

Summary
Background
Protein disulfide isomerases (PDI) may regulate thrombus formation in vivo, although the sources and targets of PDI are not fully understood.
Methods and results
Using click chemistry to link anti-CD61 and a C-terminal azido disulfide-linked peptide construct with a quenched reporter, we developed a fluorogenic platelet-targeting antibody (PDI-sAb) for thiol reductase activity detection in whole blood under flow conditions. PDI-sAb was highly responsive to various exogenous reducing agents (dithiothreitol, glutathione, and recombinant PDI) and detected thiol reductase activity on P-selectin/phosphatidylserine-positive platelets activated with convulxin/PAR1 agonist peptide, a signal partially blocked by PDI inhibitors and antibody. In a microfluidic thrombosis model using 4 μg/mL corn trypsin inhibitor-treated human blood perfused over collagen (wall shear rate = 100 s−1), PDI-sAb signal increased mostly over the first 200 sec, while platelets continually accumulated for over 500 sec, indicating primary adhesion to collagen, but not secondary aggregation, was correlated with the PDI-sAb signal. Rutin and the PDI blocking antibody RL90 reduced platelet adhesion and the PDI-sAb signal only when thrombin production was inhibited with PPACK, suggesting limited effects of platelet thiol isomerase activity on platelet aggregation on collagen in the presence of thrombin. With anti-mouse CD41 PDI-sAb used in an arteriolar laser injury model, thiol reductase activity localized in the core of growing thrombi where platelets displayed P-selectin and were in close proximity to disrupted endothelium.
Conclusion
PDI-sAb is a sensitive and real time reporter of extracellular disulfide reduction that targets clots as they form under flow to reveal spatial gradients.
Keywords: blood platelets, microfluidics, platelet aggregation, protein disulfide isomerase, thrombosis
Introduction
Thiol isomerases constitute a family of enzymes that are commonly found in endoplasmic reticulum (ER). These enzymes mediate disulfide bond formation, reduction, and rearrangement which are essential for protein folding. Platelets do not have an ER, but store and secrete protein disulfide isomerase (PDI) and other thiol isomerases upon activation [1–4]. Among the family of thiol isomerases, PDI, ERp57, and ERp5 have been implicated in thrombus formation [5–12]. Thiol isomerases have two catalytically active thioredoxin-like domains which can be in either a reduced (dithiol) or oxidized (disulfide) form depending upon surrounding redox condition. Activated platelets primarily express PDI in dithiol form, which catalyzes reduction or isomerization of disulfide bonds and has been proposed to induce free thiol exposure on activated platelet surface and conformational changes in GP1bα [13]. PDI expression upon platelet activation facilitates redox remodeling of αIIbβ3 and promotes platelet aggregation [14–17]. Once secreted, PDI likely accumulates in the thrombus by directly interacting with β3 integrin [15]. α2β1 is also a potential substrate for PDI since disulfide exchange is necessary for α2β1 but not GPVI mediated platelet adhesion [18]. With the promising antithrombotic efficacy of PDI inhibition both in vitro and in vivo, PDI inhibitors like quercetin-3-rutinoside (rutin) have been tested as potential antithrombotic reagent [19–21]. PDI has also been hypothesized as a mediator of tissue factor decryption [22, 23]. The source of PDI activity in a clot is also poorly understood. Endothelium may serve as the primary source of PDI required for thrombus formation [24], although mouse platelet-derived PDI was also essential for thrombus formation on a collagen-coated surface [25].
Common reductase activity assays [26] are not easily adapted for detection under hemodynamic conditions in whole blood due to instantaneous dilution of signal by flow and distinctive redox condition in whole blood as compare to assay buffers. Fluorescent-conjugated PDI antibodies have been used to detect PDI in vivo but have not been able to detect PDI activity within thrombi [5, 24, 27]. In the present study, we developed a platelet targeting sensor for measuring total platelet-derived thiol reductase activity under flow conditions. The sensor contains a disulfide linked glutathione (GSSG) mimicking peptide conjugated to a CD61 antibody that is able to bind to platelets (Fig. 1). The peptide fluoresces upon disulfide bond breakage and the antibody localizes the fluorescent signal on the platelet surface. We characterized and used this sensor to detect thiol reductase activity in a microfluidic thrombosis model and in an in vivo laser injury model, acquiring new information about the role of platelet-derived thiol reductases in thrombus formation and spatial gradient of thiol reductase activity in vivo.
Fig. 1.

Platelet-targeting thiol isomerase sensitive fluorogenic sensor (PDI-sAb). Customized thiol containing peptides have either a quencher (CPQ2) attached at N-terminus and or a fluorophore (CP488) and an azido group attached at N-terminus and C-terminus, respectively. The fluorophore is quenched due to close proximity to quencher once the peptides were bridged correctly by disulfide bond. Dibenzylcyclooctyne-NHS ester (DBCO) labels primary amine groups on anti-CD61 antibody and reacts with azide-labeled peptide thus serving as a connector linking disulfide bridged peptides (PDI-sP) to antibody. Fluorescence increases (at 519 nm) upon disulfide bond breakage by thiol isomerase.
Materials
Customized peptides AGCGAK(CPQ2) and azidoacetyl-AGCGAK(CP488) (CPC scientific, Sunnyale, CA, USA) are conjugated with a quencher (CPQ2, peptide Q) or a fluorophore (CP488, peptide F) (M.W. 996.1 and 944.9, respectively, purity > 90%). Reagents and antibodies used in this study are listed as follows: DBCO-Sulfo-NHS Ester (Click Chemistry Tools, Scottsdale, AZ, USA), azide-free anti-human CD61 antibody (BioLegend, San Diego, CA, USA), anti-human CD41a, anti-human CD62P, anti-mouse CD62P (whole IgG; clone RB40.34), anti-mouse CD41 (F[ab]2 fragments; clone MWReg30) (BD Biosciences, San Jose, CA, USA), Alexa Fluor® 647 conjugated human fibrinogen (Life Technologies, Grand Island, NY, USA), human recombinant protein disulfide isomerase (rhPDI, Biovision, Milpitas, CA, USA), 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), dithiothreitol (DTT), glutathione (GSH), oxidized glutathione (GSSG), sodium phosphate, disodium ethylenediaminetetraacetic acid (EDTA), acetic acid, sodium citrate (Sigma-Aldrich, St. Louis, MO, USA), H-Gly-Pro-Arg-Pro-OH (GPRP, EMD Chemicals, San Diego, CA, USA), corn trypsin inhibitor (CTI, Haematologc Technologies, Essex Junction, VT, USA), D-Phe-Pro-Arg-CMK (PPACK, Santa Cruz Biotechnology, Dallas, Texas, USA), Bradford protein assay kit, HEPES (Fisher Scientific, Pittsburg, PA, USA), anti-PDI monoclonal antibody RL90 (Abcam, Cambridge, MA, USA) and BioGel P-6 gel (Bio-Rad, Hercules, CA, USA).
Methods
Production of disulfide-linked thiol isomerase sensitive hetero-peptide dimers (PDI-sP)
Hetero-peptide dimers were synthesized according to a previously developed method (Fig. S1) [28]. Briefly, peptide Q (quencher) and peptide F (fluorophore) were dissolved separately in 0.1 % (v/v) acetic acid to a concentration of 10 μg/μL. Ellman’s buffer was prepared with 100 mM sodium phosphate, 5 mM EDTA and 7.5 mM DTNB in H2O (pH = 7.4). Reaction buffer was prepared with 100 mM sodium phosphate and 5 mM EDTA in H2O (pH = 7.4). Peptide Q (10 μL) was mixed with 80 μL of reaction buffer and 10 μL of Ellman’s buffer. The reaction mixture was blanketed with N2 and incubated for 2 hours at room temperature before peptide F (10 μL) was added to the reaction, followed by another 2 hours of N2 blanketed incubation in the dark. Formation of heterodimers (PDI-sP) was verified by adding reducing reagent DTT and measuring the fluorescence increase. Addition of DTT on average caused a 10-fold increase in fluorescence (Table S1).
Synthesis of thiol isomerase sensitive antibody (PDI-sAb)
DBCO-Sulfo-NHS Ester was dissolved in 1% DMSO to a final concentration of 900 μM and was mixed with anti-human CD61 antibody and HEPES-buffered saline (HBS buffer, 20 mM HEPES, 150 mM NaCl, pH = 7.4) in 2:1:7 volume ratio. The DBCO linking reaction was quenched by addition of Tris-HCl buffer (1 M, pH = 8.0) after 30 min incubation at room temperature. PDI-sP was added to the reaction in a 1:1 volume ratio. The reaction was incubated in the dark at room temperature for 4 hours and was then purified on BioGel P-6 gel column yielding purified PDI-sAb in HBS buffer (10 μg/ml measured by Bradford protein assay). Sensitivity of PDI-sAb to reducing reagents was measured in a Flex station microplate reader (495 nm ex/519 nm em, Molecular Devices, Sunnyvale, CA, USA) by directly mixing reducing reagents with PDI-sAb. PDI-sAb fluorescence on platelets was measured by flow cytometry (Accuri C6). A mouse specific version of the sensor was generated using an anti-mCD41 antibody.
Microfluidic clotting assay on collagen surface
PDI-sAb was tested in human whole blood (WB) in a previously developed microfluidic assay [29]. Briefly, a 250 μm wide collagen strip was generated by perfusing collagen type I fibrils through a PDMS patterning device that was vacuum-bonded on ethanol cleaned glass slides. The patterning device was then replaced by an eight-channel flow device which was perpendicularly vacuum-bonded on the collagen surface, generating 8 evenly spaced prothrombotic patches (250 μm×250 μm). Human WB was anticoagulated with a low level of CTI (4 μg/mL WB) or PPACK (100 μM) and was perfused over collagen surface at venous shear rate (100 s−1). The shear rate was controlled by a syringe pump (Harvard Apparatus, Holliston, MA, USA). The dynamic change of platelet (CD41a), fibrin (ogen), and PDI-sAb (1:9 WB) signal was monitored with IX81 fluorescent microscope (Olympus, Center Valley, PA, USA).
Blood collection and preparation
Blood was collected via venipuncture from healthy donors (free of alcohol and medication 72 hrs prior to donation) in accordance with the University of Pennsylvania IRB approval. Blood was collected directly into CTI (4 μg/mL) or PPACK (100 μM) for microfluidic experiment or into sodium citrate (1:9 WB) or PPACK for platelet rich plasma preparation (300×g, 10 min).
Mouse intravital microscopy
Intravital imaging was done as previously described [30]. Briefly, male mice 8–12 weeks of age were anesthetized with an intraperitoneal injection of sodium pentobarbital (90 mg/kg), and their jugular vein was cannulated for the introduction of PDI-sAb (3 μg/mouse) and anti-CD62P AF-647 (5 μg/mouse). The mouse cremaster muscle was prepared for viewing, and maintained under a constant flow of bicarbonate buffer (37 °C) bubbled with 95%/5% N2/CO2. Mouse arterioles of 30–50 μm diameter were visualized with a BX61WI microscope (Olympus, St. Louis, MO, USA) with a 60X (0.9 NA) water immersion objective, and a CSUX1 spinning disk confocal scanner (Yokogawa, Sugar Land, TX, USA). Fluorescence imaging was done using diode pumped solid state lasers (405 nm, 488 nm, 561 nm, 647 nm) with acousto-optic tunable filter control as an excitation source (LaserStack, Intelligent Imaging Innovations, Denver, CO, USA). Images were captured using an Evolve digital camera (Photometrics, Tucson, AZ, USA).
Penetrating injuries were produced with a pulsed nitrogen dye laser (SRS NL100, 440 nm) focused on the vessel wall through the microscope objective. The laser was fired 1–10 times until red blood cells either escaped into the extravascular space or became trapped within the layers of the vessel wall. The University of Pennsylvania Institutional Animal Care and Use Committee approved all procedures. After injury, platelet deposition was monitored using brightfield imaging, core development with anti-CD62P, and PDI activity with PDI-sAb. Background fluorescence was measured within the vessel for both PDI-sAb and anti-CD62P, and subtracted from the images to determine positive signal. In representative images, the core is shown as binary based on the background fluorescence and PDI-sAb signal is shown as the indicated gradient. Microscope control, image capture, and analysis were performed by Slidebook 5.0 (Intelligent Imaging Innovations, Denver, CO, USA).
Results
Sensitivity of PDI-sAb to reducing reagents
Rapid disulfide cleavage of PDI-sAb was observed upon addition of high dosages of DTT (Fig. 2A) or GSH (Fig. 2B). Peak fluorescence was quickly achieved within 30 sec. PDI-sAb exhibited a threshold response to DTT cleavage. It remained in disulfide form at low DTT concentrations (5 and 50 μM) but was robustly cleaved by higher concentrations of DTT and reached 8-fold increase in fluorescence with the highest tested DTT concentration (30 mM). Compared to DTT, GSH was more potent in cleaving PDI-sAb at 50 μM. However, GSH exhibited much lower reductive activity at higher dosages (5 and 30 mM), indicating the different electron transfer potentials of the two reducing reagents.
Fig. 2.

PDI-sAb sensitivity to reducing reagents. To determine the sensitivity of synthesized PDI-sAb to reducing reagents, various concentrations of DTT (A) or GSH (B) were added to PDI-sAb (○ 5 μM, ■ 50 μM, □ 500 μM, ◆ 5 mM, ● 30 mM). Fluorescence increased immediately after the addition of reducing reagents and reached peak value within the first 30 seconds. Fold increase in RFU at 300 sec (C) reveals a difference in potency of the reducing reagents. (*, p < 0.05; **, p < 0.01, ***, p < 0.005)
Sensitivity of PDI-sAb to rhPDI and psPDI
Platelet-rich plasma was isolated from citrated human whole blood and was incubated with PDI-sAb (2 μg/ml) for 5 min before dilution to 2% PRP in HBS buffer. Human recombinant PDI (0, 70, or 700 nM rhPDI) was then added to initiate PDI-sAb cleavage. The time and dose-dependent to the increase in PDI-sAb signal indicated sensitivity to reductase activity of nanomolar concentrations of rhPDI (Fig. 3A). In these measurements, a low concentration of 50 μM DTT was added to provide a favored redox condition and to prevent dithiol reformation before sampling. Such low DTT concentration did not lead to appreciable increase in fluorescent signal (Fig. 2A and Fig. 3) indicating the shift in PDI-sAb signal was solely dependent on the thiol reductase activity provided by rhPDI (Fig. 3B, C).
Fig. 3.

PDI-sAb sensitivity to exogenously added rhPDI. Real time flow cytometry of unstimulated platelets labeled with PDI-sAb (A). Recombinant human PDI (rhPDI) was added to initiate sensor cleavage. Mean fluorescence increased rapidly and reached maximum value by 300 sec at [rhPDI] = 700 nM. A histogram of all collected events for one donor shows a shift in mean PDI-sAb fluorescence by 700 nM rhPDI (B). Data from all donors (mean ± SD) shows 70 nM of rhPDI was sufficient to cause a significant increase in PDI-sAb cleavage after 5 min incubation (C). (***, p<0.005; ****, p<0.001)
The sensitivity of PDI-sAb to endogenous platelet reductase activity was also measured by flow cytometry with citrated 2% PRP. PRP was incubated with PDI-sAb in HBS buffer for 5 minutes before recalcification (5 mM Ca2+) and addition of agonists (60 μM TRAP and 2 nM convulxin). Unstimulated platelets did not express significant reductase activity within the first 300 sec (Fig. 4A). A slight increase in PDI-sAb signal was observed at later times, which was probably caused by platelet activation and granule release indicated by slight increase in P-selectin expression after 400 sec post recalcification (Fig. 4A, B). Both of detected thiol reductase activity and P-selectin expression significantly increased upon platelet cellular activation (Fig. 4C). A significant increase in PDI-sAb signal was observed at similar time points with that in P-selectin signal suggesting expression of thiol reductase activity on platelet surface occurred concurrently with α-granule exocytosis at the tested high dosage of agonists.
Fig. 4.

Sensitivity of PDI-sAb to platelet-derived thiol reductase activity. Real-time flow cytometry of PDI-sAb labeled resting (A) or stimulated platelets (B). A rapid increase in PDI-sAb mean fluorescence was only observed when platelets were stimulated by TRAP (60 μM) and convulxin (2 nM). Data from all donors (mean ± SD) shows a significant increase in both thiol reductase activity and P-selectin expression on activated platelets after 10 min incubation (C). (**, p<0.01; ***, p<0.005)
With the observation of concurrent expression of P-selectin and thiol reductase activity on the platelet surface, correlation between thiol reductase activity expression and phosphatidylserine (PS) exposure was also tested. A total of 104 events were collected after incubating 2% PRP (isolated from PPACK treated whole blood) with PDI-sAb (5 μg/ml) and agonists (60 μM TRAP and 2 nM convulxin) for 15 minutes. Baseline activity was measured in PDI-sAb labeled non-stimulated PRP (Fig. 5A, C). By the end of 15 minutes incubation, a majority (94.1%) of the stimulated platelets expressed P-selectin on their surfaces (Fig. 5B). About half of the P-selectin expressing population exhibited cell surface-associated thiol reductase activity (96% of the total reductase activity). Representative two-dimensional dot profile shows a clear linear dependence of thiol reductase activity with P-selectin expression (Fig. 5B). Only slightly over than half (54.7%) of the stimulated platelets became PS positive, but 76% of the reductase activity was expressed on PS positive platelets (Fig. 5D). Our data indicates the expression of thiol reductase activity was directly associated with the level of platelet activation and granule release.
Fig. 5.

Platelet expression of thiol reductase activity is associated with granule release and PS expression. Examples of two dimensional dot profiles for one representative donor and data for all donors (mean ± SD) show the relationship between thiol reductase activity expression and granule release on resting (A) or stimulated (B) platelets and the relationship between thiol reductase activity expression and PS expression on resting (C) or stimulated (D) platelets. The vertical axis represents PDI-sAb fluorescence while the horizontal axis represents P-selectin (A, B) or PS (C, D) fluorescence. When stimulated by TRAP (60 μM) and convulxin (2 nM), 96% of the PDI-sAb positive platelets are P-selectin positive while 76% of the PDI-sAb positive platelets are exposing PS.
Sensitivity of PDI-sAb to PDI inhibition
To test the sensitivity of PDI-sAb to PDI inhibition, 2% PRP (isolated from PPACK treated whole blood) was incubated with the PDI inhibitors rutin (cell impermeable, IC50 = 6.1 μM)[20] or PACMA 31(cell permeable, IC50 = 10 μM)[31] or anti-PDI monoclonal antibody RL 90 along with platelet agonists (60 μM TRAP and 2 nM convulxin) and PDI-sAb (5 μg/ml). A total of 104 events were collected after a 15 min incubation. Normalized signals (to the maximum signals collected on non-inhibited stimulated platelets) show that neither rutin nor PACMA 31 was a perfect inhibitor of platelet-derived thiol reductase activity. The maximum inhibition achieved without causing severe disturbance on P-selectin or PS exposure was less than 50% (Fig. 6A, B). RL 90 only caused partial reduction in reductase activity on stimulated platelets suggesting other platelet-derived thiol isomerases are also important sources of platelet surface reductase activity (Fig. 6C). At the highest tested dosages, PACMA 31 and RL 90 individually caused a reduction of over 60% in PDI-sAb signal while rutin blocked roughly half of the PDI-sAb signal. Severe reductions in both α-granule release and PS exposure were however observed suggesting possible off-target effects of high dosages of the inhibitors and antibody [25]. It is also possible that platelet-derived thiol reductase activity is essential for normal granule secretion. But the observation that PDI-null platelets display normal P-selectin exposure made this less likely [25]. We also found when platelets were treated with rutin or PACMA31, much less reductase activity was observed even in the P-selectin positive population, which indicates that the decreased PDI-sAb signal on inhibited platelets was not simply due to reduced thiol isomerase expression caused by disturbed granule release (Fig. S2). It has also been reported PDI inhibition upregulates PS exposure on endothelial cells [32], but we failed to detect similar effect of rutin on platelets PS exposure (Fig. 6).
Fig. 6.
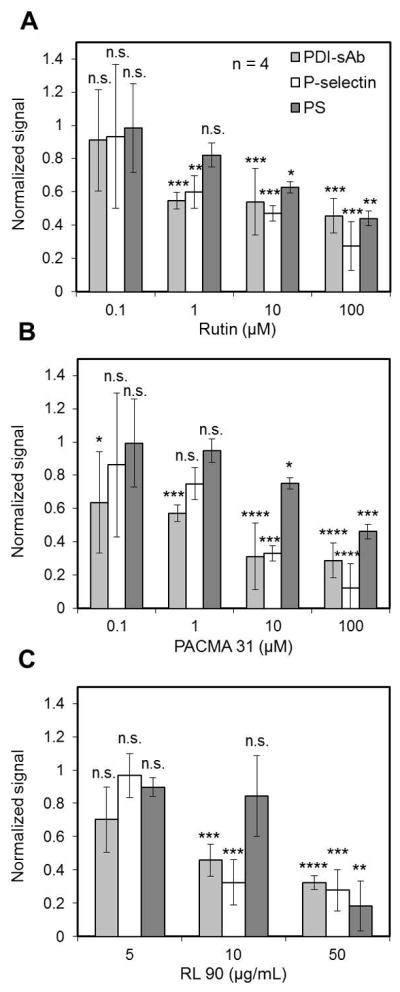
PDI-sAb sensitivity to PDI inhibitors and anti-PDI antibody. Data from all donors (normalized to stimulated but non-inhibited platelets, mean ± SD) shows inhibition of PDI-sAb cleavage on TRAP (60 μM) and convulxin (2 nM) stimulated platelets by PDI inhibitors rutin (A) and PACMA 31 (B) and anti-PDI antibody (RL 90) at various dosages. Both inhibitors and antibody caused significant reduction in P-selectin and PS expression at higher tested dosages. (**, p<0.01; ***, p<0.005; ****, p<0.001)
Detection of PDI reductase activity in a microfluidic thrombosis model
CTI- (4 μg/ml) or PPACK- (100 μM) treated human whole blood was labeled with PDI-sAb (1:10 WB, 1 μg/ml) before it was perfused over a patterned collagen surface at 100 s−1 initial wall shear rate. Platelet deposition and reductase activity in growing thrombi were monitored simultaneously. The rapid increase of PDI-sAb signal was predominately observed over the first 200 sec whereas platelets continued to accumulate for over 500 sec (Fig. 7). In the presence of thrombin, adding rutin (82 μM) did not cause reduction in platelet aggregation or in PDI-sAb cleavage (Fig. 7A–C). A similar effect was observed with a monoclonal function-blocking PDI antibody (RL90, 20 μg/mL); platelet aggregation on collagen was only affected by PDI inhibition when thrombin was inhibited by PPACK (Fig. S3). Interestingly, rutin and RL90 did not cause reduction in PDI-sAb and CD41 fluorescence during the first 60 sec, which is when primary aggregation of platelets on collagen occurred.
Fig. 7.

Detection of thiol reductase activity in vitro. Low level of CTI (4 μg/ml, A–C) or PPACK (100 μM, D–F) treated whole blood was perfused over collagen at venous shear rate (100 s−1). Platelet deposition (A) and thiol reductase activity (B) were unaffected by rutin (82 μM) in the presence of thrombin. Images of platelet and PDI-sAb signals on collagen at 500 seconds show no distinguishable difference between control and rutin treated thrombi (C). Rutin caused significant reduction in platelet deposition (D) and detected thiol reductase activity (E) when thrombin was inhibited by PPACK. Real time images at 500 seconds show a severe reduction in platelet and PDI-sAb signals in rutin treated thrombus (F). Scale bars represent 100 μm.
Detection of PDI reductase activity in vivo
Laser injury of mouse cremaster arterioles consistently produces a hemostatic clot that prevents continued red cell loss, and develops a characteristic architecture consisting of a core of highly activated platelets covered by a shell of loosely-packed and less activated platelets [33]. Using a mouse anti-platelet scaffold we produced a mouse PDI-sAb to investigate PDI reductase activity in the context of this injury model. We consistently observed a gradient of PDI-sAb signal within the thrombus, which was strongest in the core region proximal to the injury site (Fig. 8). These results are consistent with our in vitro results showing increased PDI reductase activity associated with highly activated, P-selectin positive platelets.
Fig. 8.

Detection of thiol reductase activity in vivo. Thiol reductase activity was detected in thrombus formed after laser-induced arteriolar wall injury in mouse. The site of the injury was observed using confocal fluorescence microscopy. Arrows indicate flow direction. Platelets (blue) were outlined with bright field images (A–C). Thiol reductase activity (green) was more concentrated in the core area (B,C) and was co-localized with P-selectin positive (outlined in red) area (C). Pseudo-color images (D–F) also show that PDI-sAb signal accumulated in the core area where P-selectin was expressed (outlined in white).
Discussion
In vivo studies have suggested that endothelial PDI plays a role in thrombus formation. [24], but the importance of platelet-derived PDI has been questioned. To address this issue, in this study we have developed a platelet-targeted PDI sensor (PDI-sAb) that is suitable for sensing thiol reductase activity on or near the surface of human platelets in a microfluidic thrombosis model that does not include endothelial cells. Additionally, we generated a version of the sensor that targets mouse platelets and used it to visualize sensor fluorescence dynamics within a growing thrombus in an in vivo laser injury model.
In flow cytometry, we found that PDI-sAb was a sensitive marker for thiol reductase activity and was capable of detecting the activity of nanomolar levels of rhPDI. Upon activation, platelets displayed rapidly increasing PDI-sAb signal, which was consistent with the dynamics of α-granule exocytosis. We subsequently found the majority of the detectable activity was localized on PS positive platelets, suggesting that expression of the reductase activity is dependent on the level of platelet activation. However, the dose-response of α-granule exocytosis and thiol reductase activity expression was not the same. α-granule release required lower dosage of agonists than thiol reductase activity expression (Fig. S4).
In a microfluidic clotting assay, we were able to visualize the development of PDI-sAb signal in growing thrombi on collagen surface. Most of the PDI-sAb signal increase was detected during the first 200 sec suggesting initial platelet deposition on collagen and proximity to the surface, instead of secondary platelet aggregation during clot buildup, was correlated with platelet thiol reductase activity expression. Despite the high micromolar levels of GSH that is naturally present in whole blood [34], neither anticoagulated whole blood nor anticoagulated PRP exhibited appreciable reductase activity for at least 15 minutes. Moreover, PDI-sAb remained in disulfide form in tissue factor (TF, 400 pM) stimulated pooled platelet free plasma or in solution with concentrated pure thrombin (Fig. S5 and S6). Thus, in the flow experiments, thrombus incorporated PDI-sAb signal was caused by endogenous thiol reductase activity instead of non-specific cleavage by blood enzymes. Several in vitro studies have shown the importance of thiol isomerase activity for normal platelet aggregation [6, 8, 10, 20, 24]. However, neither PDI inhibitor rutin nor PDI antibody RL90 caused reduction in platelet accumulation on collagen surface in the presence of thrombin in our hands. A previous study has shown PDI-null platelets exhibit normal aggregation and granule release when stimulated by high dosages of agonists [25]. In our microfluidic assay, the abundant surface-immobilized collagen and locally-generated secondary aggregation agonists (i.e. thrombin, ADP, and thromboxane) were probably sufficient to overcome PDI inhibition by rutin or RL90. Thus, we attribute the lack of efficacy of PDI inhibition to high level of both primary and secondary agonists for platelet aggregation and the compensating effect from other platelet-expressed thiol isomerases. We also found that when thrombin is inhibited, PDI inhibition only disturbed platelet aggregation after the initiation phase (~60 sec). In this assay, immobilized collagen vigorously and rapidly activates and recruits platelets during the initiation phase. It is unlikely PDI can further promote this process given both of the potency and surface density of collagen is high. In this microfluidic system, PDI was most likely taking part in platelet secondary aggregation by either facilitating α2bβ3 remodeling or other unknown mechanisms and its effect is only detectable in the absence of thrombin. However, we cannot tell if this was because PPACK neutralized the masking effect of thrombin on PDI function or thrombin inhibition caused deficient expression of other endogenous thiol isomerases as alternative sources of thiol isomerase activity.
We have also presented, to our knowledge, the first sensor capable of measuring the spatial distribution of thiol reductase activity in vivo. We observed a gradient of PDI-sAb signal emanating away from the in vivo injury site. Thiol reductase activity was predominately associated with highly activated P-selectin positive platelets which were localized in the core of thrombus. Both endothelium and platelets are sources of extracellular PDI in vivo [5, 6, 24]. Endothelium- and platelet-derived PDI may differ in function in that endothelium PDI may be more critical for initial platelet recruitment while platelet PDI may be more important for continued thrombus propagation [5, 20, 35]. The finding that PDI inhibition had no effect on initial platelet aggregation on collagen in the microfluidic thrombosis model lacking endothelium is in agreement with this scenario. Sequential PDI secretion by endothelium and then activated platelets would introduce a time dependency of PDI availability from different cellular sources [5, 24], which could also contribute to a less dominant role of platelet PDI on initial platelet adhesion in vivo.
A previous study suggested that β3 integrins are essential for capturing PDI at site of vascular injury [15]. We found with mouse PDI-sAb that after the core-shell structure of thrombus was fully developed, thiol reductase activity was well retained in the P-selectin positive core area but not in the shell area where likely platelet αIIbβ3 are also activated. The mouse PDI-sAb can detect endogenous platelet reductase activity as well as reductase activity derived from other cellular sources (i.e. endothelial cells) but the sensor is clearly sensing the platelet surface. The availability of thiol reductase activity is likely dependent on the level of cellular activation and physical proximity to endothelium. While the β3 integrins in the shell area can serve as substrates for PDI, the core area with more activated platelets and close proximity to endothelium is probably a more favored site for thiol reductase activity accumulation, which is consistent with flow cytometry results that reductase activity was mostly localized on more activated platelets.
In summary, we used a novel approach to visualize extracellular thiol reductase activity during thrombus formation both in vitro and in vivo. We found the distribution of thiol reductase activity was strongly correlated with P-selectin display and somewhat less correlated with PS exposure even though most of thiol reductase activity was localized on PS exposed platelets. Expression of thiol reductase activity was mostly correlated with initial platelet aggregation on collagen in the microfluidic thrombosis model. PDI inhibition showed limited effect in the present of thrombin suggesting dependency of platelet PDI function on the combination and level of triggers for platelet activation. Thiol reductase activity was concentrated in the thrombus core area in vivo and was co-localized with P-selectin display, which could be a result of more platelet activation in the core and physical proximity of the core to endothelium, an alternative cellular source of thiol reductase activity.
Supplementary Material
Essentials.
Protein disulfide isomerases (PDI) may play essential role in thrombus formation.
A platelet-binding senor (PDI-sAb) was developed to detect thiol reductase activity under flow.
Primary human platelet adhesion to collagen at 200 s-1 was correlated with PDI-sAb signal.
Detected thiol reductase activity was localized in the core of growing thrombi at injury site in mice.
Acknowledgments
This work was supported by the National Institutes of Health, National Heart, Lung and Blood Institute (P01HL120846 and P01HL040387 [L.F.B.] and R01HL103419 [S.L.D. and L.F.B.]). J.D.W. was supported by American Heart Association predoctoral fellowship 14PRE19560005.
Footnotes
Addendum
S. Zhu and J. D. Welsh designed and performed the experiments, analyzed data and contributed to the manuscript. L. F. Brass analyzed data and contributed to the manuscript. S. L. Diamond designed the study, analyzed data and contributed to the manuscript.
Disclosure of Conflict of Interest
S. L. Diamond and J. D. Welsh report grants from NIH, during the conduct of the study.
L. F. Brass reports grants from NIH during the conduct of the study and other support from Merck Pharmaceuticals, outside the submitted work.
Other authors state that they have no conflict of interest.
References
- 1.Thon JN, Peters CG, Machlus KR, Aslam R, Rowley J, Macleod H, Devine MT, Fuchs TA, Weyrich AS, Semple JW, Flaumenhaft R, Italiano JE. T granules in human platelets function in TLR9 organization and signaling. J Cell Biol. 2012;198:561–74. doi: 10.1083/jcb.201111136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen K, Detwiler TC, Essex DW. Characterization of protein disulphide isomerase released from activated platelets. Br J Haematol. 1995;90:425–31. doi: 10.1111/j.1365-2141.1995.tb05169.x. [DOI] [PubMed] [Google Scholar]
- 3.Essex DW, Chen K, Swiatkowska M. Localization of protein disulfide isomerase to the external surface of the platelet plasma membrane. Blood. 1995;86:2168–73. [PubMed] [Google Scholar]
- 4.Holbrook LM, Watkins NA, Simmonds AD, Jones CI, Ouwehand WH, Gibbins JM. Platelet release novel thiol isomerase enzymes which are recruited to the cell surface following activation. Br J Haematol. 2010;148:627–37. doi: 10.1111/j.1365-2141.2009.07994.x. [DOI] [PubMed] [Google Scholar]
- 5.Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. 2008;118:1123–31. doi: 10.1172/JCI34134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharda A, Kim SH, Jasuja R, Gopal S, Flaumenhaft R, Furie BC, Furie B. Defective PDI release from platelets and endothelial cells impairs thrombus formation in Hermansky-Pudlak syndrome. Blood. 2015;125:1633–42. doi: 10.1182/blood-2014-08-597419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Y, Ahmad SS, Zhou J, Wang L, Cully MP, Essex DW. The disulfide isomerase ERp57 mediates platelet aggregation, hemostasis, and thrombosis. Blood. 2012;119:1737–46. doi: 10.1182/blood-2011-06-360685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou J, Wu Y, Wang L, Rauova L, Hayes VM, Poncz M, Essex DW. The disulfide isomerase ERp57 is required for fibrin deposition in vivo. J Thromb Haemost. 2014;12:1890–7. doi: 10.1111/jth.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L, Wu Y, Zhou J, Ahmad SS, Mutus B, Garbi N, Hammerling G, Liu J, Essex DW. Platelet-derived ERp57 mediates platelet incorporation into a growing thrombus by regulating of the αIIbβ3 integrin. Blood. 2013;122:3642–50. doi: 10.1182/blood-2013-06-506691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Passam FH, Lin L, Gopal S, Stopa JD, Bellido-Martin L, Huang M, Furie BC, Furie B. Both platelet- and endothelial cell-derived ERp5 supports thrombus formation in a laser-induced mouse model of thrombosis. Blood. 2015;125:2276–85. doi: 10.1182/blood-2013-12-547208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jordan PA, Stevens JM, Hubbard GP, Barrett NE, Sage T, Authi KS, Gibbins JM. A role for the thiol isomerase protein ERp5 in platelet function. Blood. 2005;105:1500–7. doi: 10.1182/blood-2004-02-0608. [DOI] [PubMed] [Google Scholar]
- 12.Zhou J, Wu Y, Wang L, Rauova L, Hayes VM, Poncz M, Essex DW. The C-terminal CGHC motif of protein disulfide isomerase supports thrombosis. J Clin Invest. 2015:2015. doi: 10.1172/JCI80319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burgess JK, Hotchkiss KA, Suter C, Dudman NPB, Szollosi J, Chesterman CN, Chong BH, Hogg PJ. Physical proximity and functional association of glycoprotein 1bα and protein-disulfide isomerase on the platelet plasma membrane. J Biol Chem. 2000;275:9758–66. doi: 10.1074/jbc.275.13.9758. [DOI] [PubMed] [Google Scholar]
- 14.Manickam N, Sun X, Li M, Gazitt Y, Essex DW. Protein disulphide isomerase in platelet function. Br J Haematol. 2008;140:2168–73. doi: 10.1111/j.1365-2141.2007.06898.x. [DOI] [PubMed] [Google Scholar]
- 15.Cho J, Kennedy DR, Lin L, Huang M, Merrill-Skoloff G, Furies BC, Furie B. Protein disulfide isomerase capture during thrombus formation in vivo depends on the presence of β3 integrins. Blood. 2012;120:647–55. doi: 10.1182/blood-2011-08-372532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lahav J, Jurk K, Hess O, Barnes MJ, Farndale RW, Luboshitz J, Kehrel BE. Sustained integrin ligation involves extracellular free sulfhydryls and enzymatically catalyzed disulfide exchange. Blood. 2002;100:2472–8. doi: 10.1182/blood-2001-12-0339. [DOI] [PubMed] [Google Scholar]
- 17.Essex DW, Li M, Miller A, Feinman RD. Protein Disulfide Isomerase and Sulfhydryl-Dependent Pathways in Platelet Activation. Biochemistry. 2001;40:6070–5. doi: 10.1021/bi002454e. [DOI] [PubMed] [Google Scholar]
- 18.Lahav J, Wijnen EM, Hess O, Hamaia SW, Griffiths D, Makris M, Knight CG, Essex DW, Farndale RW. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood. 2003;102:2085–92. doi: 10.1182/blood-2002-06-1646. [DOI] [PubMed] [Google Scholar]
- 19.Khodier C, VerPlank L, Nag PP, Pu J, Wurst J, Pilyugina T, Dockendorff C, Galinski CN, Scalise AA, Passam F, van Hessem L, Dilks J, Kennedy DR, Flaumenhaft R, Palmer MAJ, Dandapani S, Munoz B, Schrieber SL. Probe Reports from the NIH Molecular Libraries Program. Bethesda MD: 2010. Identification of ML359 as a Small Molecule Inhibitor of Protein Disulfide Isomerase. [PubMed] [Google Scholar]
- 20.Jasuja R, Passam FH, Kennedy DR, Kim SH, van Hessem L, Lin L, Bowley SR, Joshi SS, Dilks JR, Furie B, Furies BC, Flaumenhaft R. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest. 2012;122:2104–13. doi: 10.1172/JCI61228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flaumenhaft R, Furie B, Zwicker JI. Therapeutic Implications of Protein Disulfide Isomerase Inhibition in Thrombotic Disease. Arterioscler Thromb Vasc Biol. 2015;35:16–23. doi: 10.1161/ATVBAHA.114.303410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen VM, Ahamed J, Versteeq HH, Berndt MC, Ruf W, Hoqq PJ. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry. 2006;45:12020–8. doi: 10.1021/bi061271a. [DOI] [PubMed] [Google Scholar]
- 23.Ahamed J, Versteeq HH, Kerver M, Chen VM, Mueller BM, Hoqq PJ, Ruf W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci. 2006;103:13932–7. doi: 10.1073/pnas.0606411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jasuja R, Furie B, Furie BC. Endothelium-derived but not platelet-derived protein disulfide isomerase is required for thrombus formation in vivo. Blood. 2010;116:4665–74. doi: 10.1182/blood-2010-04-278184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim K, Hahm E, Li J, Holbrook LM, Sasikumar P, Stanley RG, Ushio-Fukai M, Gibbins JM, Cho J. Platelet protein disulfide isomerase is required for thrombus formation but not for hemostasis in mice. Blood. 2013;122:1052–61. doi: 10.1182/blood-2013-03-492504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu S, Sankar S, Neamati N. Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today. 2014;19:222–40. doi: 10.1016/j.drudis.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 27.Reinhardt C, von Bruhl ML, Manukyan D, Grahl L, Lorenz L, Altmann B, Dlugai S, Hess S, Konrad I, Orschiedt L, Mackman N, Ruddock L, Massberg S, Engelmann B. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118:1110–22. doi: 10.1172/JCI32376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crimmins DL. Analysis of disulfide-linked homo- and hetero-peptide dimers with a strong cation-exchange sulfoethyl aspartamide column. Pept Res. 1989;2:395–401. [PubMed] [Google Scholar]
- 29.Maloney SF, Brass LF, Diamond SL. P2Y12 or P2Y1 inhibitors reduce platelet deposition in a microfluidic model of thrombosis while apyrase lacks efficacy under flow conditions. Integr Biol. 2009;2:183–92. doi: 10.1039/b919728a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welsh JD, Stalker TJ, Voronov R, Muthard RW, Tomaiuolo M, Diamond SL, Brass LF. A system approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood. 2014;124:1808–15. doi: 10.1182/blood-2014-01-550335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R, Zandi E, Petasis NA, Neamati N. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc Natl Acad Sci. 2012;109:16348–53. doi: 10.1073/pnas.1205226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popescu NI, Lupu C, Lupu F. Extracellular protein disulfide isomerase regulates coagulation on endothelial cells through modulation of phosphatidylserine exposure. Blood. 2010;116:993–1001. doi: 10.1182/blood-2009-10-249607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stalker TJ, Traxler EA, Wu J, Wannemacher KM, Cermignano SL, Voronov R, Diamond SL, Brass LF. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 2013;121:1875–85. doi: 10.1182/blood-2012-09-457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michelet F, Guequen R, Leroy P, Wellman M, Nicolas A, Siest G. Blood and plasma glutathione measured in healthy subjects by HPLC: relation to sex, aging, biological variables, and life habits. Clin Chem. 1995;41:1509–17. [PubMed] [Google Scholar]
- 35.Furie B, Flaumenhaft R. Thiol isomerases in thrombus formation. Circ Res. 2014;114:1162–73. doi: 10.1161/CIRCRESAHA.114.301808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.