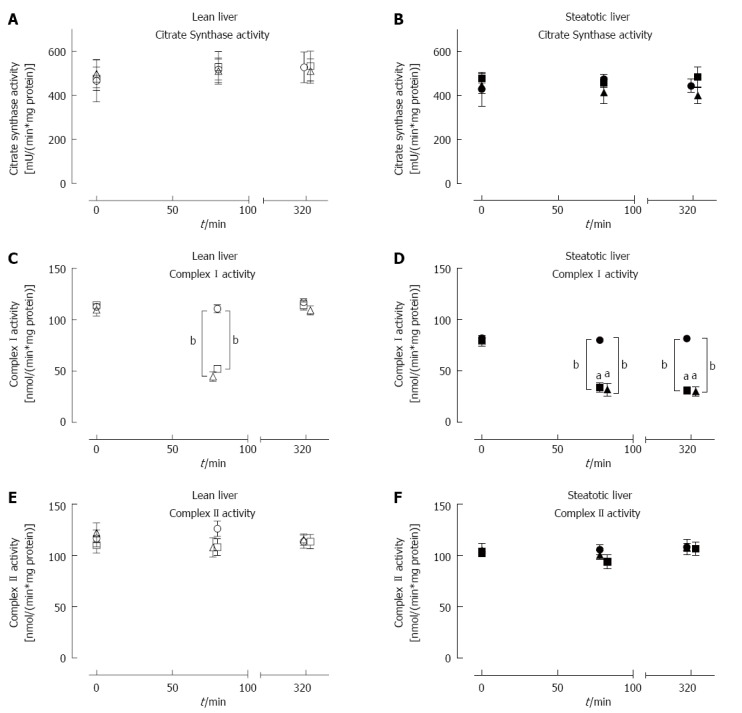
Citrate synthase, Complex I and Complex II activity at baseline, following ischemia and post-reperfusion in lean and steatotic livers. CS activity was similar between lean (A) and steatotic livers (B) throughout the procedure and was not affected by IRI or IPC. Baseline CI enzyme activity in steatotic livers were lower than lean livers (C, D). After ischemia, CI enzyme activity was significantly lower in lean and steatotic livers compared to sham livers. Additionally, CI enzyme activity (D) was lower in steatotic livers post-ischemia compared to lean livers. After reperfusion, CI activity remained lower in Steatotic-IRI and Steatotic-IPC livers (D) compared to Steatotic-Sham or lean livers. CII activity (E, F) remained stable throughout the procedure and was similar between both types of livers. IPC did not have a significant effect on CI or CII activity in both types of livers. Data are expressed as mean ± SE (n = 10 rat/group; Lean-Sham, open circle; Lean-IRI, open square; Lean-IPC, open triangle; Steatotic-Sham, closed circle; Steatotic-IRI, closed square; Steatotic-IPC, closed triangle). aP < 0.05, bP < 0.001 vs time- and group-matched lean livers. CS: Citrate synthase; CI: Complex I; CII: Complex II; IRI: Ischemia-reperfusion injury; IPC: Ischemic preconditioning.
Decreased Complex I but not Complex II enzymatic activity following reperfusion in steatotic livers
Baseline CI activity was significantly lower in steatotic livers compared to lean livers [78.4 ± 2.5 vs 116.4 ± 6.0 nmol/(min.mg protein), P < 0.001], while baseline CII activity was similar between the two groups [104.9 ± 3.3 vs 116.8 ± 6.1 nmol/(min.mg protein), P = 0.08; Figure 5C and D]. Following 60 min of ischemia, both types of liver demonstrated significantly lower CI activity (Figure 5C and D) compared to pre-ischemic or sham livers. Steatotic liver CI activity was also observed to be lower post-ischemia compared to lean livers. Following reperfusion, CI activity returned to pre-ischemic levels in lean livers but remained significantly lower in steatotic livers by approximately 65% (Figure 5D). CII activity (Figure 5E and F) was stable throughout the procedure, and there was no difference in CII activity between steatotic and lean livers. IPC did not have any significant beneficial effect on CI and CII activity in both types of livers. These activity results showed that IRI led to decreased CI activity in steatotic livers and also that IPC was not able to influence CI or CII activities; all of which was consistent with the earlier oxygraph functional analysis (above).
DISCUSSION
In this study, we used Sprague-Dawley rats with diet-induced hepatic steatosis. Compared to normal lean livers, steatotic livers demonstrated increased parenchymal injury following IRI, as indicated by their raised serum ALT and histology injury scores. Steatotic livers had lower baseline CI activity but similar baseline CS and CII activity compared to lean livers. The steatotic livers were also observed to have decreased CI activity and function following IRI, which unlike lean livers, showed no recovery of either activity or function even after prolonged reperfusion times. This finding indicated that CI is a particularly vulnerable site of IRI-induced damage in steatotic livers. Our results demonstrated that IPC was effective in decreasing liver injury in both lean and steatotic livers according to ALT levels. However, this protective effect was not translated to measures of CI function or CI activity. In summary, these data demonstrate that steatotic livers developed significant underlying mitochondrial impairment that was worsened by IRI and not able to be recovered by IPC.
In this study we developed a novel methodology for the repeated procurement of liver samples from the same animal over time, which has not been published before. This technique not only decreased the number of animals used but also presents statistical advantages by enabling repeated measures analyses to identify study effects. The theoretical disadvantage of the progressive hepatectomy samples altering the status of the subsequent samples did no eventuate. In particular, when the Sham groups were examined, there were no significant changes found in serum ALT, histological scores, or MF using the progressive sampling approach.
MF and complex enzyme activities
Steatotic livers had impaired MF, which is thought to contribute to the increased steatotic liver susceptibility to IRI[5]. CS activity was similar across groups and time-points, indicating that MF differences in this study are due to alterations in mitochondria activity and not a difference in mitochondrial mass.
A key dysfunction was found in CI, which is a large protein (about 1 MDa) comprising 45-47 subunits. It is embedded in the mitochondrial inner membrane to form an essential component of the mitochondrial ETS[20]. Impaired CI function has a substantial effect on ATP generation and contributes to a wide range of pathologies[21]. Our MF findings are consistent with a previous study that reported similar baseline CI function between steatotic and control livers and lower post-reperfusion CI function in isolated mitochondria from steatotic livers[5]. We have now extended this finding to show that the individual CI activity in tissue homogenates was also affected in steatotic livers post-IRI. Steatotic liver CI has previously been shown to be susceptible to oxidative damage from decreased mitochondrial antioxidants[5]. Further exacerbating this effect, CI can be a major site of reactive oxygen species production[22], and steatotic livers produce more reactive oxygen species in vivo than lean livers[5]. Oxygen reperfusion post-ischemia also leads to greater superoxide generation in steatotic livers relative to lean livers[23]. The lower CI activity observed in steatotic livers may be due to damage from IRI or may be a physiological response of steatotic livers to limit oxidative mitochondrial damage; which may be contributing to the decrease in tolerance of steatotic livers to IRI.
Complex II is the only mitochondrial membrane-bound enzyme that is also involved in the citric acid cycle, as it oxidizes succinate and transfers electrons to co-enzyme Q[24]. CII function has been reported to be similar at baseline and post-IRI in steatotic and lean rat livers[5], and our results corroborate this finding. CII abnormalities are infrequently reported in the literature[19], and its function in steatotic livers are seldom reported. We observed that CII activity post-ischemia and post-reperfusion was similar between steatotic and lean livers, which had not been previously described in this context. CII activity and function appears to be unaffected by IRI, suggesting that CII is more resistant to damage than CI. CII may even contribute to superoxide production through an apparent reverse electron flow to CI instead[24], although this scenario is somewhat controversial, as it appears to defy thermodynamics and redox potentials[21]. Despite intact CII function, CII-driven flux is less effective in ATP synthesis (coupled to OXPHOS at Complex III and Complex IV) compared to CI-driven flux (coupled to OXPHOS at CI, Complex III and Complex IV), and this reduction in CCI-driven flux should impair reconstitution of ATP pools on reperfusion.
Effect of ischemic preconditioning
MF recovery following IRI is thought to be essential, as it generates the majority of cellular ATP[6]. Inadequate MF post-IRI would lead to decreased or delayed ATP generation during the critical period of reperfusion and could impair liver recovery. In this study, we hypothesized that IPC protects the steatotic liver from IRI-induced damage by protecting MF. We showed that IPC was partially protective against normothermic IRI (biochemical and histological indices) in both lean and steatotic livers, consistent with other studies[9,11]. In our study, liver transaminase levels were improved in preconditioned steatotic livers, which had not been previously investigated in diet-based models of obesity. These results were consistent with the limited clinical data on the effect of IPC on biochemical markers from steatotic livers subjected to normothermic IRI during liver resection[10,25].
Our results also indicated that despite some improvement in conventional liver injury markers, IPC did not improve MF or key enzyme activities over the duration of the study. This finding was in contrast to results from a previous study in choline-deficient rats that showed improved conventional liver injury markers and MF in preconditioned steatotic livers post-IRI[9]. Importantly, there were substantial differences in our study design to that of the only other previous experimental study. In that study, Rolo et al[9] performed MF analysis on isolated mitochondria at 25 °C and demonstrated that RCR was lower in both lean and steatotic livers post-IRI, and these effects were normalized by IPC in both groups of livers. However, mitochondrial respiration and particularly State 4 respiration are sensitive to assay temperature[26], and the results may not be truly representative of physiological MF at 37 °C. Furthermore, the process of mitochondrial isolation results in the loss of fragile and/or damaged mitochondrial sub-populations. Here, we decided to use tissue homogenates as a means to lessen the potential for any mitochondrial selection bias[15]. Additionally, animals fed the choline-deficient diet used by Rolo et al showed weight loss, which is in contrast to the obesity often seen clinically in patients with hepatic steatosis[27]. In this study, the combination of a dietary model, tissue homogenates, and undertaking MF analysis at 37 °C represented the first advancement to a more physiological and clinically relevant MF analysis of the interaction of steatosis, IRI, and IPC. It is of note that most other previous studies on IPC in steatotic livers were performed in genetic models of hepatic steatosis[28]. However, the underlying mutations in these models are not prevalent in clinical hepatic steatosis pathophysiology, and the high-fat/high-carbohydrate used in our study more closely resembles the clinical setting[27].
The mechanism of IPC has been extensively reviewed elsewhere[29], but the direct impact of IPC on mitochondria is not as well characterized. In experimental lean rat livers subjected to IRI, ATP recovery was unaffected by IRI; but in steatotic livers, ATP recovery was found to be impaired post-IRI compared to lean livers[30]. In other studies, IPC was reported to preserve ATP recovery in lean livers post-IRI[29], while other studies have suggested that IPC was also effective in improving ATP recovery in steatotic livers post-IRI[11]. For our study, we investigated MF and complex activities. We chose a 10’ + 10’ IPC protocol, as this was similar to the first clinical protocol described by Clavien et al[10]. In their study, IPC led to an improvement in biochemical and histological markers of injuries. This finding was then replicated in a further prospective trial[25], which demonstrated that IPC improved ATP levels in younger patient’s post-reperfusion. In older patients, however, IPC decreased ATP levels post-reperfusion when compared to control livers. We found no improvement in MF with IPC, and although we were not able to measure ATP in this study, our findings suggest that the protective effect of IPC in this model was not likely to be mediated through increased mitochondrial ATP production. Therefore, there may be other mechanisms underlying the effect of IPC on hepatic ATP recovery. These mechanisms may include decreased cellular metabolism in preconditioned livers leading to conservation of ATP or reduced microcirculatory dysfunction. Alternatively, some of the IPC benefit may be due to increased production of nitric oxide and to opening of ATP-dependent potassium channels in preconditioned livers with a subsequent decrease in energy consumption[29].
The lack of full protection and liver function recovery from IPC observed by us and others in clinical and animal studies may reflect persistent underlying mitochondrial dysfunction, as demonstrated in our present study[3]. This observation supports future investigation of other IPC protocols or combinatorial use with mitochondrial-targeted therapies, as these may provide further clinical improvements. However, it could also potentially be influenced by our animal model, as our model differs from those published in the literature[9]. For completeness, liver weights should have been measured, but samples were obtained in “piece-meal” fashion and its was not possible.
As the prevalence of metabolic syndrome continues to rise in the population, hepatic steatosis has become the most common hepatic abnormality[1]. Therefore, it is important to identify new ways to improve outcomes from steatotic liver surgery. In this study, we investigated the impact of IPC on MF in a steatosis setting. We demonstrated that IRI was associated with increased liver injury in steatotic livers. Although the precise mechanisms underlying the increased susceptibility of steatotic liver to IRI remain unclear, we have shown for the first time, using a clinically relevant diet model and MF analysis at physiological temperatures, that there was an inherent decreased in CI activity in steatotic livers, which worsened following IRI. Our results also showed that the IPC protocol used in our study, while improving liver biomarkers and histology, did not influence MF directly. If we are to improve further the clinical benefit of IPC on livers with steatosis, then testing alternative IPC protocols or adjunct mitochondrial therapies will also be needed.
COMMENTS
Background
Steatotic livers are encountered with increasing frequency in liver surgery. It has been associated with poor outcome following warm ischemia-reperfusion injury (IRI). One possible proposed mechanism was mitochondrial dysfunction. However, the relationship between hepatic steatosis and mitochondrial dysfunction in warm IRI has not been clearly defined. Ischemic preconditioning has been touted as a possible therapeutic option for attenuating the harmful effect of IRI.
Research frontiers
Mechanisms of injury in steatotic livers are poorly understood, and further understanding will improve patient outcome.
Innovations and breakthroughs
This study is the first to investigate mitochondrial function, mitochondrial Complex I and Complex II activities; and the impact of ischemic preconditioning on mitochondrial function in warm IRI, in a dietary-induced model of hepatic steatosis.
Applications
Steatotic livers have decreased baseline Complex I activity. After reperfusion injury, Complex I function and activity were impaired in steatotic livers compared to lean livers. Ischemic preconditioning did not influence mitochondrial function in this setting.
Terminology
Mitochondrial Complex I is paramount for intact mitochondrial function, and impairment of Complex I leads to impaired ATP (cellular energy currency) production and consequently cell death.
Peer-review
It is well known that hepatic steatosis increases susceptibility to IRI and that this effect is linked to mitochondrial dysfunction. This study explored alterations of mitochondrial Complexes I and II in lean and high-fat, high sucrose diet–induced steatotic rat livers after 1 h-warm ischemia plus 4 h reperfusion. The authors showed that there was a significant decrease in Complex I in steatotic livers compared to lean livers but there was no difference in Complex II between these two groups. IPC decreased alanine aminotransferase release and histological changes after IRI but did not blunt decreases in Complex I in steatotic livers. This study obtained some interesting data.
Footnotes
Supported by University of Auckland Faculty Research Development Fund.
Institutional animal care and use committee statement: All procedures were reviewed and approved by the University of Auckland Animal Ethics Committee (R965).
Conflict-of-interest statement: The authors do not have any conflict of interest to declare.
Data sharing statement: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: January 25, 2016
First decision: February 18, 2016
Article in press: March 18, 2016
P- Reviewer: Nagarajan P, Zhong Z S- Editor: Ma YJ L- Editor: Filipodia E- Editor: Zhang DN
References
- 1.McCormack L, Dutkowski P, El-Badry AM, Clavien PA. Liver transplantation using fatty livers: always feasible? J Hepatol. 2011;54:1055–1062. doi: 10.1016/j.jhep.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Behrns KE, Tsiotos GG, DeSouza NF, Krishna MK, Ludwig J, Nagorney DM. Hepatic steatosis as a potential risk factor for major hepatic resection. J Gastrointest Surg. 1998;2:292–298. doi: 10.1016/s1091-255x(98)80025-5. [DOI] [PubMed] [Google Scholar]
- 3.Veteläinen R, van Vliet A, Gouma DJ, van Gulik TM. Steatosis as a risk factor in liver surgery. Ann Surg. 2007;245:20–30. doi: 10.1097/01.sla.0000225113.88433.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selzner M, Rüdiger HA, Sindram D, Madden J, Clavien PA. Mechanisms of ischemic injury are different in the steatotic and normal rat liver. Hepatology. 2000;32:1280–1288. doi: 10.1053/jhep.2000.20528. [DOI] [PubMed] [Google Scholar]
- 5.Caraceni P, Domenicali M, Vendemiale G, Grattagliano I, Pertosa A, Nardo B, Morselli-Labate AM, Trevisani F, Palasciano G, Altomare E, et al. The reduced tolerance of rat fatty liver to ischemia reperfusion is associated with mitochondrial oxidative injury. J Surg Res. 2005;124:160–168. doi: 10.1016/j.jss.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol Rev Camb Philos Soc. 1966;41:445–502. doi: 10.1111/j.1469-185x.1966.tb01501.x. [DOI] [PubMed] [Google Scholar]
- 7.Hand SC, Menze MA. Mitochondria in energy-limited states: mechanisms that blunt the signaling of cell death. J Exp Biol. 2008;211:1829–1840. doi: 10.1242/jeb.000299. [DOI] [PubMed] [Google Scholar]
- 8.Lloris-Carsí JM, Cejalvo D, Toledo-Pereyra LH, Calvo MA, Suzuki S. Preconditioning: effect upon lesion modulation in warm liver ischemia. Transplant Proc. 1993;25:3303–3304. [PubMed] [Google Scholar]
- 9.Rolo AP, Teodoro JS, Peralta C, Rosello-Catafau J, Palmeira CM. Prevention of I/R injury in fatty livers by ischemic preconditioning is associated with increased mitochondrial tolerance: the key role of ATPsynthase and mitochondrial permeability transition. Transpl Int. 2009;22:1081–1090. doi: 10.1111/j.1432-2277.2009.00916.x. [DOI] [PubMed] [Google Scholar]
- 10.Clavien PA, Yadav S, Sindram D, Bentley RC. Protective effects of ischemic preconditioning for liver resection performed under inflow occlusion in humans. Ann Surg. 2000;232:155–162. doi: 10.1097/00000658-200008000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selzner N, Selzner M, Jochum W, Clavien PA. Ischemic preconditioning protects the steatotic mouse liver against reperfusion injury: an ATP dependent mechanism. J Hepatol. 2003;39:55–61. doi: 10.1016/s0168-8278(03)00147-8. [DOI] [PubMed] [Google Scholar]
- 12.Smith GC, Vickers MH, Shepherd PR. Olanzapine effects on body composition, food preference, glucose metabolism and insulin sensitivity in the rat. Arch Physiol Biochem. 2011;117:241–249. doi: 10.3109/13813455.2011.576681. [DOI] [PubMed] [Google Scholar]
- 13.D’Alessandro AM, Kalayoglu M, Sollinger HW, Hoffmann RM, Reed A, Knechtle SJ, Pirsch JD, Hafez GR, Lorentzen D, Belzer FO. The predictive value of donor liver biopsies for the development of primary nonfunction after orthotopic liver transplantation. Transplantation. 1991;51:157–163. doi: 10.1097/00007890-199101000-00024. [DOI] [PubMed] [Google Scholar]
- 14.Abu-Amara M, Yang SY, Quaglia A, Rowley P, Tapuria N, Seifalian AM, Fuller BJ, Davidson BR. Effect of remote ischemic preconditioning on liver ischemia/reperfusion injury using a new mouse model. Liver Transpl. 2011;17:70–82. doi: 10.1002/lt.22204. [DOI] [PubMed] [Google Scholar]
- 15.Mittal A, Hickey AJ, Chai CC, Loveday BP, Thompson N, Dare A, Delahunt B, Cooper GJ, Windsor JA, Phillips AR. Early organ-specific mitochondrial dysfunction of jejunum and lung found in rats with experimental acute pancreatitis. HPB (Oxford) 2011;13:332–341. doi: 10.1111/j.1477-2574.2010.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012;590:3349–3360. doi: 10.1113/jphysiol.2012.230185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gnaiger E. Mitochondrial pathways and respiratory control. An introduction to OXPHOS analysis. 4th ed. Mitochondr Physiol Network 19.12. Innsbruck: OROBOROS MiPNet Publications; 2014. p. 80. [Google Scholar]
- 18.Hickey AJ, Chai CC, Choong SY, de Freitas Costa S, Skea GL, Phillips AR, Cooper GJ. Impaired ATP turnover and ADP supply depress cardiac mitochondrial respiration and elevate superoxide in nonfailing spontaneously hypertensive rat hearts. Am J Physiol Cell Physiol. 2009;297:C766–C774. doi: 10.1152/ajpcell.00111.2009. [DOI] [PubMed] [Google Scholar]
- 19.Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assays of respiratory chain complex activity. Methods Cell Biol. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- 20.Hickey AJ, Jüllig M, Aitken J, Loomes K, Hauber ME, Phillips AR. Birds and longevity: does flight driven aerobicity provide an oxidative sink? Ageing Res Rev. 2012;11:242–253. doi: 10.1016/j.arr.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Duchen MR. Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol Aspects Med. 2004;25:365–451. doi: 10.1016/j.mam.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene. 2002;286:135–141. doi: 10.1016/s0378-1119(01)00814-9. [DOI] [PubMed] [Google Scholar]
- 23.Nardo B, Caraceni P, Pasini P, Domenicali M, Catena F, Cavallari G, Santoni B, Maiolini E, Grattagliano I, Vendemiale G, et al. Increased generation of reactive oxygen species in isolated rat fatty liver during postischemic reoxygenation. Transplantation. 2001;71:1816–1820. doi: 10.1097/00007890-200106270-00018. [DOI] [PubMed] [Google Scholar]
- 24.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clavien PA, Selzner M, Rüdiger HA, Graf R, Kadry Z, Rousson V, Jochum W. A prospective randomized study in 100 consecutive patients undergoing major liver resection with versus without ischemic preconditioning. Ann Surg. 2003;238:843–850; discussion 851-852. doi: 10.1097/01.sla.0000098620.27623.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemieux H, Garedew A, Blier P, Tardif JC, Gnaiger E. Temperature effects on the control and capacity of mitochondrial respiration in permeabilized fibers of the mouse heart. Biochim Biophys Acta. 2006;1757:201–202. [Google Scholar]
- 27.Panchal SK, Brown L. Rodent models for metabolic syndrome research. J Biomed Biotechnol. 2011;2011:351982. doi: 10.1155/2011/351982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chu MJ, Vather R, Hickey AJ, Phillips AR, Bartlett AS. Impact of ischaemic preconditioning on experimental steatotic livers following hepatic ischaemia-reperfusion injury: a systematic review. HPB (Oxford) 2015;17:1–10. doi: 10.1111/hpb.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montalvo-Jave EE, Piña E, Montalvo-Arenas C, Urrutia R, Benavente-Chenhalls L, Peña-Sanchez J, Geller DA. Role of ischemic preconditioning in liver surgery and hepatic transplantation. J Gastrointest Surg. 2009;13:2074–2083. doi: 10.1007/s11605-009-0878-7. [DOI] [PubMed] [Google Scholar]
- 30.Hui AM, Kawasaki S, Makuuchi M, Nakayama J, Ikegami T, Miyagawa S. Liver injury following normothermic ischemia in steatotic rat liver. Hepatology. 1994;20:1287–1293. doi: 10.1002/hep.1840200528. [DOI] [PubMed] [Google Scholar]