

Abstract
Neurotransmitter uncaging, especially that of glutamate, has been used to study synaptic function for over 30 years. One limitation of caged glutamate probes is the blockade of GABA-A receptor function. This problem comes to the fore when the probes are applied at the high concentrations required for effective 2-photon photolysis. To mitigate such problems one could improve the photochemical properties of caging chromophores and/or remove receptor blockade. We show that addition of a dicarboxylate unit to the widely used MNI-Glu reduced the off-target effects by about 50–70%. When the same strategy was applied to an electron rich 2-(p-phenyl-o-nitrophenyl)-propyl (PNPP) cage, the pharmacological improvements were not as significant as for MNI. Finally, we used very extensive biological testing of the PNPP-caged Glu (more than 250 uncaging currents at single dendritic spines) to show that nitro-biphenyl caging chromophores have a 2-photon uncaging efficacy similar to that of MNI-Glu.
Photochemically protected neurotransmitters offer physiologists a powerful means of controlling important aspects of chemical and electrical signaling in cells from the central nervous system[1–3]. Such optical probes, called caged compounds, initially used the ortho-nitrobenzyl[4] and ortho-nitroveratryl[5] chromophores developed, respectively, by Baltrop and Patchornik. Whilst such chromophores have proved the most widely used of all photochemical protecting groups in both chemistry and biology[6–8], benzyl ester derivatives of biomolecules such as Glu and GABA have some predictable (e.g. hydrolysis at physiological pH), and some unpredictable weaknesses (e.g. GABA-A antagonism) in the context of synaptic physiology[3].
Several studies have sought to address these issues. For example, in 1999 Corrie and co-workers showed that a 7-nitroindolinyl-caged Glu[9] is very stable at physiological pH (i.e. 7.4). Subsequently several studies published by the Corrie/Ogden and Ellis-Davies/Kasai groups improved the photochemical properties of the probes made in 1999. In particular 4-methoxy-7-nitroindolinyl-Glu[10, 11] (MNI-Glu) has proved useful, as starting in 2001[12] it has been used for 2-photon (2P) photolysis by more than 25 laboratories in almost 100 studies[13, 14]. Much to our surprise this caged Glu probe turned out to be quite antagonistic towards GABA-A receptors[15]. Since this initial report, several studies have verified our preliminary findings that caged Glu probes have significant GABA-A receptor antagonistic effects[16–19]. Compared to improvements in hydrolytic stability, attempts to reduce such antagonism have proved less successful. For example, the Corrie/Ogden team developed a bisphosphate derivative of MNI-GABA, hoping that the high density of negative charge would eliminate antagonism[20]. Since the synthesis of this compound was quite laborious[21], it was disappointing to see that significant relief of antagonism was only seen at relatively low probe concentrations (0.10 mM range), ones that are really only useful for 1P photolysis[22]. Interestingly we have found that a single carboxylate can also be quite effective in this context[16], so we hypothesized that dicarboxylate (dc) derivatives would be even more effective in reducing GABA-A receptor antagonism of MNI-Glu (note, dc moieties have also been used in other studies[23, 24] but no detailed GABA-A receptor pharmacology of these probes was presented). And since such molecules looked synthetically much more tractable than the bisphosphates, we chose to make such derivatives of MNI-Glu and another caged Glu probe, based around the p-phenyl-o-nitrophenpropyl (PNPP) photochemical protecting group, originally developed by Pfleiderer and co-workers in 2004[25] (applied to Glu in 2008[26]). Here we present the synthesis of such caged Glu probes (called “dcMNI-Glu” (1) and “dcPNPP-Glu” (2), respectively). We benchmark the photochemical and pharmacological properties against MNI-Glu on pyramidal neurons in acutely isolated brain slices. Importantly, the dicarboxylate substitutions do not hinder the photochemical properties of the caged Glu probes for 2P photolysis at single spine heads and do improve the off-target GABA-A antagonism vis-à-vis MNI-Glu. But we find that such improvements are still not sufficient for use of caged Glu at the high concentrations required for 2P uncaging in reasonable power domains with complete removal of inhibitory blockade, suggesting there is further scope for probe development.
Results and discussion
Synthesis
The synthesis of compound 1 started with the know acid 3[27]. Carbodiimide coupling of di-tert-butyl-L-Asp gave compound 4 in 77% yield (Figure 1A). We chose to nitrate this compound using NaNO3 in TFA to give a 1:1 mixture of our first target, dcMNI-Glu and the 5-nitro regiomer 5 as their TFA salts in an 77% overall yield after HPLC purification. Our original synthesis of MNI-Glu also delivered the TFA salt as we used water/acetonitrile/TFA (25% v/v acetonitrile and 0.1% TFA) to purify the product[12]. Subsequently we also desalted this material by purification by RP HPLC without the traditional TFA counter ion[28]. For physiological experiments we have previously found both versions of MNI-Glu to be indistinguishable[12, 28], and so both versions used in the experiments below (see Experimental Section for details). Whilst a higher yield of 1 was potentially possible using the Claycop reagent[29], we selected this nitration method as we hoped larger amounts of 5 could be used as the substrate for a second nitration to give the 5,7-dinitro indoline 6. This caged Glu was an attractive target as we have established that similar compounds (e.g. MDNI-Glu[30] and CDNI-Glu[31]) are 5–6 times more photoactive than mono nitroindoles such as MNI-Glu. (Recently our 2005 study of MDNI-Glu has been independently confirmed by four groups[32–35] in 2014–15, though it should be noted DNI-Glu was used as the acronym for MDNI-Glu in these reports.) To our surprise, unlike the mono carboxyl analog, this nitration reaction did not proceed cleanly, with none of compound 6 being apparent under forcing conditions (i.e. 20 equivalents of NaNO3 in TFA for 3 d, Figure 1A).
Figure 1.

Synthesis of dcMNI-Glu and dcPNPP-Glu.
Reagents and conditions: a) EDC, DMAP; b) TFA, 4 h then NaNO3 (1.2 equ) for 10 min; c) TFA, NaNO3 (20 equ) for 3 d; d) NaI, K2HCO3; e) N-BOC-L-glutamate acid α-tert-butyl ester, EDC, DMAP; f) TFA/DCM (20% v/v).
The synthesis of 2 started with phenol 7[24], which was alkylated with bromide 8 to give alcohol 9 in 56% yield (Figure 1B). Carbodiimide coupling was used to synthesize the fully protected caged compound 10 in 76% yield after flash chromatography purification. This intermediate was deprotected with TFA (20% v/v in DCM) and purified by HPLC to give the target compound 2 as the TFA salt in an 81% yield. We found that the purification step was important, as when the crude reaction mixture was used for physiological experiments neurons were not healthy enough for long term experiments.
We tested the stability of the new caged Glu probes in physiological buffer. Both compounds were completely stable at room temperature (20–25°C) for 4 hours. HPLC analysis of freshly made ACSF perfusate solutions showed no hydrolysis during the course of daily physiological experiments. In some cases we froze such solutions at −40°C overnight and found we could reuse the same solution for brain slice experiments the next day. HPLC analysis showed no hydrolysis by the end of the second day of experiments. These data are consistent with previous reports of similar caged compounds[24, 36]. Finally, at 37°C we found that 2 showed 2.7% hydrolysis after 4 h and 15% after 24 hours (Supplemental Figure 1).
Photochemical properties
The absorption spectra of compounds 1 and 2 are quite similar. Both caged neurotransmitters absorb moderately well in the 340–410 nm range. Figure 2A shows the biphenyl cage absorbs about twice as much light at 350 nm and 3-fold more at 405 nm. These wavelengths are often used for uncaging with flash lamps and UV-visible lasers, as such light are compatible with standard microscope objectives. Though it should be noted that many lenses adapted for 2P microscopy, such as those used on our microscopes (see Experimental Section), are not parfocal at 350 nm. Further, some scan head mirrors have coatings optimized for wavelengths in the 400–1300 nm-range, so reflect 350-nm light poorly. However some microscopes are optimized for UV-visible light[37], thus caged Glu probes with larger extinction coefficients in the UV range (around 350 nm) could be useful complements to the DEAC450 caging[37] chromophore we developed recently, as this has an absorption minimum at 350 nm.
Figure 2.

Photochemical properties of caged glutamates probes.
A) Absorption spectra of dcMNI-Glu (red), MNI-Glu (red dash) and dcPNPP-Glu (orange) in physiological buffer at pH 7.4. B) Change in the absorption spectrum of dcMNI-Glu during photolysis in physiological buffer at pH 7.4. Red curve is t = 0, pink is fully photolyzed material. C) Putative photochemistry of dcPNPP-Glu according to ref. [39]. LC-MS analysis confirmed these structures (Supplemental Figure 1). D) HPLC of the reaction mixture from photolysis of dcPNPP-Glu; E) Absorption spectra of dcPNPP-Glu (orange) and peaks at 14.04 (green) and 20.45 (pink) min. The latter is typical of the o-nitrobenzyl chromophore and the former shows longer wavelength absorption characteristic of aromatic nitroso species. F) Photochemical reaction of model dcPNPP-caged thymine derivative 13.
The quantum yield of photolysis of 1 was measured using the change in the UV-visible absorption spectrum (Figure 2B). Corrie and co-workers have carefully characterized such changes and have found that the quantum yield of photolysis and Glu release correlates with the growth of absorption at 400 nm[11, 38]. We compared the time course of photolysis of MNI-Glu and dcMNI-Glu and found the time-course of the evolution of changes in the absorption spectra were indistinguishable, corresponding to a quantum yield of photolysis of dcMNI-Glu of 0.085.
Photolysis of 2 is more complex than 1, as would be expected from the careful studies of similar systems published by Steiner, Pfleiderer and co-workers[39–42]. HPLC analysis of the photolysis of 2 revealed that it decreased with a similar rate to MNI-Glu. The chromatogram from photolysis of 2 (Figure 2C) showed two major photoproducts that had similar relative retention times when compared with the starting materials to those reported in other systems by Steiner and co-workers. The UV-visible absorption spectra are also analogous to those reported by Steiner and co-workers in their uncaging studies[39, 42]. LC-MS analysis of the reaction confirmed these structural assignments (Supplemental Figure 2). Analysis of irradiation of solutions of compound 13 revealed that this second peak still had the thymine chromophore, along with appearance of the aromatic nitroso absorption (for synthesis details of 13 see Supplemental Figure 3). We used the fact that the thymine chromophore total optical density must, in principle, be preserved during irradiation to obtain an estimate of the chemical yield of acid and therefore the nitroso side-product. We determined the extinction coefficients of pure 9 and 15 in order to estimate their relative contribution to the absorption of 13 at 280 nm. With these data, compound 12 yielded 60% free acid and 30% nitroso side product based upon the assumption that the nitroso chromophore (e.g. 14) absorbs similarly to 9 at 280 nm. Taken together these data suggest that about 30% of the uncaging leads to a product that “hijacks” glutamate release. Goeldner and co-workers have reported similar results with some of their NPP-caged Glu compounds[24, 36].
Comparison of the physiological properties of dcMNI-Glu with MNI-Glu
Two-photon uncaging at single spine heads
We hypothesized that the photochemical properties of the new nitroindolinyl caged compound 1 would be very similar to the widely used parent MNI-Glu probe. Such proved to be the case (Figure 3). Local perfusion was used to apply both caged Glu probes to the same area of layer 2/3 pyramidal neurons in succession (Figure 3A). Previously we had used 10–12 mM MNI-Glu for similar experiments, as the relatively low 2P cross section of the MNI chromophore necessitates high concentration for effective 2P uncaging at non-toxic photon fluxes[12, 28, 31]. We found that the currents evoked with 2P uncaging of MNI-Glu and dcMNI-Glu with the same power at the same spine head (Figure 3B), both locally applied at 10 mM, were indistinguishable (Fig. 3C, n = 6, P > 0.05). This result is consistent with the similar 1P photochemical properties of the two caged compounds.
Figure 3.

Comparative photochemical and physiological properties of dcMNI-Glu and MNI-Glu.
A) Cartoon illustrating local perfusion of caged Glu probes on a patch-clamped layer 2/3 pyramidal neuron. B) High-resolution fluorescence image of a dendritic spine used for comparative uncaging of MNI-Glu and dcMNI-Glu at a 10 mM concentration; C) Representative examples of current traces evoked by 2P uncaging of MNI-Glu (blue) and dcMNI-Glu (red) at the same spine head; D) Summary of several comparative uncaging experiments showing that dcMNI-Glu and MNI-Glu are equally effective for 2P uncaging (n = 6); E) Representative examples of measurements of inhibitory postsynaptic currents during local perfusion of 10 mM MNI-Glu or dcMNI-Glu onto layer 2/3 neurons. Note full recovery of the miniature currents after wash out; F) Summary of several experiments measuring the blockade of inhibitory currents during local perfusion of MNI-Glu or dcMNI-Glu onto layer 2/3 pyramidal neurons. At both concentrations tested dcMNI-Glu was significantly less antagonistic (n = 7, P < 0.05).
Antagonism of GABA-A receptors
Since local perfusion of MNI-Glu onto neurons in brain slices has been used by several groups[12, 28, 43], we tested the antagonism of dcMNI-Glu using this technique. This approach has several advantages in that it: 1. conserves the caged compound; 2. allows study of the same synapses with different drugs and/or probes and 3. enables perfusion of fresh ACSF onto the brain slice rather than recirculation of a small volume of buffer. Thus, we tested the blockade of miniature inhibitory postsynaptic currents (mIPSCs) in layer 2/3 pyramidal neurons (Figure 3D). We chose these cells as they are relatively small compared to layer 5 and CA1 hippocampal neurons. We found that the high concentrations (10 mM) required for successful 2P uncaging of MNI-Glu blocked most of the mIPSCs (Figure 3E, n = 7). However the same concentration of dcMNI-Glu was significantly less antagonistic (Figure 3E, n = 7). A ten-fold lower concentration of each caged compound was even less antagonistic (Fig. 3E). It should be noted that Yuste and co-workers reported[17] that bath application of 0.3 mM MNI-Glu caused 80% reduction of evoked IPSCs. Whereas in 2001 Corrie and co-workers stated that bath application of 1 mM MNI-Glu had no effect on mIPSCs[44] (however in 2012 this assessment was changed somewhat[18]). Thus we also tested MNI-Glu pharmacology during bath application of the caged compound (see below).
Comparison of the physiological properties of dcPNPP-Glu with MNI-Glu
Two-photon uncaging at single spine heads
Even though dcPNPP-Glu and MNI-Glu have similar 1P photochemical properties (see above), 2P excitation is often not as easy to predict by analogy to the 1P properties (e.g. the λmax for 2P excitation is not always double the 1P maximum[45]). Thus, we extensively tested the non-linear photochemical properties of dcPNPP-Glu in a biological context, and compared these results to MNI-Glu. For both compounds we uncaged at five spine heads (Figure 4A,B left) on ten neurons using various powers for each spine head (Figure 4A,B right). As expected for 2P uncaging, the evoked currents correlated linearly with the square of the incident power (measured at the exit of the objective), and representative examples for individual cells are shown in Figure 4C,D. Importantly, we found that dcPNPP-Glu had no effect on sEPSCs (MNI-Glu 9.390 +/− 0.047 pA (n=520) and dcPNPP-Glu 9.300 +/− 0.050 pA (n=918), Figure 4E). The average slopes from all tested spines (Figure 4F–H) revealed that both caged compounds were similarly effective for 2P stimulation of receptors in living brain slices and are summarized in Fig. 4I. Note the slope for dcPNPP-Glu was about 30% shallower than MNI-Glu, the “hijack” mechanism of the former (Figure 2) presumably accounts for this difference, but the slopes were not statistically different (P = 0.27, unpaired t-test). Thus our biological data implies that the PNPP-caged Glu has a 2P-uncaging cross-section similar to that of MNI-Glu (0.06GM).
Figure 4.

Comparative uncaging physiology of dcPNPP-Glu and MNI-Glu at single spine heads.
A) Fluorescent image of a dendritic segment of a patch clamped CA1 neuron (left), uncaging was effected at five spines (stars). Right, representative traces of currents evoked by 2P uncaging of MNI-Glu at a single spine with increasing powers. B) Fluorescent image of a dendritic segment of a patch clamped CA1 neuron (left), uncaging was effected at five spines (stars). Right, representative traces of currents evoked by 2P uncaging of dcPNPP-Glu at a single spine with increasing powers. C,D) Summary plots of the correlation of photolytic input from uncaging MNI-Glu or dcPNPP-Glu at five spines on one neuron. Different cells were used for each compound. The blue and orange lines in C) and D) show the averages from each neuron for MNI-Glu and dcPNPP-Glu, respectively. Note that the linear relationship of p2 (p = power measured at the exit of objective lens) implies that photolysis arises from 2P excitation of both probes (t = shutter open time, c = probe concentration). Plotting the product (p2 × t × c) allows conditions to be normalized across many experiments; E) Summary of the relative effect of dcPNPP-Glu (n = 6) on the amplitude of sEPSCs compared to MNI-Glu (n = 7; set to 100%). dcPNPP-Glu did not affect the amplitude of sEPSCs (P = 0.90, unpaired t-test); F-G) Power series from uncaging at five spines from ten neurons for both MNI-Glu and dcPNPP-Glu evoked a similar range of currents as seen from the groups of slopes (note for one cell with dcPNPP-Glu only four spines were tested); I) The average slopes from all spines for MNI-Glu (n = 50) and dcPNPP-Glu (n = 49) were not significantly different (P = 0.27).
Measurement of absolute 2P absorbances is, perhaps, a more preferable method for comparison of cross-sections but such equipment is highly specialized[46] and thus, unfortunately, not readily available to us. Since cuvette 2P photolysis of a PNPP-caged Glu has been reported to be at least 50× better than MNI-Glu[24], or perhaps even 500× better for different acids in another related report[47], thus we would expect such large differences to be detected by the biology very easily. For example, the 5-fold difference in photochemical properties between CDNI-Glu and MNI-Glu is readily apparent in the biology[31]. Thus, we believe that the large sample of spines (50 for MNI and 49 for dcPNPP) and the number of neurons analyzed, 10 for each compound, provides a reliable basis for comparison of the true relative effectiveness of the PNPP caging chromophore in comparison to the widely used MNI cage. And since the biological efficacy of caged glutamates is the property neuroscientists are really interested in, our large sample size (49 spines using 5–9 powers for each spine) provides detailed and direct information for the biology research community about the physiological effectiveness of the PNPP cage for the first time.
Antagonism of GABA-A receptors
We recorded mIPSCs during bath application of 0.5 – 1 mM caged compounds and found that all three optical probes (MNI, dcMNI and dcPNPP) significantly antagonized mIPSCs compared to controls (Figure 5). As reported recently by Ogden and co-workers[18], our data showed that MNI-Glu bath applied at 1 mM strongly reduced the amplitude and frequency of mIPSCs (n = 8; ANOVA with post-hoc Tukey test, P < 0.05). Although, the presence of 0.5 or 1 mM dcMNI-Glu (n = 8, respectively) likewise reduced the amplitude and frequency of mIPSCs compared to control conditions, the effect was significantly less pronounced (ANOVA with post-hoc Tukey test, P < 0.05). Application of dcPNPP (n = 6) reduced the amplitude and frequency of mIPSCs (ANOVA with post-hoc Tukey test, P < 0.05). However, compared to MNI-Glu, the effect of dcPNPP on the mIPSC frequency was significantly less strong, further demonstrating the protective effect of dicarboxylate decoration of caged neurotransmitters. Interestingly bath application of both MNI-Glu and dcMNI-Glu had a more pronounced effect than local perfusion (Figure 3E), probably because the latter technique does not enable full equilibration of a well defined probe concentration across the entire neuron. But even though we could use 2P uncaging to evoke postsynaptic currents that were similar to synaptic events with 1 mM caged Glu (Figure 4A,B), we still feel that the power dosage required for these inputs is probably too large for long term experiments with dcMNI-Glu or dcPNPP-Glu. Thus, such studies should still use either 10 mM and 5 mM concentrations during local or bath application, respectively. However, our data suggest that caged Glu probes with good extinction coefficients and low GABA-A receptor antagonism could be useful for 1P uncaging on neurons in a manner similar to caged inhibitory transmitters such as DPNI-GABA[20, 22].
Figure 5.
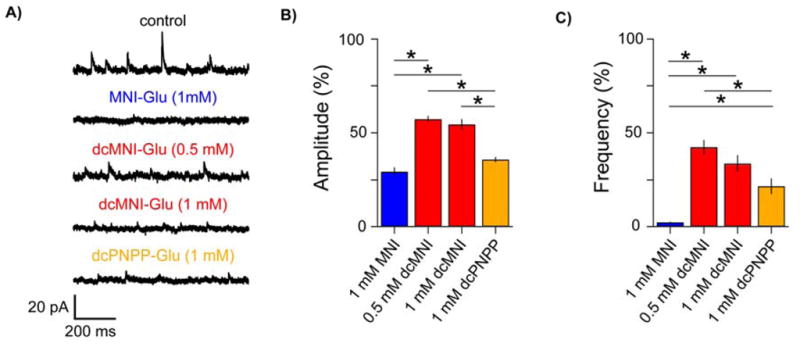
Comparative pharmacology of dcPNPP-Glu and MNI-Glu.
A) Whole-cell recordings from CA1 pyramidal neurons clamped at 10 mV were used to assess the blockade of GABA-A receptors during bath application of caged Glu probes. The amplitude and frequency of miniature inhibitory postsynaptic currents (mIPSCs) from neurons without any probes applied were recorded and compared to those with various caged Glu probes (MNI, blue; dcMNI, red; dcPNPP, orange). Representative segments of the total recording duration of 2–4 minutes/cell are shown. B,C) Summaries of the effects of caged Glu probes on the amplitude and frequency of mIPSCs. All compounds reduced the amplitude and frequency of mIPSCs compared to control cells (n = 13; ≥373 mIPSCs/cell were analyzed), the effect was less pronounced for dcMNI (0.5 and 1 mM: n = 8, respectively; ≥348 and ≥197 mIPSCs/cell were analyzed, respectively) compared to MNI (n = 8; 17–43 mIPSCs/cell were analyzed). The effect of dcPNPP (n = 6; ≥139 mIPSCs/cell were analyzed) on the amplitude was similar to MNI, but its effect on frequency was significantly less strong. * p < 0.05, ANOVA with post-hoc Tukey test.
Conclusions
Since the invention of caged glutamate by Hess and co-workers in 1990[48], the development of new probes for glutamate uncaging has been a focus for chemical biologists[3]. Whilst much interesting chemistry has been discovered, relatively few dramatic improvements in caged glutamate probe technology have appeared[11, 12, 19, 49]. One such probe, MNI-Glu, is important as it allowed diffraction-limited neurotransmitter 2P uncaging for the first time[12], and thus enabled new avenues of biology to be explored[13, 14]. Our data indicates that very extensive comparative physiological testing against MNI-Glu is an extremely revealing method to benchmark new caging chromophores in order to confirm any potential improvements in the photophysical and pharmacological properties of any novel photochemical protecting group[19, 31]. We suggest that this method should be adopted where possible as, in principle, caging chromophores with large 2P cross sections[24, 50–54] should allow application of optical probes at much lower concentrations to neurons than MNI-Glu and open new directions in neurophysiology.
Experimental Section
Synthesis
Di-tert-butyl 2-(2-((1-(5-(tert-butoxy)-4-((tert-butoxycarbonyl)amino)-5-oxopentanoyl)indolin-4-yl)oxy)acetamido)succinate 4. To a solution of 2-((1-(5-(tert-butoxy)-4-((tert-butoxycarbonyl)amino)-5-oxopentanoyl)indolin-4-yl)oxy)acetic acid 3 (0.502 g, 1.105 mmol) and L-Asp di-tert-butyl ester (0.50 g, 1.80 mmol) in dichloromethane was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.346 g, 1.8 mmol) and the RM was stirred for 18 h. The RM was diluted with methylene chloride, washed with water, citric acid (0.5 N), saturated NaHCO3, and brine. The organic phase was concentrated in vacuo, and purified by flash chromatography with 50% ethyl acetate in hexane gave 4 (0.60 g) in 77% yield.
1H NMR: δ (300 MHz, CDCl3) 7.89 (d, J = 8.3 Hz, 1H), 7.63 (d, J = 8.3 Hz, 1H), 7.16 (t, J = 8.3 Hz, 2H), 6.51 (d, J = 8.3, Hz, 1H), 5.2 (m, 1H), 4.75 (dt, J = 8.4, 4.3 Hz, 2H), 4.54 (s, 2H), 3.27 (m, 1H), 2.97-2.69 (ABq, J = 64.3, 17.3, 4.6, 2H), 1.54 (s, 9H), 1.48 (s, 9H), 1.43 (s, 9H), 1.41 (s, 9H).
13C NMR: δ (70 MHz, CDCl3) 170.04, 169.94, 169.18, 169.09, 167.61, 153.14, 152.17, 144.57, 129.06, 118.63, 111.21, 106.85, 82.89, 82.42, 81.54, 81.29, 67.18, 58.37, 48.54, 48.39, 37.35, 32.66, 31.96, 29.74, 28.32, 27.95.
HR-MS C36H55N3O11 requires 705.3837, 706.5959 M+1 found.
2-(2-((1-(4-amino-4-carboxybutanoyl)-7-nitroindolin-4-yl)oxy)acetamido)succinic acid 1. A solution of 4 (0.60 g, 0.85 mmol) in TFA (5 mL) was stirred at RT for 4 h, then NaNO3 was added (0.085 g, 0.97 mmol) and the RM was stirred for 10 mins. The reaction mixture was concentrated, dissolved in water/acetonitrile (25% v/v, with 0.1% TFA) and filtered through Celite, then 0.4 micron filter and purified by HPLC to give 1 (0.195g) and 5 (0.195 g) as their TFA salts in an 77% overall yield. 1H NMR: δ (300 MHz, MeOH-d4) 8.35 (d, J = 8.9 Hz, 1H), 7.43 (d, J = 8.9 Hz, 1H), 6.83 (d, J = 8.9 Hz, 1H), 4.90 (m, 1H), 4.85 (s, 2H), 4.32 (t, J = 8.9 Hz, 2H), 4.03 (t, J = 6.3 Hz, 1H), 3.24 (t, J = 8.9 Hz, 2H), 2.93-2.83 (m, 4H), 2.30-2.21 (m, 2H).
13C NMR: δ (70 MHz, MeOH-d4) 175.47, 174.86, 173.43, 172.88, 171.12, 159.75, 138.88, 138.54, 127.55, 126.78, 110.78, 69.62, 54.58, 52.56, 51.09, 37.86, 33.57, 28.56, 28.13.
HR-MS. C19H22N4O11 requires 482.1285, 483.1038 M+1 found.
4′-{2-[(1S)-1,2-Bis(tert-butoxycarbonyl)ethylamino]-2-oxoethoxy-4-nitro[1,1′-biphenyl]-3-yl)propanol 9.
A solution of di-tert-butyl-L-Asp (0.565 g, 2.0 mmol), bromoacetic acid (0.33 g, 2.2 mmol), N-methylmorpholine (0.5 mL, 5 mmol) and DCC (0.46 g, 2.5 mmol) was stirred at RT for 18 h. The RM was concentrated in vacuo, redissolved in ether (50 mL) and washed with water, saturated NaHCO3, and citric acid (0.5 N) to give 8 (0.236 g) in a crude yield of 32%. This material was used without purification for the next step in which a solution of 7 and 8 in acetone with sodium iodide and K2HCO3 was heated at reflux for 17 h. The RM was cooled, filtered, concentrated in vacuo, dissolved in methylene chloride, washed with water, citric acid (0.5 N) and concentrated in vacuo. Flash chromatography with 50% ethyl acetate in hexane gave 9 (0.205 g) in 56% yield.
1H NMR: δ (300 MHz, CDCl3) 7.86 (d, J = 8.3 Hz, 1H), 7.61 (d, J = 1.9 Hz, 1H), 7.55 (dt, J = 6.7, 2.2 Hz, 2H), 7.49 (dd, J = 8.3, 1.9 Hz, 1H), 7.05 (dt, J = 6.7, 2.2 Hz, 2H), 4.78 (dt, J = 8.4, 4.2 Hz, 1H), 4.57 (d, J = 0.7 Hz, 2H), 3.91-3.80 (m, 2H), (3.66 (m, 1H), 2.95 (dd, J = 17, 4.4 Hz, 1H), 2.75 (dd, J = 17, 4.4 Hz, 1H), 1.67 (brs, 1H), 1.47 (s, 9H), 1.43 (s, 9H), 1.38 (d, J = 6.9 Hz, 3H).
13C NMR: δ (70 MHz, CDCl3) 169.60, 169.39, 167.78, 157.94, 149.27, 145.17, 139.28, 133.14, 128.96, 126.63, 125.49, 125.22, 115.63, 82.91, 82.02, 68.12, 67.70, 49.04, 37.86, 36.81, 28.38, 28.27, 18.05.
MS: C29H38N2O9 requires 558.2577, 558.2573 found (M+).
4′-{2-[(1S)-1,2-Bis(tert-butoxycarbonyl)ethylamino]-2-oxoethoxy-4-nitro[1,1′-biphenyl]-3-[(4S)-4-tert-butoxycarbonyl-4-(tert-butoxycarbonylamino)-butanoyloxy)-2-propylester] 10.
To a solution of 9 (0.056 g, 0.10 mmol) and N-BOC-L-glutamate acid α-tert-butyl ester (0.030 g, 0.10 mmol) in methylene chloride, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.025 g, 0.12 mmol) and dimethylaminopyridine (0.015 g, 0.12 mmol) were added and the RM was stirred for 18 h. The RM was diluted with methylene chloride, washed with water, citric acid (0.5 N), saturated NaHCO3, and brine. The organic phase was concentrated in vacuo, and purified by flash chromatography with 50% ethyl acetate in hexane gave 10 (0.064 g) in 76% yield.
1H NMR: δ (300 MHz, CDCl3) 7.85 (dd, J = 8.5, 1.4 Hz, 1H), 7.58-7.48 (m, 5H), 7.06, (dt, J = 6.7, 2.2 Hz, 2H), 5.03 (brs, 1H), 4.79 (dt, J = 8.4, 4.4, 1H), 4.58 (s, 2H), 4.34-4.20 (m, 2H), 4.20-4.10 (brs, 1H), 3.9 (m, 1H), 2.95 (dd, J = 17.2, 5.4 Hz, 2H), 2.77 (dd, J = 17.2, 5.4 Hz, 2H), 2.40-2.24 (m, 2H), 2.15-2.00 (m, 1H), 1.90-1.78 (m, 1H), 1.48-1.42 (m, 36 H).
13C NMR: δ (70 MHz, CDCl3) 172.31, 171.03, 169.54, 168.99, 167.25, 157.60, 148.61, 144.84, 137.83, 132.60, 128.53, 125.99, 125.40, 124.87, 115.28, 82.49, 82.05, 81.60, 77.08, 68.34, 67.31, 53.21, 48,63, 37.45, 33.07, 30.11, 28.26, 27.98, 27.92, 27.87, 17.83.
C43H61N3O14 requires 843.4154, found 866.3957 (M+Na).
4′-{2-[(1S)-1,2-Bis(carboxy)ethylamino]-2-oxoethoxy-4-nitro[1,1′-biphenyl]-3-[(4S)-carboxy-4-amino-butanoyloxy)-2-propylester] 2.
Compound 10 (0.030 g, 0.036 mmol) was carefully deprotected using 20% TFA in methylene chloride. The course of the reaction was monitored by HPLC, and after 20 h no SM remained. The solvents were removed under a stream of nitrogen then high vacuum. The product was insoluble in D2O, but addition of a small amount of NaHCO3 solubilised the product making it amenable for NMR. For preparative HPLC the crude RM was dissolved in 40% acetonitrile in water (0.1% TFA), and purified with the same solvents to give 2 (0.020 g, 0.029 mmol) as its TFA salt in an 81% yield.
1H NMR: δ (300 MHz, CD3OD) 7.86 (d, J = 7.5 Hz, 1H), 7.76 (d, J = 1.8 Hz, 1H), 7.69-7.62 (m, 3H), 7.13 (dt, J = 8.8, 2.4 Hz, ), 4.87 (m, 1H), 4.64 (s, 2H), 4.42-4.30 (m, 2H), 3.90 (T, J = 6.9 Hz, 1H), 3.81-3.70 (m, 1H), 3.00-2.86 (m, 2H), 2.60-2.40 (m, 2H), 2.20-1.98 (m, 2H) 1.41 (d, J = 6.9 Hz, 3H).
13C NMR: δ (70 MHz, CD3OD) 174.76, 174.25, 174.10, 172.27, 171.30, 160.41, 151.16, 147.11, 139.78, 134.44, 130.44, 127.93, 127.43, 126.65, 117.34, 111.93, 70.69, 70.65, 69.02, 37.53, 35.46, 31.40, 27.60, 18.89.
C26H29N3O12 requires 575.1751, found 598.1674 (M+Na).
Photochemistry
The quantum yields for photolysis of 1 and 2 were measured by comparing the time of photolysis with MNI-Glu. The rate of change of absorption of 0.1 mM solutions of MNI-Glu and 1 in HEPES buffer (40 mM, pH 7.4, 100 mM KCl) were measured in a 1-cm cuvette and found to be identical. For compound 2 the filtered (350 +/− 5 nm) output of a 500 W medium pressure Hg lamp was used and concentrations were set such that absorptions were equal at 350 nm ([MNI] = 0.046 mM, [2] = 0.02 mM). Inosine was used as an inert internal standard. The path-length of the cuvette was 0.1 mm. The extent of reaction was monitored by HPLC using a Beckman System Gold fitted with a Beckman 134 diode array detector. Spectra (250–550 nm) of the eluates were captured ever second, and the chromatograms were continously monitored at 254 and 350 nm.
Physiology
All animal experiments were approved and performed according intuitional IACUC rules. Brain slices were prepared acutely and two-photon microscopy was performed as previously described[55, 56]. Brain slices were transferred to the recording chamber and perfused with gassed ACSF at room temperature. Whole-cell recordings were made from hippocampal CA1 or cortical layer 2/3 pyramidal neurons. Patch pipettes with a resistance of 3–5 MΩ were filled with one of three internal solutions. For uncaging experiments and sEPSC recordings cells were clamped at −60 mV and pipettes were filled with a solution consisting of (in mM): 135 K-gluconate, 10 HEPES, 4 Mg2Cl2, 5 EGTA, 10 Na-phosphocreatine, 4 Na2-GTP, 0.4 Na-GTP, 0.05 Alexa-594 (pH 7.35). For mIPSC recordings cells were clamped at +10 mV and pipettes were filled with a solution consisting of (in mM): 135 CsMeSO3, 10 HEPES, 4 Mg2Cl2, 5 EGTA, 10 Na-phosphocreatine, 4 Na2-GTP, 0.4 Na-GTP (pH 7.35) or 120 CsMeSO3, 8 NaCl, 15 CsCl2, 10 TEA·Cl, 10 HEPES, 2 QX-314, 4 MgATP and 0.3 Na2GTP (pH 7.3). mIPSCs were recorded in the presence of 1 μM TTX, 10 μM CNQX and 50 μM D-AP5 applied via the perfusion system, sEPSCs were recorded in ACSF containing the caged compound. mIPSCs and sEPSCs were isolated using the template search of pClamp (Moleculare Devices, Sunnyvale, CA, USA) and analyzed using custom-written IGOR Pro procedures (WaveMetrics, Lake Oswego, OR, USA). MNI-Glu used for Figure 3 was from Tocris and MNI-Glu. TFA salt used for Figures 4 and 5 was made by us as described previously[12].
For comparative uncaging experiments in Figure 3 each caged compound was locally applied to the same dendrite at a concentration of 10 mM. Uncaging at 720 nm was effected at the same spine with the same power on six cells. For experiments in Figure 4 five isolated spines on a proximate basal or oblique dendrite of a hippocampal CA1 pyramidal neuron were selected for uncaging at 720 nm. Laser pulses of 1–3 ms were applied to each spine with an inter-pulse interval of 250–500 ms. Power was increased in 10 mW steps (ranging between 0–80 mW). To analyze the power-current relationship, the average current response (in pA) of the five spines per cell was calculated for each laser power and plotted against the product (p2 × t × c), where “p” is uncaging laser power in mW measured at the exit of the objective lens, “t” the uncaging duration in ms, and “c” the concentration of the compound in mM. The average linear fit was R2 > 0.95 for both compounds and the slopes were compared between the conditions tested. Analysis of power-current relationships was carried out using FitMaster (HEKA), Excel and IGOR Pro.
Supplementary Material
Acknowledgments
This work was supported by NIH grants to GCRED (GM53395 and NS69720) and BLS (NS046579). All authors have approved the submitted version of the MS.
References
- 1.Ellis-Davies GCR. In: Photolysis of Caged Glutamate for Use in the CNS, Encyclopedia of Neuroscience. Squire LR, editor. Elsevier/Academic Press; New York: 2009. [Google Scholar]
- 2.Eder M, Zieglgansberger W, Dodt HU. Rev Neurosci. 2004;15(3):167–183. doi: 10.1515/revneuro.2004.15.3.167. [DOI] [PubMed] [Google Scholar]
- 3.Ellis-Davies GCR. Beilstein J Org Chem. 2013;9:64–73. doi: 10.3762/bjoc.9.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barltrop JA, Plant PJ, Schofield P. Chem Commun. 1966:822–823. [Google Scholar]
- 5.Patchornik A, Amit B, Woodward RB. J Am Chem Soc. 1970;92(21):6333–6335. [Google Scholar]
- 6.Mayer G, Heckel A. Angew Chem Int Ed Engl. 2006;45(30):4900–4921. doi: 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]
- 7.Ellis-Davies GCR. Nat Methods. 2007;4(8):619–628. doi: 10.1038/nmeth1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee HM, Larson DR, Lawrence DS. ACS Chem Biol. 2009;4(6):409–427. doi: 10.1021/cb900036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papageorgiou G, Ogden DC, Barth A, Corrie JET. J Am Chem Soc. 1999;121(27):6503–6504. [Google Scholar]
- 10.Matsuzaki M, Ellis-Davies GCR, Kasai H. Society for Neuroscience Annual Conference; 2000; p. 426.12. [Google Scholar]
- 11.Papageorgiou G, Corrie J. Tetrahedron. 2000;56:8197–8205. [Google Scholar]
- 12.Matsuzaki M, Ellis-Davies GCR, Nemoto T, Miyashita Y, Iino M, Kasai H. Nat Neurosci. 2001;4(11):1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellis-Davies GCR. ACS Chem Neurosci. 2011;2:185–197. doi: 10.1021/cn100111a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alvarez VA, Sabatini BL. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- 15.Ellis-Davies GCR, Meucci O, Shimizu S. Society for Neuroscience Annual Conference; 2007; p. 480.6/S14. [Google Scholar]
- 16.Matsuzaki M, Hayama T, Kasai H, Ellis-Davies GCR. Nat Chem Biol. 2010;6(4):255–257. doi: 10.1038/nchembio.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fino E, Araya R, Peterka DS, Salierno M, Etchenique R, Yuste R. Front Neural Circuits. 2009;3:2. doi: 10.3389/neuro.04.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palma-Cerda F, Auger C, Crawford DJ, Hodgson ACC, Reynolds SJ, Cowell JK, Swift KAD, Cais O, Vyklicky L, Corrie JET, Ogden D. Neuropharmacology. 2012;63(4):624–634. doi: 10.1016/j.neuropharm.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 19.Olson JP, Kwon HB, Takasaki KT, Chiu CQ, Higley MJ, Sabatini BL, Ellis-Davies GCR. J Am Chem Soc. 2013;135(16):5954–5957. doi: 10.1021/ja4019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trigo FF, Papageorgiou G, Corrie JET, Ogden D. J Neurosci Meth. 2009;181(2):159–169. doi: 10.1016/j.jneumeth.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 21.Papageorgiou G, Corrie JET. Tetrahedron. 2007;63(39):9668–9676. [Google Scholar]
- 22.Trigo FF, Bouhours B, Rostaing P, Papageorgiou G, Corrie JET, Triller A, Ogden D, Marty A. Neuron. 2010;66(2):235–247. doi: 10.1016/j.neuron.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 23.Papageorgiou G, Ogden D, Corrie JET. Photochem Photobiol Sci. 2008;7(4):423–432. doi: 10.1039/b800683k. [DOI] [PubMed] [Google Scholar]
- 24.Specht A, Bolze F, Donato L, Herbivo C, Charon S, Warther D, Gug S, Nicoud JF, Goeldner M. Photochem Photobiol Sci. 2012;11(3):578–586. doi: 10.1039/c2pp05360h. [DOI] [PubMed] [Google Scholar]
- 25.Buhler S, Lagoja I, Giegrich H, Stengele K, Pfleiderer W. Helv Chim Acta. 2004;87(3):620–659. [Google Scholar]
- 26.Gug S, Charon S, Specht A, Alarcon K, Ogden D, Zietz B, Léonard J, Haacke S, Bolze F, Nicoud JF, Goeldner M. Chembiochem. 2008;9(8):1303–1307. doi: 10.1002/cbic.200700651. [DOI] [PubMed] [Google Scholar]
- 27.Ellis-Davies GCR. Nat Protoc. 2011;6(3):314–326. doi: 10.1038/nprot.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith MA, Ellis-Davies GC, Magee JC. J Physiol. 2003;548(Pt 1):245–258. doi: 10.1113/jphysiol.2002.036376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gigante B, PraZeres AO, Marcelo-Curto MJ, Cornélis A, Laszlo PJ. J Org Chem. 1995;60:3445–3447. [Google Scholar]
- 30.Fedoryak OD, Sul JY, Haydon PG, Ellis-Davies GCR. Chem Commun. 2005:3664–3666. doi: 10.1039/b504922a. [DOI] [PubMed]
- 31.Ellis-Davies GCR, Matsuzaki M, Paukert M, Kasai H, Bergles DE. J Neurosci. 2007;27(25):6601–6604. doi: 10.1523/JNEUROSCI.1519-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiovini B, Turi GF, Katona G, Kaszas A, Palfi D, Maak P, Szalay G, Szabo MF, Szabo G, Szadai Z, Kali S, Rozsa B. Neuron. 2014;82(4):908–924. doi: 10.1016/j.neuron.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Bywalez WG, Patirniche D, Rupprecht V, Stemmler M, Herz AV, Palfi D, Rozsa B, Egger V. Neuron. 2015;85(3):590–601. doi: 10.1016/j.neuron.2014.12.051. [DOI] [PubMed] [Google Scholar]
- 34.Tonnesen J, Katona G, Rozsa B, Nagerl UV. Nature Neurosci. 2014;17(5):678. doi: 10.1038/nn.3682. [DOI] [PubMed] [Google Scholar]
- 35.Popovic MA, Carnevale N, Rozsa B, Zecevic D. Nature Commun. 2015;6 doi: 10.1038/ncomms9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Specht A, Thomann JS, Alarcon K, Wittayanan W, Ogden D, Furuta T, Kurakawa Y, Goeldner M. Chembiochem. 2006;7(11):1690–1695. doi: 10.1002/cbic.200600111. [DOI] [PubMed] [Google Scholar]
- 37.Olson JP, Banghart MR, Sabatini BL, Ellis-Davies GCR. J Am Chem Soc. 2013;135(42):15948–15954. doi: 10.1021/ja408225k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papageorgiou G, Ogden D, Corrie JE. J Org Chem. 2004;69(21):7228–7233. doi: 10.1021/jo049071x. [DOI] [PubMed] [Google Scholar]
- 39.Walbert S, Pfleiderer W, Steiner U. Helv Chim Acta. 2001;84(6):1601–1611. [Google Scholar]
- 40.Smirnova J, Woll D, Pfleiderer W, Steiner U. Helv Chim Acta. 2005;88(4):891–904. [Google Scholar]
- 41.Woell D, Laimgruber S, Galetskaya M, Smirnova J, Pfleiderer W, Heinz B, Gilch P, Steiner UE. J Am Chem Soc. 2007;129(40):12148–12158. doi: 10.1021/ja072355p. [DOI] [PubMed] [Google Scholar]
- 42.Woell D, Smirnova J, Galetskaya M, Prykota T, Buehler J, Stengele KP, Pfleiderer W, Steiner UE. Chem-Eur J. 2008;14(21):6490–6497. doi: 10.1002/chem.200800613. [DOI] [PubMed] [Google Scholar]
- 43.Abrahamsson T, Cathala L, Matsui K, Shigemoto R, DiGregorio DA. Neuron. 2012;73(6):1159–1172. doi: 10.1016/j.neuron.2012.01.027. [DOI] [PubMed] [Google Scholar]
- 44.Canepari M, Nelson L, Papageorgiou G, Corrie J, Ogden D. J Neurosci Meth. 2001;112:29–42. doi: 10.1016/s0165-0270(01)00451-4. [DOI] [PubMed] [Google Scholar]
- 45.Zipfel WR, Williams RM, Webb WW. Nat Biotechnol. 2003;21(11):1369–1377. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]
- 46.He GS, Tan LS, Zheng Q, Prasad PN. Chem Rev. 2008;108(4):1245–1330. doi: 10.1021/cr050054x. [DOI] [PubMed] [Google Scholar]
- 47.Aujard I, Benbrahim C, Gouget M, Ruel O, Baudin JB, Neveu P, Jullien L. Chemistry- Eur J. 2006;12(26):6865–6879. doi: 10.1002/chem.200501393. [DOI] [PubMed] [Google Scholar]
- 48.Wilcox M, Viola R, Johnson K, Billington A, Carpenter B, McCray J, Guizikowski A, Hess G. J Org Chem. 1990;55:1585–1589. [Google Scholar]
- 49.Wieboldt R, Gee KR, Niu L, Ramesh D, Carpenter BK, Hess GP. Proc Natl Acad Sci U S A. 1994;91(19):8752–8756. doi: 10.1073/pnas.91.19.8752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tran C, Gallavardin T, Petit M, Slimi R, Dhimane H, Blanchard-Desce M, Acher FC, Ogden D, Dalko PI. Org Lett. 2015;17(3):402–405. doi: 10.1021/ol5035035. [DOI] [PubMed] [Google Scholar]
- 51.Komori N, Jakkampudi S, Motoishi R, Abe M, Kamada K, Furukawa K, Katan C, Sawada W, Takahashi N, Kasai H, Xue B, Kobayashi T. Chem Commun. 2016;52:331–334. doi: 10.1039/c5cc07664a. [DOI] [PubMed] [Google Scholar]
- 52.Ciuciu AI, Korzycka KA, Lewis WJM, Bennett PM, Anderson HL, Flamigni L. Phys Chem Chem Phys. 2015;17:6554–6564. doi: 10.1039/c4cp05812g. [DOI] [PubMed] [Google Scholar]
- 53.Cueto Diaz EJ, Picard S, Chevasson V, Daniel J, Hugues V, Mongin O, Genin E, Blanchard-Desce M. Org Lett. 2015;17(1):102–105. doi: 10.1021/ol5033046. [DOI] [PubMed] [Google Scholar]
- 54.Boinapally S, Huang B, Abe M, Katan C, Noguchi J, Watanabe S, Kasai H, Xue B, Kobayashi T. Journal of Organic Chemistry. 2014;79(17):7822–7830. doi: 10.1021/jo501425p. [DOI] [PubMed] [Google Scholar]
- 55.Kwon H-B, Kozorovitskiy Y, Oh W-J, Peixoto RT, Akhtar N, Saulnier JL, Gu C, Sabatini BL. Nat Neurosci. 2012;15(12):1667–1674. doi: 10.1038/nn.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amatrudo JM, Olson JP, Lur G, Chiu CQ, Higley MJ, Ellis-Davies GCR. ACS Chem Neurosci. 2014;5(1):64–70. doi: 10.1021/cn400185r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.