

Abstract
Lipid peroxidation can be broadly defined as the process of inserting a hydroperoxy group into a lipid. Polyunsaturated fatty acids present in the phospholipids are often the targets for peroxidation. Phospholipids are indispensable for normal structure of membranes. The other important function of phospholipids stems from their role as a source of lipid mediators – oxygenated free fatty acids that are derived from lipid peroxidation. In the CNS, excessive accumulation of either oxidized phospholipids or oxygenated free fatty acids may be associated with damage occurring during acute brain injury and subsequent inflammatory responses. There is a growing body of evidence that lipid peroxidation occurs after severe traumatic brain injury in humans and correlates with the injury severity and mortality. Identification of the products and sources of lipid peroxidation and its enzymatic or non-enzymatic nature is essential for the design of mechanism-based therapies. Recent progress in mass spectrometry-based lipidomics/oxidative lipidomics offers remarkable opportunities for quantitative characterization of lipid peroxidation products, providing guidance for targeted development of specific therapeutic modalities. In this review, we critically evaluate previous attempts to use non-specific antioxidants as neuroprotectors and emphasize new approaches based on recent breakthroughs in understanding of enzymatic mechanisms of lipid peroxidation associated with specific death pathways, particularly apoptosis. We also emphasize the role of different phospholipases (calcium-dependent and -independent) in hydrolysis of peroxidized phospholipids and generation of pro- and anti-inflammatory lipid mediators.
Keywords: Phospholipase A2, cyclooxygenase, lipoxygenase, cytochrome c, cardiolipin, glutathione
1. Introduction
Oxygenated lipid mediators and reactive oxygen species play important roles in maintaining homeostatic balance under normal conditions (Halliwell, 1991; Niki, 2009). However excessive generation of reactive oxygen species with depletion of antioxidant reserves resulting in lipid peroxidation have been implicated in programmed cell death pathways and inflammation (Niki, 2009). After the identification of soybean lipoxygenase, lipid peroxidation has been studied extensively to understand its mechanisms, dynamics and effects on biological systems (Bergstrom, 1945; Halliwell and Chirico, 1993; Khan, 1955). Apart from producing local effects such as altering membrane structure, integrity, fluidity, permeability and functionality, the products of lipid peroxidation are effective in initiating secondary cellular responses (Ernster et al., 1968; Sevanian and Hochstein, 1985).
The central nervous system (CNS) is particularly vulnerable to uncontrolled lipid peroxidation upon injury for several reasons. First, the brain exhibits high metabolic activity receiving 20% of the oxygen despite accounting for 2% of the body weight (Pratico et al., 2002). With high rates of oxidative metabolic activity, the brain generates large quantities of reactive oxygen metabolites (Ames et al., 1993). Second, neurons lack the capacity to regenerate. Third, neurons exhibit a high membrane-to-cytoplasm ratio (Evans, 1993). Fourth, the brain has high levels of redox transition metals such as iron that can catalyze reactive metabolite generation (Halliwell, 1992). While catalytically-active iron is typically sequestered intracellularly by ferritin and extracellularly by transferrin, low pH conditions observed after traumatic brain injury (TBI) might facilitate iron release for oxidative reactions (Hall et al., 2010). Furthermore, disruption of the vasculature by mechanical forces can lead to release of free hemoglobin into the brain parenchyma providing an additional source of redox-active iron. Finally, despite high oxidative metabolic activity, the brain has a relatively low antioxidant capacity compared to other tissues (Markesbery and Lovell, 2007). Generation of lipid mediators from lipid hydroperoxides has important functions in normal CNS physiology (Niki, 2009). However their uncontrolled and excessive production plays an important role in secondary injury mechanisms that are set into motion after TBI (Kasprzak et al., 2001).
Lipid peroxidation can be broadly defined as the process of inserting a hydroperoxy group into a lipid. Polyunsaturated fatty acids (PUFA) present in the glycerolipids, phospholipids and cholesterols are often the targets for peroxidation, where the peroxyl groups are derived from an oxygen molecule or from hydrogen peroxide. PUFAs are fatty acids containing two or more double bonds each separated by a methylene bridge (-CH2-) at their aliphatic carbon back bone. The bis-allelic hydrogen or the hydrogen attached to the methylene bridge are very easy to remove, thus making these lipids highly susceptible to lipid peroxidation (Gardner, 1989). Due to their ability to form a flexible membrane structure (Feller et al., 2002), PUFAs are an important and major fatty acid class accounting for approximately 50% of total membrane fatty acids (Tinoco, 1982). Among the PUFAs, various species are observed in different locations. For example, the major ω-6 fatty acids linoleic acid and arachidonic acid are seen across the membrane systems, whereas the ω-3 fatty acid docosahexaenoic acid is concentrated in the brain cortex and organelles such as synaptosomes, synaptic vesicles, mitochondria and microsomes (Bradbury, 2011; Tinoco, 1982). The omnipresence, high abundance and sensitivity towards oxidation makes PUFAs one of the major targets of oxidative stress through lipid peroxidation in the cellular system.
In TBI, lipid peroxidation is marked by two major pathways: enzymatic and non-enzymatic. The enzymatic pathways involve activation of phospholipase A2 (PLA2) (Bazan et al., 1995), cyclooxygenases (COX) (Dash et al., 2000), lipoxygenases (LOX) (Zhang et al., 2006), cytochrome c and other peroxidases (Ji et al., 2012a; Mendes Arent et al., 2014). Non-enzymatic pathways primarily involve interaction of transition metals with reactive oxygen species (ROS) (Bayir et al., 2006). Moreover, the cellular defense system against lipid peroxidation is also compromised with TBI. This defense system includes various free radical removal enzymes such as catalase, superoxide distmutases (SOD) and glutathione peroxidases(Ansari et al., 2008) as well as sacrificial antioxidants such as Vitamin C and E (Bayir et al., 2006). Accumulation of many of the lipid peroxidation products are known to be toxic for the cells. Lipid mediators generated from oxidation of free or esterified PUFA have been implicated in dysregulation of cerebral blood flow, blood brain barrier damage, activation of inflammatory response and programmed cell death pathways (Williams and Higgs, 1988); (Kagan et al., 2009a; Friedmann Angeli et al., 2014). On the other hand some of the lipid mediators generated from multistage enzymatic oxidation of PUFA support the resolution of inflammatory process (Serhan, 2014).
In the following sections, we will describe the mechanisms of lipid peroxidation; summarize detection of lipid peroxidation in clinical TBI, and we will review previous attempts to use non-specific antioxidants as neuroprotectors and emphasize new approaches based on recent breakthroughs in understanding of enzymatic mechanisms of lipid peroxidation.
2. Mechanism of Lipid peroxidation
Classic description of the chain reaction of lipid peroxidation involves three main steps: initiation, prolongation and termination as shown in Figure 1. Initiation is the removal of a hydrogen atom from the PUFA to form a carbon-centered lipid free radical (Pederson and Aust, 1975). After the hydrogen is removed, the unpaired electron is rearranged to form a more stable radical containing conjugated dienes. The lipid radical then attracts molecular oxygen from the available dissolved oxygen pool to form lipid peroxyl radicals. The lipid peroxyl radical initiates a new cycle by extracting a hydrogen, thereby perpetuating the peroxidation reaction. The final step, or termination, is the formation of a non-radical from the combination of two radicals (Halliwell and Chirico, 1993; Porter et al., 1995; Wills, 1966; Witting, 1965). Apart from hydroperoxides, lipid peroxidation produces many products which include: lipid hydroxides, isoprostanes, neuroprostanes, aldehydes (such as 4-hydroxy-2-nonenal or 4-HNE), ketones, alkanes, lyso-phospholipids and aldehyde modified proteins (Gueraud et al., 2010; Porter, 1984). In this section, we will primarily review the peroxidation of phospholipids since phospholipids constitute 25% of brain’s dry weight (Yusuf, 1992) and have high PUFA content. The majority of brain phospholipids contain PUFAs in their sn-2 position and some of them also contain PUFA in their sn-1 position making them vulnerable to oxidation (Bayir et al., 2007b; Ji et al., 2012a; Sparvero et al., 2010).
Figure 1. Chain reaction of lipid peroxidation.

Chain reaction of lipid peroxidation involves a three step pathway: 1) initiation - removal of a hydrogen from PUFAs to form a carbon-centered lipid radical; 2) propagation - attachment of a peroxy group to the lipid radical to form a lipid peroxyl radical. This lipid peroxyl radical initiates a new cycle by extracting a hydrogen, thereby perpetuating the peroxidation reaction; and 3) termination - disappearance of free radical by forming a non-radical such as hydroxide.
2.1. Enzymatic lipid peroxidation
Phospholipid hydroperoxides are the primary product of phospholipid oxidation and are mainly generated by enzymatic pathways in the cells. Enzymes containing iron in either the heme or non-heme form can catalyze the peroxidation of phospholipids. Cyclooxygenases (prostaglandin-endoperoxide synthase or COX) are the major phospholipid-oxidizing enzymes and produce prostanoids such as prostaglandins, prostacyclin and thromboxane (Bergstroem et al., 1964; Dubois et al., 1998; Samuelsson, 1979). COX primarily acts on free arachidonic acid to produce various prostanoids; however there is evidence that it can also act on a variety of other PUFAs including linoleic acid, gamma-linolenic acid, eicosadienoic acid, eicosatrienoic acid, eicosapentanoic acid, adrenic acid and docosahexaenoic acid (Laneuville et al., 1995; Wada et al., 2007; Yuan et al., 2009). The enzymatic activity of COX was originally described to utilize free fatty acids as substrates, but recent studies report COX oxidizes fatty acids esterified to phospholipids (Hammond and O'Donnell, 2012). The mechanism underlying the production of such esterified prostaglandins is not clearly understood (Aldrovandi et al., 2013). There are three types of COX enzymes present in the mammalian system: COX1, COX2 and COX3. COX1 is constitutively expressed, whereas the others are expressed upon induction (Chandrasekharan et al., 2002; Crofford, 1997).
Another major class of lipid peroxidation enzymes are the LOX. While COX generates cyclic lipid peroxidation byproducts, LOX produce linear peroxidation products (Kuhn et al., 2015). Lipoxygenases form positional, stereo- and enantio-specific lipid peroxidation products. Lipoxygenases are responsible for the production of hydroperoxy, hydroxy, epoxy and ketoxy fatty acids, leukotrienes (Hammarstrom, 1983), hepoxilins (Nigam et al., 2007) and lipoxins (Serhan et al., 1984). Lipoxygenases can effectively oxidize both free and esterified fatty acid chains (Clark et al., 2011; Maskrey et al., 2007; O'Donnell and Murphy, 2012). Based on the peroxide attachment position on the fatty acid chain, the mammalian lipoxygenases are classified as LOX3, LOX5, LOX8, LOX12 and LOX15. LOX3 mainly acts as an isomerase, while the others are primarily involved in peroxidation of phospholipids (Kuhn et al., 2015).
While COX and LOX play a primary role in phospholipid oxidation, other enzymes such as the major xenobiotic-metabolizing cytochrome p-450 family can also oxidize phospholipids (Serbinova et al., 1989). Cytochrome p-450 enzymes belong to the class of ω-hydroxylases (Kawashima et al., 1997), epoxygenases (Wang et al., 2004) and bisallylic hydroxylases (Brash et al., 1995) which generate terminal hydroxylated fatty acids, epoxy fatty acids and HETEs, respectively. The cytochrome p-450 family consists of 500 genes and 78 different membrane-bound, heme-containing, NADPH-dependent oxidases (Roman, 2002).
Aside from COX, LOX and cytochrome p-450, various iron-containing proteins can gain lipid peroxidase activity. One such protein that has gained attention in the field of lipid peroxidation is the mitochondrial electron transport chain complex protein, cytochrome c. Upon pro-apoptotic stimulation, cytochrome c selectively oxidizes the anionic phospholipid cardiolipin in mitochondria and phosphatidylserine in the plasma membrane (Bayir et al., 2007b; Ji et al., 2012b; Kagan et al., 2005a; Tyurina et al., 2006). Similar to cytochrome c, a number of other membrane proteins with heme catalytic centers have been implicated in lipid peroxidation: myeloperoxidase (Davidenkova et al., 1982; Goldman et al., 1999), cytoglobin (Reeder et al., 2011), myoglobin (Grisham, 1985) and hemoglobin (Szebeni et al., 1984).
2.2. Non-enzymatic lipid peroxidation
Non-enzymatic lipid peroxidation is primarily derived from free radical reactions. Hydroxyl radical formed from the interaction of transition metals with reactive oxygen species (ROS) is the major radical involved in this process (Kappus, 1987). ROS are derived from molecular oxygen, hydrogen peroxide (Fridovich and Porter, 1981) and organic peroxides (Girotti, 1985). Although molecular oxygen is thought to be a powerful oxidizing agent, it is not directly involved in free radical-mediated lipid peroxidation reactions. Acceptance of a single electron by molecular oxygen forms a superoxide anion radical (O2•−) (Kellogg and Fridovich, 1975). Under normal conditions, superoxide is reduced by SOD to form hydrogen peroxide (Fridovich, 1989). Hydrogen peroxide in turn reacts with ferrous ion via Fenton reaction to form hydroxyl radical, hydroxyl anion and ferric ions (Repetto et al., 2010) (Figure 2). Superoxide can also be utilized for the reduction of ferric ion to ferrous ion as the first step of Haber–Weiss reaction (Thomas et al., 1985).
Figure 2.

Reactions involved in the production of free radicals.
In addition to ferrous iron, other transition metal ions such as Cu2+, Co2+, Ni2+ can generate hydroxyl radicals through Fenton-like reaction, albeit with much slower reaction rates. Both iron and copper ions are also involved in the degradation of organic hydroperoxides into organic peroxyl radicals and hydroxyl radicals (Repetto et al., 2010). While the amount of free ionic iron or copper is extremely low under normal conditions due to effective handling of these transition metals within and outside of the cellular environment (MacKenzie et al., 2008), there are conditions where transition metals are elevated as a result of disturbed handling. In disorders characterized by disturbed handling of transition metals, such as hemochromatosis (Papanikolaou and Pantopoulos, 2005) and neurodegenerative conditions (Lee et al., 2006), free transition metals may play a significant role in the generation of lipid hydroperoxides.
Another pathway that can generate hydroxyl radicals is the decomposition of peroxynitrous acid (Beckman et al., 1990). Reaction of superoxide anion radical with nitric oxide (NO) radical forms peroxynitrite anion (Robak and Gryglewski, 1993). The hydroxyl radicals produced by this pathway can act as the initiation step of the lipid peroxidation reaction (Beckman et al., 1990; Beckman et al., 1994; Pryor and Squadrito, 1995). Overall, the non-enzymatic generation of lipid hyrodperoxides is difficult to target therapeutically due to the high reaction rates and non-specific nature of generated products.
3. Detection of lipid peroxidation in clinical TBI
Oxidative stress and lipid peroxidation have been widely studied in experimental TBI (Bazan, 1976; Bazan et al., 2005; Chan, 1996; Hall, 1989; Hall, 1993; Hall et al., 1996). In this section we will briefly review detection of lipid peroxidation in clinical TBI. Lipid peroxidation end products such as malondialdehyde, 4-HNE and isoprostanes have been shown to increase in bio-fluids and brain tissue samples from TBI patients (Al Nimer et al., 2013; Cristofori et al., 2001; Cristofori et al., 2005; Kasprzak et al., 2001; Lin et al., 2014b; Lorente et al., 2015; Niki, 2014). Despite criticism for lack of specificity and artefactual production during analytical processing, malondialdehyde (MDA) and thiobarbituric acid reactive substances (TBARS) remain the most commonly used markers of lipid peroxidation in clinical TBI, and their increase in cerebrospinal fluid (CSF) and serum have been associated with severity of injury and mortality (Cristofori et al., 2001; Cristofori et al., 2005; Kasprzak et al., 2001; Lin et al., 2014b; Lorente et al., 2015). Thiobarbituric acid (TBA) is not specific for MDA but can react with other aldehydes and decomposition products of hydroperoxides (Yoshida et al., 2010). In a heated sample at low pH, interaction of MDA and TBA generates pink chromogen that can be measured by spectrophotometry or spectrofluorophotometry (Niki, 2014) . In order to improve the specificity for lipid oxidation measurements, High Performance Liquid Chromatography (HPLC) methods have been developed and applied to CSF (Cristofori et al., 2001; Cristofori et al., 2005). Another lipid oxidation marker, 4- HNE, is an aldehyde end product of lipid hydroperoxide break down. It forms covalent adducts with proteins and can be detected by immunohistochemistry (Niki, 2014). A recent study reported that 4-HNE-conjugates are present in neurons in human TBI but were not detected in CSF (Al Nimer et al., 2013).
Isoprostanes are a series of prostaglandin-like compounds produced by free radical-mediated lipid peroxidation of arachidonic acid and can be quantified with Enzyme Linked Immunosorbent Assay (ELISA), Radio Immuno Assay (RIA), HPLC, Gas Chromatography-Mass Spectrometry (GC-MS) or Liquid Chromatography-Mass Spectrometry LC-MS (Roberts and Morrow, 2002). Isoprostanes can be formed by cyclooxygenase-catalyzed oxidation of arachidonic acid, however the enzyme does not normally utilize arachidonyl phospholipids as substrates (Bayir et al., 2006). Increases in F2 isoprostane or 8-epi prostaglandin F2α generation have been observed in the CSF of adult TBI patients (Bayir et al., 2004; Wagner et al., 2004). Interestingly, F2-isoprostane levels were two-fold higher in males than females on day 1 post-injury and were reduced with therapeutic hypothermia in the male TBI population (Bayir et al., 2004). Studies in the infant and child TBI population also demonstrated elevation of CSF F2 isoprostane concurrent with other biomarkers of oxidative stress (Bayir et al., 2002). Assessment of end product of lipid hydroperoxides can give a general idea about the involvement of lipid peroxidation in TBI; however, it does not identify the origin and identity of oxidized phospholipids. The latter is important for identification of causal links between lipid peroxidation and mechanisms of secondary injury after TBI.
The primary product of lipid peroxidation is phospholipid hydroperoxides (Niki, 2014). Their short half-life makes them difficult to detect in biological samples. Nevertheless, recent developments in mass spectrometry-based oxidative lipidomics approaches make it possible to detect phospholipid hydroperoxides in plasma and tissue samples (Ji et al., 2012a); (Tyurin et al., 2010; Tyurina et al., 2015). Using this oxidative lipidomics approach, we have shown that lipid peroxidation is a nonrandom event after experimental and clinical TBI, and it is most prominent in the mitochondria early after injury as cytochrome c-catalyzed cardiolipin oxidation (Bayir et al., 2007b; Ji et al., 2012a). Accumulation of cardiolipin oxidation products is essential for the release of pro-apoptotic factors such as cytochrome c into cytosol (Kagan et al., 2005b; Tyurin et al., 2010). Furthermore, cytochrome c-mediated oxidation of cardiolipin is involved in the synthesis of various lipid mediators through hydrolysis by calcium independent iPLA2γ (Tyurina et al., 2014). Overall there is accumulating evidence demonstrating that lipid peroxidation occurs after severe TBI in humans and correlates with injury severity and mortality. Thus the secondary injury cascades leading to lipid peroxidation, particularly enzyme-mediated lipid peroxidation, constitute viable therapeutic targets for neuroprotection.
4. Therapeutic strategies targeting lipid peroxidation
In this section we will briefly review therapies targeting specific enzymatic pathways of lipid peroxidation including mitochondria-targeted electron scavengers and general antioxidant strategies in TBI. For the therapeutic strategies with limited data in TBI, we will also present available information from other acute CNS insults such as stroke and spinal cord injury.
4.1. Phospholipase A2
Phospholipase A2 (PLA2) is an esterase that specifically cleaves the fatty acyl group attached to the sn-2 position in phospholipids. PLA2 enzymes are grouped into four classes based on their activity and subcellular localization, namely secretory PLA2 (sPLA2), cytoplasmic PLA2 (cPLA2), plasmalogen-selective PLA2 (Pls-PLA2) and calcium-independent PLA2 (iPLA2) (Dennis, 1994). Even though PLA2 is not directly involved in the peroxidation of lipids, it is the first rate-limiting step in the production of multiple oxygenated lipid mediators (Farooqui and Horrocks, 2006).
PLA2 activity has been shown to increase after TBI (Shohami et al., 1987), stroke (Yagami et al., 2014) , and SCI (Horrocks et al., 1985). PLA2 generates lysophospholipid and free fatty acid, primarily arachidonic acid. Accordingly, the increase in arachidonic acid and its oxidized derivatives such as eicosanoids and leukotrienes observed after closed head injury were shown to be due to increased cPLA2 activity (Shohami et al., 1989). On the other hand increased activities of both cPLA2 and Pls-PLA2 contribute to increased free fatty acid content after cerebral ischemia (Bazan et al., 1993) (Farooqui and Horrocks, 1994). It was shown that cPLA2 elevation initiate the neural injury and Pls-PLA2 triggers secondary injury (Farooqui et al., 2003; Farooqui and Horrocks, 2004; Fujishima et al., 1999; Sapirstein and Bonventre, 2000). Activation of Pls-PLA2 as well as loss of plasmalogen-containing phospholipids were also reported after compression and sheer stress spinal cord injury (Lukacova et al., 1996).
A number of experimental studies utilizing PLA2 inhibitors have shown benefit in acute CNS injury models. Examples include Cytidine-5'-diphosphocholine (CDP-choline) in TBI and stroke (Hurtado et al., 2007; Jacotte-Simancas et al., 2015; Secades and Frontera, 1995), quinacrine in middle cerebral artery occlusion (MCAO) (Estevez and Phillis, 1997), and arachidonyl trifluoromethyl ketone in SCI (Huang et al., 2009). CDP-choline is also investigated for its neuroprotective potential in clinical TBI (Adibhatla, 2013; Zafonte et al., 2009). Apart from inhibition of PLA2 activity, CDP-choline also stimulates synthesis of phosphatidylcholine (Grieb, 2014). Despite promising results in TBI and stroke models and in small clinical studies (Calatayud Maldonado et al., 1991; Clark et al., 1997; Clark et al., 1999; Clark et al., 2001; Davalos et al., 2002; Tazaki et al., 1988) two recent large clinical trials showed no benefit in TBI and ischemic stroke (Zafonte et al., 2012; Davalos et al., 2012). It is worth mentioning that glycosphingolipids, primarily gangliosides, strongly inhibit PLA2 activity ex vivo (Bianco et al., 1989), thus they have been tested for their neuroprotective potential in a number of CNS injury models (Sabel et al., 1984) (Toffano et al., 1984) as well as in patients with spinal cord injury. Although the results of the initial single center randomized control trial (RCT) with GM1 ganglioside were protective (Geisler et al., 1991) subsequent multicenter RCT failed to show a significant difference between GM1 treatment and placebo (Geisler et al., 2001). Nevertheless, there was a trend favoring treatment in the subgroup analysis of those with ASIA Impairment Scale-B (AIS B) classification.
4.2. Cyclooxygenase
Cyclooxygenase or prostaglandin-endoperoxide synthase catalyzes the production of prostaglandin H2 (PGH2), which is a precursor of many prostanoids and thromboxanes formed from arachidonic acid (DeWitt, 1991). Among the COX enzymes, COX1 is widely expressed in neurons and microglia, whereas COX2 is expressed constitutively in hippocampal and cortical neurons (Yermakova and O'Banion, 2000). A number of studies reported increased COX2 activity after TBI (Hickey et al., 2007; Morrison et al., 2000; Strauss et al., 2000). Higher levels of COX2 expression are associated with functional deficit and worse outcome after TBI.
Table 1 summarizes experimental studies evaluating COX inhibitors in TBI models. Overall the studies in TBI models suggest that selective inhibitors of COX-2 are not effective in improving outcome. Nonselective COX inhibitors on the other hand might be beneficial depending on the model and timing of the therapy after TBI. But the antiplatelet effects of nonselective COX inhibitors might overweigh the benefit that can be obtained from them in TBI. Notably long-term use of selective COX-2 inhibitors has been associated with an increase in ischemic cerebrovascular events (Bresalier et al., 2005). In contrast, inhibition of COX1 by aspirin has been associated with a reduction in adverse cardiovascular events (Berger et al., 2006; Bhatt et al., 2006). Evidence suggests that beneficial effects of aspirin, particularly low dose aspirin (72–162 mg/day), could be in part related to acetylation of COX2. COX2 acetylation triggers synthesis of novel specialized pro-resolving lipid mediators (SPMs) with anti-inflammatory actions such as 15-epi-lipoxin A4 (Serhan et al., 2000). In agreement with this concept, a recent study in fluid percussion injury model showed improved motor and cognitive function when rats were pretreated with aspirin-triggered SMP, 17(R)-Resolvin D1 (Harrison et al., 2015). Although aspirin may have beneficial effects by preventing secondary injury from ischemia in subarachnoid hemorrhage (Dorhout Mees et al., 2003), pre-injury intake of aspirin was shown to increase the risk of lesion progression and unfavorable outcome after TBI in a recent large multicenter study (Fabbri et al., 2013).
Table 1.
Cyclooxygenase inhibitors in acute brain injury
| Sl. No | Compound | Species | Injury | Effect | Ref |
|---|---|---|---|---|---|
| 1 | Ibuprofen | Mice | TBI, Fluid percussion | No effect | (Harrison et al., 2014) |
| 2 | Indomethacin | Rats | TBI, Fluid percussion | Reduced mortality due to more rapid return to normal breathing after the apneic period post-TBI. | (Kim et al., 1989) |
| 3 | Nimesulide | Rat | TBI, impact acceleration diffuse TBI | Substantial improvement in cognitive function | (Cernak et al., 2002) |
| 4 | Meloxicam | Rat | TBI, Mild weight drop | Reduced MDA, cerebral edema, blood brain barrier (BBB) permeability, restored glutathione levels, improved neurological score at 48h. | (Hakan et al., 2010) |
| 5 | Indomethacin | Mice | TBI, Weight drop | Reduction in brain 6-Keto PGF1α levels (6 h) and neurodeficit, no effect on brain edema | (Girgis et al., 2013) |
| 6 | Carpofen | Mice | TBI, Weight Drop | Reduced lesion size; water content and number of microglia in the lesioned cortex; lowered the levels of proinflammatory cytokines and improved functional outcome | (Thau-Zuchman et al., 2012) |
| 7 | Celecoxib | Rat | TBI, Weight drop | No effect on the neurological score or the brain water content | (Girgis et al., 2013) |
| 8 | Meloxicam | Mice | TBI, Weight drop | No effect on the neurological score or the brain water content | (Girgis et al., 2013) |
| 9 | Lornoxicam | Rats | TBI, weight drop diffuse TBI | Reduced brain edema but did not affect BBB permeability | (Topcu et al., 2013) |
| 10 | Nimesulide | Rat | Trauma, Impact acceleration | Improved memory (Barnes maze) and motor (rotarod) performance after neurotrauma | (Cernak et al., 2001) |
| 11 | Roficoxib | Rat | TBI, FPI | No effect on hippocampal neuronal death | (Kunz et al., 2006) |
| 12 | Indomethacin | Human | TBI, Head-injured patients | Decreased ICP as well as cerebral blood flow and temperature | (Jensen et al., 1991) |
4.3. Lipoxygenases
Lipoxygenases catalyze the di-oxygenation of PUFA containing a 1, 4 diene structure. Lipoxygenases are involved in the production of octanoids, eicosanoids and docosanoids (Brash, 1999; Kuhn et al., 2015). Among several LOX enzymes, 5 LOX protein levels were reported to increase in glial cells and neutrophils after TBI (Zhang et. al, 2006). Following fluid percussion injury, plasma levels of 12-HETEand PGE-2 were found to increase during the first hour after injury with peak levels at 5 minutes (Ellis et al., 1989). Among these products 12-HETE is produced from Arachidonic acid by 12-LOX and PGE-2 by COX2. Corroborating the findings in experimental TBI, 12-HETE and 5-HETE levels in the CSF of patients with head injury were found to increase 10–17 fold vs. control (Farias et al., 2011). The source of increased 12-HETE and 5-HETE production after TBI is likely glia, neutrophils and to a lesser extent neurons (Hariri et al., 1989; Zhang et al., 2006). Among several LOX enzymes 12/15 LOX protein levels were reported to increase primarily in neurons in the peri-infarct area after transient MCAO in mice (van Leyen et al., 2006). Furthermore baicalein, a nonspecific 12/15 LOX inhibitor, ameliorated brain edema and improved neurological deficits after transient focal ischemia (Cui et al., 2010) (Jin et al., 2008; Moskowitz et al., 1984). Leukotriene synthesis indicative of 5 LOX activity was also reported during ischemia and reperfusion and was thought to play an important role in the development of cerebral edema (Ban et al., 1989; Moskowitz et al., 1984). The lipid mediators generated by LOX activity have strong effects on neutrophil recruitment and vascular permeability, thus LOX are thought be important therapeutic targets. Table 2 summarizes the studies evaluating therapeutic potential of LOX inhibition in TBI and other CNS injuries. Recent studies utilized more specific inhibitors of LOX, whereas earlier studies used less selective inhibitors with activity against both COX and LOX.
Table 2.
Lipoxygenase inhibitors in acute brain injury
| Sl. No | Compound | Species | Injury | Effect | Ref |
|---|---|---|---|---|---|
| 1 | MK-886, Boscari ® | Rat | TBI, controlled cortical impact | Reduced lesion volume and neuronal death | (Voigt et al., 2012) |
| 2 | MK-886, Boscari ® | Rat | TBI, controlled cortical impact | Reduced inflammatory response in a region specific manner | (Hartig et al., 2013) |
| 3 | MK-886 | Rat | TBI, unilateral moderate fluid percussion injury | Reduced brain edema, attenuated blood brain barrier | (Corser-Jensen et al., 2014) |
| 4 | SK&F 105809 | Rat | TBI, weight drop | High dose attenuated brain edema | (Shohami et al., 1992) |
| 5 | LOXBlock-1,3 | HT22 Cell, Rat primary neuron, Oligodendrog lial Cells | Cell, glutamate toxicity | Protection Against Oxidative Glutamate Toxicity | (van Leyen et al., 2008) |
| 6 | Baicalein | HT22 | Cell, Iodoacetic acid toxicity | Attenuated cell death | (Lapchak et al., 2007) |
| 7 | MCI-186 | Rat | Ischemia, permanent left internal carotid artery occlusion | Attenuated brain edema | (Nishi et al., 1989) |
| 8 | Baicalein | Rabbit | Ischemia, small clot embolic stroke model | Reduced behavioral deficits | (Lapchak et al., 2007) |
| 9 | Nordihydroguai aretic acid | Rat | Ischemia, transient 4- vessel occlusion | Reduced the post ischemic neuronal death , improved behavioral deficits | (Shishido et al., 2001) |
| 10 | Caffeic acid | Rat | Ischemia, transient MCAO | Attenuated brain atrophy, infarct volume, and astrocyte proliferation | (Zhou et al., 2006) |
| 11 | Baicalein | Mice, primary cortical neurons | Ischemia, transient MCAO | Reduced glutamate-induced death of primary neurons. Decreased infarct size in mice. ischemia | (van Leyen et al., 2006) |
| 12 | Baicalein | Rat | Ischemia,, permanent MCAO | Improved neurological deficit, reduced brain water content and infarct size | (Cui et al., 2010) |
| 13 | Baicalein, CDC, AA861 | organotypic cultures of rat hippocam pal slices | Oxygen glucose deprivation | Attenuation of neuronal damage | (Arai et al., 2001) |
| 14 | BW755C | Rat | SCI | Improved neurological recovery | (Faden et al., 1988) |
| 15 | Phenidone | Rat | Stroke-prone spontaneously hypertensive rats | Prolonged stroke free survival | (Munsiff et al., 1992) |
Interestingly several lipid mediators generated by LOX from arachidonic, eicosapentaenoic and docosahexaenoic acids called SPMs (Figure 3) play an important role in the resolution of inflammation primarily by attenuating neutrophil infiltration and enhancing phagocyte efferocytosis (Dennis and Norris, 2015). Among the SPMs generated as depicted in the figure 3., the arachidonic acid derivative, lipoxin A4, and its derivatives, ATLA4 (aspirin-triggered lipoxin A4) and BMA-11, were shown to exert neuroprotective effects in stroke models (Hawkins et al., 2014; Wu et al., 2010; Wu et al., 2012; Ye et al., 2010). Intracerebroventricular administration of lipoxin A4 10 minutes after weight drop model of experimental TBI was reported to decrease BBB breakdown and brain edema and attenuate brain levels of TNFα, IL1β and IL6 mRNA (Luo et al., 2013). Similarly, lipid mediators derived from ω3-fatty acids docosahexaenoic acid and eicosapentanoic acid, namely resolvins and neuroprotectins, were also shown to exert beneficial effects in cerebral ischemia reperfusion injury (Bazan, 2005; Marcheselli et al., 2003; Serhan et al., 2004). Figure 4. Summarizes all the LOX and COX dependent mediators and its role in inflammation. Taken together, the beneficial vs. detrimental effects of LOX may depend on the spatial (including subcellular localization) and temporal profiles of the lipid mediators they generate after injury.
Figure 3. Biosynthetic pathways of specialized pro-resolving lipid mediators (SPMs).
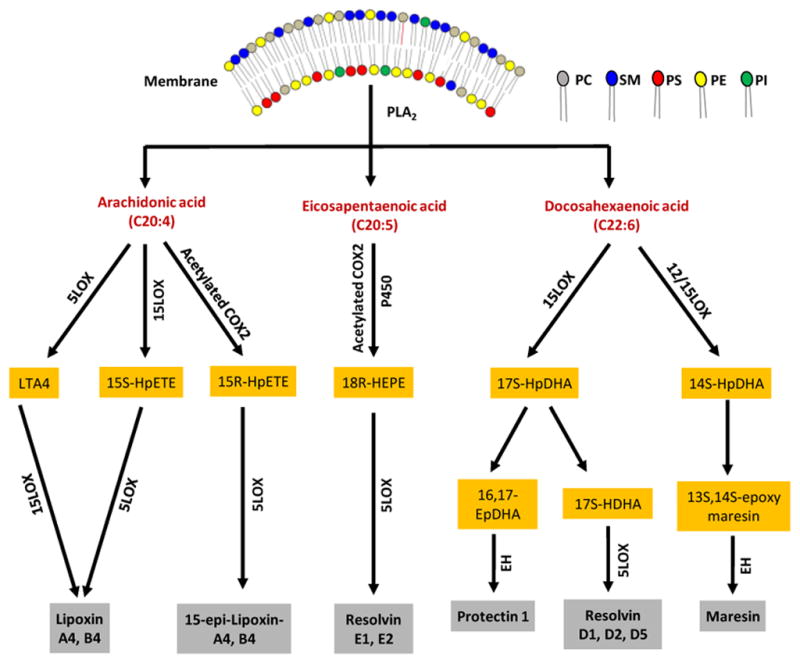
Free fatty acids produced after the hydrolysis of phospholipids such as phosphatidylcholine (PC), phosphatidylserine (PS), phosphatidylethanolamine (PE) and phosphatidylinositol (PI) are modified by either LOX or acetylated COX to produce the hydroperoxy fatty acids. Hydroperoxy arachidonic acids are directly converted into lipoxin and epi lipoxin by the LOX enzyme, and Hydroperoxy EPA is converted into Resolvin E1, and E2. In the case of hydroperoxy DHA, further conversion to epoxy products and consequently into maresins and resolvin D1, D2 occurs.
Figure 4.
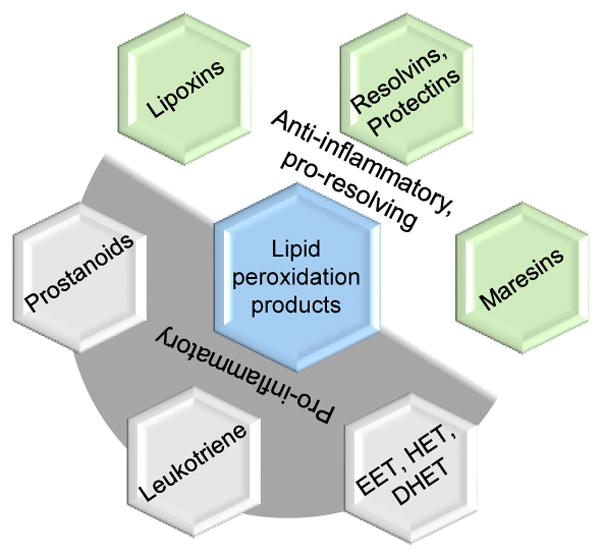
Lipid mediators and their roles in inflammation
4.4. Mitochondrial targeted lipid peroxidation inhibitors
The classic pathway of lipid mediator synthesis includes (Figures 3 and 4) hydrolysis of esterified PUFA by one of the Ca2+-dependent PLA2, followed by oxygenation of the released free PUFA by COX, LOX and P450 enzymes. Recently an alternative Ca2+-independent pathway was described whereby oxygenation of esterified PUFA within the phospholipid molecule is followed by Ca2+-independent PLA2-mediated hydrolysis to yield lipid mediators (Figure 5) (Tyurina et al., 2014). It has been shown that peroxidized cardiolipin can be hydrolyzed by mitochondrial Ca2+-independent iPLA2γ to release linoleic- and arachidonic acid-derived lipid mediators (HODE and HETE) after controlled cortical impact (CCI) and global cerebral ischemia (Ji et al., 2015; Tyurina et al., 2014). Cardiolipin oxidation is catalyzed by cytochrome c, an electron transport protein in mitochondria (Kagan et al., 2005b). Normally all six of the coordination positions in the heme iron of cytochrome c are occupied, thus preventing its interactions with small ligands such as hydrogen peroxide (Stellwagen, 1968). Upon interaction with cardiolipin, cytochrome c loses its tertiary structure resulting in loss of ability to participate in electron transfer and gain peroxidase function (Kapralov et al., 2007); (Basova et al., 2007). The resultant cytochrome c/cardiolipin complex leads to cardiolipin oxidation (Kagan et al., 2005b). It has been shown that in addition to being a source for lipid mediator synthesis, cardiolipin peroxidation is essential for the release of pro-apoptotic factors, including cytochrome c, from mitochondria into the cytosol (Ji et al., 2012a; Kagan et al., 2005b). Subsequently, the released cytochrome c participates in apoptosome formation and catalysis of phosphatidylserine oxidation ultimately leading to externalization of phosphatidylserine, a recognition signal for phagocytosis of apoptotic cells by microglia (Fadeel et al., 2010).
Figure 5. Lipid peroxidation mechanisms after TBI and possible therapeutic targets.

Lipid peroxidation after TBI follows two main pathways: 1) Calcium-dependent pathway in which PUFAs are initially hydrolyzed from phospholipids by PLA2 and subsequently oxidized by various enzymes; 2) Calcium-independent mitochondrial pathway in which mitochondria specific phospholipid cardiolipin is oxidized by cytochrome c/H2O2 and further hydrolyzed by calcium independent PLA2 to form lipid peroxides. Inhibition or reduction of marked components or enhanced activity of marked components in lipid peroxidation pathways are possible therapeutic options after TBI.
Several different mitochondria-targeted inhibitors of the cardiolipin oxidation pathway have been developed based on: i) inhibition of production of hydrogen peroxide, which is utilized as an oxidizing equivalent in the peroxidase reaction; ii) inhibition of the peroxidase activity of cytochrome c in the complex (Kagan et al., 2009b). Among the first category, mitochondria-targeted electron acceptor XJB-5-131 was shown to concentrate >500-fold in neuronal mitochondria. Mitochondrial accumulation of XJB-5-131 prevents formation of superoxide and thus hydrogen peroxide, resulting in improved neurocognitive outcome after TBI and asphyxial cardiac arrest in PND17 rats (Ji et al., 2012a; Ji et al., 2015). Among the second category, triphenylphosphonium conjugated imidazole oleic acid (TPP-IOA) and stearic acid (TPP-ISA) block peroxidase activity of cytochrome c/cardiolipin complex by specifically binding to its heme-iron. Both compounds inhibit pro-apoptotic oxidative events, suppress cytochrome c release and prevent cell death (Atkinson et al., 2011). Another target in the cardiolipin oxidation pathway is Ca2+-independent iPLA2γ. Inhibition of Ca2+-independent iPLA2γ by its selective inhibitor R-BEL (R)-(E)-6-(bromomethylene)-3-(1-naphthalenyl)-2H-tetrahydropyran-2-one prevents generation of lipid mediators from oxidized cardiolipin in the intestine (Tyurina et al., 2014). There has not been a study evaluating the neuroprotective potential of R-BEL administration after TBI. It is possible that the lipid mediators generated upon cardiolipin oxidation during apoptosis, such as 9-HODE and 13-HODE, are modulating the inflammatory process after injury. It is possible that timing of therapy could be important after injury, as constitutive genetic ablation of Ca2+-independent iPLA2γ was shown to alter hippocampal cardiolipin content and molecular species and lead to mitochondrial degeneration, autophagy and cognitive dysfunction (Mancuso et al., 2009).
4.5. General inhibitors of lipid peroxidation
Apart from specific inhibitors of enzymatic lipid oxidation, a number of therapies can attenuate lipid peroxidation and have shown neuroprotective potential in experimental studies after TBI. These therapies generally fall under the following categories; a) general, nonspecific antioxidants that act as a free radical scavengers in the biological system including endogenous antioxidants and b) antioxidant enzymes. In this section we will briefly discuss therapies in these two categories.
4.5.1. Vitamin E
Vitamin E family is composed of 8 compounds (4 tocopherols (d-α, d-β, d-γ, d-δ) and 4 tocotrienols (d-α, d-β, d-γ, d-δ)) that exhibit the biological activity of naturally occurring vitamin E, d-α-tocopherol. Tocopherols are lipid-soluble and scavenge lipid peroxyl radicals, inhibiting the chain reaction of lipid peroxidation (Figure 1). The resultant tocopherol radical is reduced back to tocopherol by ascorbic acid and lipoic acid via the vitamin E recycling process (Chan, 1993). Vitamin E levels have been shown to decrease concomitant with enhanced accumulation of MDA in jugular venous blood samples of patients with TBI (Paolin et al., 2002). Interestingly, there was no change in the peripheral venous blood levels of vitamin E and MDA in these patients. Experimental studies have shown beneficial effects of Vitamin E treatment in TBI. Dietary vitamin E supplementation attenuated elevated brain free fatty acid levels induced by sudden decompression-mediated vasogenic cerebral edema (Yoshida et al., 1985) and improved cerebral blood flow in the cortex after epidural compression injury (Busto et al., 1984). Furthermore, improvement in functional outcome has been reported with Vitamin E post-treatment after fluid percussion injury (Clifton et al., 1989) and with Vitamin E pre-treatment after repetitive mild TBI (Conte et al., 2004). Accumulating evidence in experimental stroke suggests that the neuroprotective potential of tocotrienols may be greater than tocopherols (Patel et al., 2011), but no studies in TBI have evaluated the effect of tocotrienols.
The only published RCT of Vitamin E (at a dose of 400 IU/d IM for 7 days) in severe TBI showed decreased hospital mortality and improved 6 month Glasgow Outcome Scale Score (GOS) vs. placebo in adults (Razmkon et al., 2011). Due to the small size of this trial (n = 23–27 patients/group), known antiplatelet effects of Vitamin E, and associated risk of hemorrhagic stroke with Vitamin E supplementation (Schurks et al., 2010), a multicenter RCT is necessary before Vitamin E can be recommended for routine use in severe TBI .
4.5.2. Vitamin C
Ascorbate or Vitamin C acts as a primary defense against aqueous oxygen, nitrogen and sulfhydryl radicals in the blood. Vitamin C is very effective in trapping free radicals, preventing formation of lipid hydroperoxyides and reducing membrane damage (Frei et al., 1989; Frei, 1991). There is a high concentration of ascorbate in both gray and white matter of the brain Additionally, the choroid plexus has a specific active transport system that increases the ascorbate concentrations in CSF to a level approximately 10-times higher than in the plasma (Spector and Eells, 1984). Ascorbate level in the brain is decreased after traumatic brain injury (Awasthi et al., 1997; Tyurin et al., 2000) , however the extracellular ascorbate increases after ischemia (Hillered et al., 1988) and trauma (Hillered et al., 1990).
A 2–3-fold decrease in CSF ascorbate levels was reported in clinical TBI in infants, children and adults (Bayir et al., 2002) (Cristofori et al., 2001). Similarly, plasma ascorbate levels are decreased after stroke (Yokoyama et al., 2000), intracranial hemorrhage and head trauma (Polidori et al., 2001). Razmkon and colleagues (Razmkon et al., 2011) compared treatment with low-dose (500 mg/d IV for 7 days) and high-dose Vitamin C (10 g IV on the 1st and 4th days followed by 4 g/d IV for 3 days) with placebo after severe TBI in adults. While high-dose Vitamin C decreased the diameter of perilesional hypodense region on head CT (defined as edema), low-dose Vitamin C increased the diameter of this region compared with placebo. There was no effect of either dose of Vitamin C on mortality or 6 month GOS scores. Unfortunately the effect of Vitamin C or E supplementation on plasma, CSF or interstitial fluid ascorbate and Vitamin E levels was not evaluated in this trial. The results of this placebo-controlled randomized single center trial are interesting and further studies should evaluate the safety and efficacy of high-dose Vitamin C in severe TBI patients.
4.5.3. Glutathione (GSH)
GSH is the most abundant intracellular non-protein thiol. Along with Vitamin E and C, GSH completes the major elements of endogenous antioxidant reserve. GSH is a co-substrate in the reduction of hydrogen peroxide and lipid hydroperoxides by glutathione peroxidase (GPx). When it is involved in direct scavenging of free radicals, the antioxidant function of GSH is largely achieved as thiyl radicals are formed. Decrease in GSH is implicated in TBI (Xiong et al., 1999), SCI (Azbill et al., 1997) and cerebral infarction (Frithz et al., 1982). Brain tissue GSH levels decrease at 6–24 h after controlled cortical impact (CCI) with restoration of normal levels at 72 h in adult rats (Tyurin et al., 2000). In contrast, GSH levels increase in CSF on day 1 after injury with normalization by day 7 in infants and children with severe TBI compared to controls (Bayir et al., 2002).
GSH is synthesized from glutamate, glycine, and cysteine. N-acetylcysteine (NAC) is a cysteine donor, and when administered within 1 h of CCI in rats, it was shown to restore brain and mitochondrial GSH levels and improve mitochondrial function (Xiong et al, 1999). NAC has limited permeability through the intact BBB. A novel cell-permeable amide form of NAC has better bioavailability and was shown to decrease lesion volume and improve cognitive function compared to NAC or vehicle treatment when given 30 minutes after CCI in rats (Pandya et al., 2014). Another GSH-replenishing compound, gamma-glutamylcysteinyl ethyl ester (GCEE) provides glutamate and cysteine and bypasses the rate-limiting step in the GSH synthesis. Treatment with GCEE 10 minutes after CCI in mice reduced autophagy and led to partial improvement in behavioral and histologic outcome compared to vehicle (Lai et al., 2008). NAC has been evaluated in a double-blind, placebo-controlled randomized study after blast-induced mild TBI. The results of this trial showed that supplementation of standard therapy with oral NAC decreased number of symptoms such as dizziness, hearing loss, headache, memory loss, sleep disturbances, and neurocognitive dysfunction ,assessed during the first 7 days after injury in the combat setting (Hoffer et al., 2013). To date, there has not been a clinical study evaluating the efficacy of NAC in severe TBI.
4.5.4. Other antioxidants
A number of other antioxidants have also been evaluated in experimental studies for their neuroprotective role in TBI, including curcumin(Wu et al., 2011; Zhu et al., 2014), epigallocatechin gallate (Itoh et al., 2011), 2-methylaminochromans (Hall, 1995), resveratrol (Lin et al., 2014a), melatonin (Ding et al., 2015) and alpha lipoic acid (Ozbal et al., 2015) . Although the multicenter trial with tirilizad showed no overall benefit (Marshall et al., 1998), subgroup analysis suggested that it may be effective in reducing mortality in male patients suffering from severe TBI with accompanying traumatic subarachnoid hemorrhage. Progesterone, a pleiotropic agent, was shown to be neuroprotective in part through attenuation of lipid peroxidation in experimental TBI (Djebaili et al., 2005; Roof et al., 1997). Despite initial promising results in single center studies, multicenter clinical trials of progesterone failed to show benefit relative to placebo in severe, moderate-to-severe, or moderate acute TBI (Skolnick et al., 2014; Wright et al., 2014).
4.5.5. Superoxide Dismutase (SOD)
Similar to the biological antioxidant reserve, some of the endogenous enzymes also play an important role in removing ROS and the associated lipid peroxidation and toxic effects. One such enzyme is SOD, which converts superoxide into hydrogen peroxide. Although small fluctuations in the steady state concentrations of superoxide play an important role in learning and memory, its enhanced production after injury is deleterious (Knapp and Klann, 2002; Thiels et al., 2000). Numerous sources including the mitochondrial electron transport chain and NADPH oxidase generate excess superoxide after injury (Brennan et al., 2009). Excess superoxide production combined with the decreased SOD activity (both cytosolic CuZnSOD and mitochondrial MnSOD (Bayir et al., 2007a; Wang et al., 2015) results in elevated superoxide levels following TBI.
The reaction rate for formation of peroxynitrite from superoxide and nitric oxide is comparable to the reaction rate of SOD (Gray and Carmichael, 1992; Kissner et al., 2003), thus it is important to maintain high activity of SOD. In agreement with this, mice expressing 50% of the normal complement of MnSOD display worse outcome after CCI (Gray and Carmichael, 1992; Kissner et al., 2003). Similarly, treatment with SOD or overexpression of CuZnSOD was associated with beneficial effects including decreased brain edema and improved motor function after diffuse TBI (Kontos and Wei, 1986; Mikawa et al., 1996). However the beneficial effects of increasing SOD activity may depend on the SOD variant, injury type and age. Beni et al., has shown that CuZnSOD knock out mice but not CuZnSOD transgenics showed better neurological scores than the wild type mice after contusion head injury (Beni et al., 2006). Similarly, immature CuZnSOD transgenic mice sustain worse injury than their wild type littermates after hypoxia-ischemia (Ditelberg et al., 1996). It is likely that increasing SOD activity without a concomitant increase in the activities of catalase or GPx will result in enhanced hydrogen peroxide accumulation, which can activate apoptotic death pathways (Kagan et al., 2005a) as well as NFκB-mediated inflammation (Beni et al., 2006). Administration of SOD-mimics has been evaluated in clinical TBI. A large clinical trial with polyethylene glycol (PEG)-conjugated SOD showed no improvement in survival or neurological outcome after moderate to severe TBI (Young et al., 1996), although an earlier small phase II study reported a positive trend (Muizelaar, 1993).
4.5.6. Catalase
Catalase converts hydrogen peroxide into water and molecular oxygen. It has a low affinity for hydrogen peroxide, thus it is only effective in eliminating hydrogen peroxide at high concentrations. Catalase activity was shown to increase after TBI (Goss et al., 1997). The increase in catalase activity is region specific after controlled cortical impact, with hippocampus having a higher increase vs. cortex (DeKosky et al., 2004; Miller et al., 2014). Pre-treatment with polyethylene glycol SOD/catalase restored opioid-induced vasodilation and cyclic GMP production after fluid percussion injury, suggesting that both superoxide and hydrogen peroxide might have a causal role in decreased cerebral blood flow observed after injury (Thorogood and Armstead, 1996). Hypothermia treatment decreased catalase activity in the hippocampus after controlled cortical impact (DeKosky et al., 2004; Miller et al., 2014). In patients, this is likely secondary to attenuated endogenous antioxidant consumption and lipid peroxidation with hypothermia (Bayir et al., 2009).
4.5.7. Glutathione peroxidase (GPx)
GPx are selenoenzymes involved in the conversion of peroxides into hydroxides. There are 8 different classes of GPx enzymes that can metabolize hydrogen peroxide or lipid hydroperoxides. Among these, Gpx4 is the only GPx that accepts phospholipid hyroperoxides in membranes as substrate using GSH as hydrogen donor (Roveri et al., 1994). The critical importance of this enzyme is evident from the studies demonstrating embryonic lethality of GPx4 deficient mice (Yant et al., 2003). Gpx4 ablation in adult mice reportedly results in delayed death accompanied by neuronal loss in hippocampus and reactive gliosis (A Khan and JT Gowder, 2014). GPx4 plays an important role in apoptotic and ferroptotic cell death pathways (Friedmann Angeli et al., 2014; Yang et al., 2014). Conditions of cysteine and glutathione depletion favor ferroptosis and lead to oxidation of PUFAs in membrane phospholipids (Friedmann Angeli et al., 2014; Yang et al., 2014). GPx4 is expressed in all neuronal layers of the immature brain (Savaskan et al., 2007). In the adult brain, GPx4 expression becomes more restricted to neurons of the cerebral cortex, hippocampus and cerebellum, whereas glial cells are devoid of GPx4 (Savaskan et al., 2007). Following bilateral entorhinal cortex lesion, GPx4 expression is upregulated in reactive astrocytes of lesioned areas but not in neurons. The GPx mimic, Ebselen [2-phenyl-1,2-benzisoselenazol-3(2H)-one], is reported to reduce NO levels and modulate the TL4-mediated p38-MAPK pathway after weight drop injury in rats (Wei et al., 2014).
5. Concluding remarks and Future Directions
Lipids are indispensable for normal structure of organelles and cells. Furthermore, lipid mediators derived from oxidized phospholipids and free fatty acids play important signaling functions to maintain homeostasis (Balasubramanian et al., 2015; Chu et al., 2013; Fadeel et al., 2010). Increased ROS generation, enhanced activity of lipid peroxidation enzymes, and decreases in reductants and activity of antioxidant enzymes result in uncontrolled oxidation of phospholipids that contributes to secondary injury after TBI.
Lipid peroxidation inhibitors such as free radical scavengers have been used numerous times in TBI, but clinical trials of these agents in TBI and other free radical diseases have largely failed. The reasons for to the negative results are multiple and include: 1) limited CNS penetration of the drugs tested; 2) complex actions of mediators formed from peroxidized lipids depending on the time and the extracellular milieu after injury; and 3) inadequate understanding and identification of the sources and mechanisms of redox imbalance after TBI. Unless the true sources and mechanisms of TBI redox imbalance are identified, the use of sacrificial antioxidants and free radical scavengers will fail.
Recent improvements in LC-MS based lipidomics and oxidative lipidomics approaches offer remarkable opportunities for identification, characterization and quantification of lipid oxidation products and their source. This information is critical for targeted therapy development. Future experimental studies evaluating therapies targeting lipid peroxidation should consider assessment of therapeutic target specificity and CNS penetration of the compounds being tested in diverse experimental models of TBI. In addition, the effect of the targeted therapies on lipid peroxidation should be evaluated using contemporary methods in experimental and clinical TBI. Combined these approaches might improve the likelihood of successful translation of promising therapies targeting lipid peroxidation to clinical TBI.
Highlights.
Description of attempts to use non-specific antioxidants as neuroprotectors
Enzymatic lipid peroxidation in specific death pathways - particularly apoptosis
Generation of lipid mediators from peroxidized phospholipids by phospholipases
Acknowledgments
Supported, in part, by grants from the NIH (NS061817, U19AIO68021, NS076511, NS084604).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Khan AA, Gowder JTS. Glutathione peroxidase: A potential marker for the most common diseases and disorders. Recent Patents on Biomarkers. 2014;4:43–52. [Google Scholar]
- Adibhatla RM. Citicoline in stroke and TBI clinical trials. Nat Rev Neurol. 2013;9:173. doi: 10.1038/nrneurol.2012.166-c1. [DOI] [PubMed] [Google Scholar]
- Al Nimer F, Strom M, Lindblom R, Aeinehband S, Bellander BM, Nyengaard JR, Lidman O, Piehl F. Naturally occurring variation in the Glutathione-S-Transferase 4 gene determines neurodegeneration after traumatic brain injury. Antioxid Redox Signal. 2013;18:784–94. doi: 10.1089/ars.2011.4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrovandi M, Hammond VJ, Podmore H, Hornshaw M, Clark SR, Marnett LJ, Slatter DA, Murphy RC, Collins PW, O'Donnell VB. Human platelets generate phospholipid-esterified prostaglandins via cyclooxygenase-1 that are inhibited by low dose aspirin supplementation. J Lipid Res. 2013;54:3085–97. doi: 10.1194/jlr.M041533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci U S A. 1993;90:7915–22. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med. 2008;45:443–52. doi: 10.1016/j.freeradbiomed.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Nishiyama N, Matsuki N, Ikegaya Y. Neuroprotective effects of lipoxygenase inhibitors against ischemic injury in rat hippocampal slice cultures. Brain Res. 2001;904:167–72. doi: 10.1016/s0006-8993(01)02491-x. [DOI] [PubMed] [Google Scholar]
- Atkinson J, Kapralov AA, Yanamala N, Tyurina YY, Amoscato AA, Pearce L, Peterson J, Huang Z, Jiang J, Samhan-Arias AK. A mitochondria-targeted inhibitor of cytochrome c peroxidase mitigates radiation-induced death. Nature communications. 2011;2:497. doi: 10.1038/ncomms1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi D, Church DF, Torbati D, Carey ME, Pryor WA. Oxidative stress following traumatic brain injury in rats. Surg Neurol. 1997;47:575–81. doi: 10.1016/s0090-3019(96)00461-2. discussion 581–2. [DOI] [PubMed] [Google Scholar]
- Azbill RD, Mu X, Bruce-Keller AJ, Mattson MP, Springer JE. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. 1997;765:283–90. doi: 10.1016/s0006-8993(97)00573-8. [DOI] [PubMed] [Google Scholar]
- Balasubramanian K, Maeda A, Lee JS, Mohammadyani D, Dar HH, Jiang JF, St Croix CM, Watkins S, Tyurin VA, Tyurina YY, Kloditz K, Polimova A, Kapralova VI, Xiong Z, Ray P, Klein-Seetharaman J, Mallampalli RK, Bayir H, Fadeel B, Kagan VE. Dichotomous roles for externalized cardiolipin in extracellular signaling: Promotion of phagocytosis and attenuation of innate immunity. Sci Signal. 2015;8:ra95. doi: 10.1126/scisignal.aaa6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basova LV, Kurnikov IV, Wang L, Ritov VB, Belikova NA, Vlasova, Pacheco AA, Winnica DE, Peterson J, Bayir H, Waldeck DH, Kagan VE. Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry. 2007;46:3423–34. doi: 10.1021/bi061854k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, Graham SH, Janesko K, Clark RS, Kochanek PM. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr Res. 2002;51:571–8. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- Bayir H, Marion DW, Puccio AM, Wisniewski SR, Janesko KL, Clark RS, Kochanek PM. Marked gender effect on lipid peroxidation after severe traumatic brain injury in adult patients. J Neurotrauma. 2004;21:1–8. doi: 10.1089/089771504772695896. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kochanek PM, Kagan VE. Oxidative stress in immature brain after traumatic brain injury. Dev Neurosci. 2006;28:420–31. doi: 10.1159/000094168. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Clark RS, Janesko-Feldman K, Rafikov R, Huang Z, Zhang X, Vagni V, Billiar TR, Kochanek PM. Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J Neurochem. 2007a;101:168–81. doi: 10.1111/j.1471-4159.2006.04353.x. [DOI] [PubMed] [Google Scholar]
- Bayir H, Tyurin VA, Tyurina YY, Viner R, Ritov V, Amoscato AA, Zhao Q, Zhang XJ, Janesko-Feldman KL, Alexander H, Basova LV, Clark RS, Kochanek PM, Kagan VE. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann Neurol. 2007b;62:154–69. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- Bayir H, Adelson PD, Wisniewski SR, Shore P, Lai Y, Brown D, Janesko-Feldman KL, Kagan VE, Kochanek PM. Therapeutic hypothermia preserves antioxidant defenses after severe traumatic brain injury in infants and children. Crit Care Med. 2009;37:689–95. doi: 10.1097/CCM.0b013e318194abf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG. Free arachidonic acid and other lipids in the nervous system during early ischemia and after electroshock. Adv Exp Med Biol. 1976;72:317–35. doi: 10.1007/978-1-4684-0955-0_26. [DOI] [PubMed] [Google Scholar]
- Bazan NG, Zorumski CF, Clark GD. The activation of phospholipase A2 and release of arachidonic acid and other lipid mediators at the synapse: the role of platelet-activating factor. J Lipid Mediat. 1993;6:421–7. [PubMed] [Google Scholar]
- Bazan NG, Rodriguez de Turco EB, Allan G. Mediators of injury in neurotrauma: intracellular signal transduction and gene expression. J Neurotrauma. 1995;12:791–814. doi: 10.1089/neu.1995.12.791. [DOI] [PubMed] [Google Scholar]
- Bazan NG. Neuroprotectin D1 (NPD1): a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 2005;15:159–66. doi: 10.1111/j.1750-3639.2005.tb00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG, Marcheselli VL, Cole-Edwards K. Brain response to injury and neurodegeneration: endogenous neuroprotective signaling. Ann N Y Acad Sci. 2005;1053:137–47. doi: 10.1196/annals.1344.011. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–4. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Chen J, Ischiropoulos H, Crow JP. Oxidative chemistry of peroxynitrite. Methods Enzymol. 1994;233:229–40. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- Beni SM, Tsenter J, Alexandrovich AG, Galron-Krool N, Barzilai A, Kohen R, Grigoriadis N, Simeonidou C, Shohami E. CuZn-SOD deficiency, rather than overexpression, is associated with enhanced recovery and attenuated activation of NF-κB after brain trauma in mice. Journal of Cerebral Blood Flow & Metabolism. 2006;26:478–490. doi: 10.1038/sj.jcbfm.9600209. [DOI] [PubMed] [Google Scholar]
- Berger JS, Roncaglioni MC, Avanzini F, Pangrazzi I, Tognoni G, Brown DL. Aspirin for the primary prevention of cardiovascular events in women and men: a sex-specific meta-analysis of randomized controlled trials. JAMA. 2006;295:306–13. doi: 10.1001/jama.295.3.306. [DOI] [PubMed] [Google Scholar]
- Bergstroem S, Danielsson H, Klenberg D, Samuelsson B. The Enzymatic Conversion of Essential Fatty Acids into Prostaglandins. J Biol Chem. 1964;239:PC4006–8. [PubMed] [Google Scholar]
- Bergstrom S. Autoxidation of linoleic acid. Nature. 1945;156:717. [PubMed] [Google Scholar]
- Bhatt DL, Fox KA, Hacke W, Berger PB, Black HR, Boden WE, Cacoub P, Cohen EA, Creager MA, Easton JD, Flather MD, Haffner SM, Hamm CW, Hankey GJ, Johnston SC, Mak KH, Mas JL, Montalescot G, Pearson TA, Steg PG, Steinhubl SR, Weber MA, Brennan DM, Fabry-Ribaudo L, Booth J, Topol EJ, Investigators C. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med. 2006;354:1706–17. doi: 10.1056/NEJMoa060989. [DOI] [PubMed] [Google Scholar]
- Bianco ID, Fidelio GD, Maggio B. Modulation of phospholipase A2 activity by neutral and anionic glycosphingolipids in monolayers. Biochem J. 1989;258:95–9. doi: 10.1042/bj2580095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury J. Docosahexaenoic acid (DHA): an ancient nutrient for the modern human brain. Nutrients. 2011;3:529–54. doi: 10.3390/nu3050529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash AR, Boeglin WE, Capdevila JH, Yeola S, Blair IA. 7-HETE, 10-HETE, and 13-HETE are major products of NADPH-dependent arachidonic acid metabolism in rat liver microsomes: analysis of their stereochemistry, and the stereochemistry of their acid-catalyzed rearrangement. Arch Biochem Biophys. 1995;321:485–92. doi: 10.1006/abbi.1995.1421. [DOI] [PubMed] [Google Scholar]
- Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem. 1999;274:23679–82. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–63. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, Konstam MA, Baron JA Adenomatous Polyp Prevention on Vioxx Trial I. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- Busto R, Yoshida S, Ginsberg MD, Alonso O, Smith DW, Goldberg WJ. Regional blood flow in compression-induced brain edema in rats: effect of dietary vitamin E. Ann Neurol. 1984;15:441–8. doi: 10.1002/ana.410150507. [DOI] [PubMed] [Google Scholar]
- Calatayud Maldonado V, Calatayud Perez JB, Aso Escario J. Effects of CDP-choline on the recovery of patients with head injury. J Neurol Sci. 1991;103(Suppl):S15–8. doi: 10.1016/0022-510x(91)90003-p. [DOI] [PubMed] [Google Scholar]
- Cernak I, O'Connor C, Vink R. Activation of cyclo-oxygenase-2 contributes to motor and cognitive dysfunction following diffuse traumatic brain injury in rats. Clin Exp Pharmacol Physiol. 2001;28:922–5. doi: 10.1046/j.1440-1681.2001.03549.x. [DOI] [PubMed] [Google Scholar]
- Cernak I, O'Connor C, Vink R. Inhibition of cyclooxygenase 2 by nimesulide improves cognitive outcome more than motor outcome following diffuse traumatic brain injury in rats. Exp Brain Res. 2002;147:193–9. doi: 10.1007/s00221-002-1245-z. [DOI] [PubMed] [Google Scholar]
- Chan AC. Partners in defense, vitamin E and vitamin C. Can J Physiol Pharmacol. 1993;71:725–31. doi: 10.1139/y93-109. [DOI] [PubMed] [Google Scholar]
- Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–9. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS, Simmons DL. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A. 2002;99:13926–31. doi: 10.1073/pnas.162468699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Qiang Wang KZ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SR, Guy CJ, Scurr MJ, Taylor PR, Kift-Morgan AP, Hammond VJ, Thomas CP, Coles B, Roberts GW, Eberl M, Jones SA, Topley N, Kotecha S, O'Donnell VB. Esterified eicosanoids are acutely generated by 5-lipoxygenase in primary human neutrophils and in human and murine infection. Blood. 2011;117:2033–43. doi: 10.1182/blood-2010-04-278887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark WM, Warach SJ, Pettigrew LC, Gammans RE, Sabounjian LA. A randomized dose-response trial of citicoline in acute ischemic stroke patients. Citicoline Stroke Study Group. Neurology. 1997;49:671–8. doi: 10.1212/wnl.49.3.671. [DOI] [PubMed] [Google Scholar]
- Clark WM, Williams BJ, Selzer KA, Zweifler RM, Sabounjian LA, Gammans RE. A randomized efficacy trial of citicoline in patients with acute ischemic stroke. Stroke. 1999;30:2592–7. doi: 10.1161/01.str.30.12.2592. [DOI] [PubMed] [Google Scholar]
- Clark WM, Wechsler LR, Sabounjian LA, Schwiderski UE Citicoline Stroke Study G. A phase III randomized efficacy trial of 2000 mg citicoline in acute ischemic stroke patients. Neurology. 2001;57:1595–602. doi: 10.1212/wnl.57.9.1595. [DOI] [PubMed] [Google Scholar]
- Clifton GL, Lyeth BG, Jenkins LW, Taft WC, DeLorenzo RJ, Hayes RL. Effect of D, alpha-tocopheryl succinate and polyethylene glycol on performance tests after fluid percussion brain injury. J Neurotrauma. 1989;6:71–81. doi: 10.1089/neu.1989.6.71. [DOI] [PubMed] [Google Scholar]
- Conte V, Uryu K, Fujimoto S, Yao Y, Rokach J, Longhi L, Trojanowski JQ, Lee VM, McIntosh TK, Pratico D. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J Neurochem. 2004;90:758–64. doi: 10.1111/j.1471-4159.2004.02560.x. [DOI] [PubMed] [Google Scholar]
- Corser-Jensen CE, Goodell DJ, Freund RK, Serbedzija P, Murphy RC, Farias SE, Dell'Acqua ML, Frey LC, Serkova N, Heidenreich KA. Blocking leukotriene synthesis attenuates the pathophysiology of traumatic brain injury and associated cognitive deficits. Exp Neurol. 2014;256:7–16. doi: 10.1016/j.expneurol.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofori L, Tavazzi B, Gambin R, Vagnozzi R, Vivenza C, Amorini AM, Di Pierro D, Fazzina G, Lazzarino G. Early onset of lipid peroxidation after human traumatic brain injury: a fatal limitation for the free radical scavenger pharmacological therapy? J Investig Med. 2001;49:450–8. doi: 10.2310/6650.2001.33790. [DOI] [PubMed] [Google Scholar]
- Cristofori L, Tavazzi B, Gambin R, Vagnozzi R, Signoretti S, Amorini AM, Fazzina G, Lazzarino G. Biochemical analysis of the cerebrospinal fluid: evidence for catastrophic energy failure and oxidative damage preceding brain death in severe head injury: a case report. Clin Biochem. 2005;38:97–100. doi: 10.1016/j.clinbiochem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Crofford LJ. COX-1 and COX-2 tissue expression: implications and predictions. J Rheumatol Suppl. 1997;49:15–9. [PubMed] [Google Scholar]
- Cui L, Zhang X, Yang R, Liu L, Wang L, Li M, Du W. Baicalein is neuroprotective in rat MCAO model: role of 12/15-lipoxygenase, mitogen-activated protein kinase and cytosolic phospholipase A2. Pharmacol Biochem Behav. 2010;96:469–75. doi: 10.1016/j.pbb.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Dash PK, Mach SA, Moore AN. Regional expression and role of cyclooxygenase-2 following experimental traumatic brain injury. J Neurotrauma. 2000;17:69–81. doi: 10.1089/neu.2000.17.69. [DOI] [PubMed] [Google Scholar]
- Davalos A, Castillo J, Alvarez-Sabin J, Secades JJ, Mercadal J, Lopez S, Cobo E, Warach S, Sherman D, Clark WM, Lozano R. Oral citicoline in acute ischemic stroke: an individual patient data pooling analysis of clinical trials. Stroke. 2002;33:2850–7. doi: 10.1161/01.str.0000038691.03334.71. [DOI] [PubMed] [Google Scholar]
- Davidenkova EF, Grigor'eva VV, Shafran MG, Verlinskaia DK, Prozorova MV. Lipid peroxidation and the myeloperoxidase activity of neutrophilic leukocytes in shereshevskyi-Turner syndrome. Biull Eksp Biol Med. 1982;94:72–3. [PubMed] [Google Scholar]
- DeKosky ST, Taffe KM, Abrahamson EE, Dixon CE, Kochanek PM, Ikonomovic MD. Time course analysis of hippocampal nerve growth factor and antioxidant enzyme activity following lateral controlled cortical impact brain injury in the rat. J Neurotrauma. 2004;21:491–500. doi: 10.1089/089771504774129838. [DOI] [PubMed] [Google Scholar]
- Dennis EA. Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem. 1994;269:13057–60. [PubMed] [Google Scholar]
- Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15:511–23. doi: 10.1038/nri3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt DL. Prostaglandin endoperoxide synthase: regulation of enzyme expression. Biochim Biophys Acta. 1991;1083:121–34. doi: 10.1016/0005-2760(91)90032-d. [DOI] [PubMed] [Google Scholar]
- Ding K, Xu J, Wang H, Zhang L, Wu Y, Li T. Melatonin protects the brain from apoptosis by enhancement of autophagy after traumatic brain injury in mice. Neurochem Int. 2015;91:46–54. doi: 10.1016/j.neuint.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Ditelberg JS, Sheldon RA, Epstein CJ, Ferriero DM. Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatric research. 1996;39:204–208. doi: 10.1203/00006450-199602000-00003. [DOI] [PubMed] [Google Scholar]
- Djebaili M, Guo Q, Pettus EH, Hoffman SW, Stein DG. The neurosteroids progesterone and allopregnanolone reduce cell death, gliosis, and functional deficits after traumatic brain injury in rats. J Neurotrauma. 2005;22:106–18. doi: 10.1089/neu.2005.22.106. [DOI] [PubMed] [Google Scholar]
- Dorhout Mees SM, Rinkel GJ, Hop JW, Algra A, van Gijn J. Antiplatelet therapy in aneurysmal subarachnoid hemorrhage: a systematic review. Stroke. 2003;34:2285–9. doi: 10.1161/01.STR.0000083621.44269.3E. [DOI] [PubMed] [Google Scholar]
- Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–73. [PubMed] [Google Scholar]
- Ellis EF, Police RJ, Rice LY, Grabeel M, Holt S. Increased plasma PGE2, 6-keto-PGF1 alpha, and 12-HETE levels following experimental concussive brain injury. J Neurotrauma. 1989;6:31–7. doi: 10.1089/neu.1989.6.31. [DOI] [PubMed] [Google Scholar]
- Ernster L, Nordenbrand K, Orrenius S, Das ML. Microsomal lipid peroxidation. Hoppe Seylers Z Physiol Chem. 1968;349:1604–5. [PubMed] [Google Scholar]
- Estevez AY, Phillis JW. The phospholipase A2 inhibitor, quinacrine, reduces infarct size in rats after transient middle cerebral artery occlusion. Brain Res. 1997;752:203–8. doi: 10.1016/s0006-8993(96)01450-3. [DOI] [PubMed] [Google Scholar]
- Evans PH. Free radicals in brain metabolism and pathology. Br Med Bull. 1993;49:577–87. doi: 10.1093/oxfordjournals.bmb.a072632. [DOI] [PubMed] [Google Scholar]
- Fabbri A, Servadei F, Marchesini G, Bronzoni C, Montesi D, Arietta L Societa Italiana di Medicina d'Emergenza Urgenza Study G. Antiplatelet therapy and the outcome of subjects with intracranial injury: the Italian SIMEU study. Crit Care. 2013;17:R53. doi: 10.1186/cc12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadeel B, Xue D, Kagan V. Programmed cell clearance: molecular regulation of the elimination of apoptotic cell corpses and its role in the resolution of inflammation. Biochem Biophys Res Commun. 2010;396:7–10. doi: 10.1016/j.bbrc.2010.02.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden AI, Lemke M, Demediuk P. Effects of BW755C, a mixed cyclo-oxygenase-lipoxygenase inhibitor, following traumatic spinal cord injury in rats. Brain Res. 1988;463:63–8. doi: 10.1016/0006-8993(88)90527-6. [DOI] [PubMed] [Google Scholar]
- Farias SE, Heidenreich KA, Wohlauer MV, Murphy RC, Moore EE. Lipid mediators in cerebral spinal fluid of traumatic brain injured patients. J Trauma. 2011;71:1211–8. doi: 10.1097/TA.0b013e3182092c62. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA. Excitotoxicity and neurological disorders: involvement of membrane phospholipids. Int Rev Neurobiol. 1994;36:267–323. doi: 10.1016/s0074-7742(08)60306-2. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Ong WY, Horrocks LA. Plasmalogens, docosahexaenoic acid and neurological disorders. Adv Exp Med Biol. 2003;544:335–54. doi: 10.1007/978-1-4419-9072-3_45. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA. Brain phospholipases A2: a perspective on the history. Prostaglandins Leukot Essent Fatty Acids. 2004;71:161–9. doi: 10.1016/j.plefa.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA. Phospholipase A2-generated lipid mediators in the brain: the good, the bad, and the ugly. Neuroscientist. 2006;12:245–60. doi: 10.1177/1073858405285923. [DOI] [PubMed] [Google Scholar]
- Feller SE, Gawrisch K, MacKerell AD., Jr Polyunsaturated fatty acids in lipid bilayers: intrinsic and environmental contributions to their unique physical properties. J Am Chem Soc. 2002;124:318–26. doi: 10.1021/ja0118340. [DOI] [PubMed] [Google Scholar]
- Frei B, England L, Ames BN. Ascorbate is an outstanding antioxidant in human blood plasma. Proc Natl Acad Sci U S A. 1989;86:6377–81. doi: 10.1073/pnas.86.16.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei B. Ascorbic acid protects lipids in human plasma and low-density lipoprotein against oxidative damage. Am J Clin Nutr. 1991;54:1113S–1118S. doi: 10.1093/ajcn/54.6.1113s. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide dismutases. An adaptation to a paramagnetic gas. J Biol Chem. 1989;264:7761–4. [PubMed] [Google Scholar]
- Fridovich SE, Porter NA. Oxidation of arachidonic acid in micelles by superoxide and hydrogen peroxide. J Biol Chem. 1981;256:260–5. [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Radmark O, Kobayashi S, Seibt T, Beck H, Neff F, Esposito I, Wanke R, Forster H, Yefremova O, Heinrichmeyer M, Bornkamm GW, Geissler EK, Thomas SB, Stockwell BR, O'Donnell VB, Kagan VE, Schick JA, Conrad M. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frithz G, Ronquist G, Hugosson R. Perspectives of adenylate kinase activity and glutathione concentration in cerebrospinal fluid of patients with ischemic and neoplastic brain lesions. Eur Neurol. 1982;21:41–7. doi: 10.1159/000115452. [DOI] [PubMed] [Google Scholar]
- Fujishima H, Sanchez Mejia RO, Bingham CO, 3rd, Lam BK, Sapirstein A, Bonventre JV, Austen KF, Arm JP. Cytosolic phospholipase A2 is essential for both the immediate and the delayed phases of eicosanoid generation in mouse bone marrow-derived mast cells. Proc Natl Acad Sci U S A. 1999;96:4803–7. doi: 10.1073/pnas.96.9.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner HW. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic Biol Med. 1989;7:65–86. doi: 10.1016/0891-5849(89)90102-0. [DOI] [PubMed] [Google Scholar]
- Geisler FH, Dorsey FC, Coleman WP. Recovery of motor function after spinal-cord injury--a randomized, placebo-controlled trial with GM-1 ganglioside. N Engl J Med. 1991;324:1829–38. doi: 10.1056/NEJM199106273242601. [DOI] [PubMed] [Google Scholar]
- Geisler FH, Coleman WP, Grieco G, Poonian D Sygen Study G. The Sygen multicenter acute spinal cord injury study. Spine (Phila Pa 1976) 2001;26:S87–98. doi: 10.1097/00007632-200112151-00015. [DOI] [PubMed] [Google Scholar]
- Girgis H, Palmier B, Croci N, Soustrat M, Plotkine M, Marchand-Leroux C. Effects of selective and non-selective cyclooxygenase inhibition against neurological deficit and brain oedema following closed head injury in mice. Brain Res. 2013;1491:78–87. doi: 10.1016/j.brainres.2012.10.049. [DOI] [PubMed] [Google Scholar]
- Girotti AW. Mechanisms of lipid peroxidation. J Free Radic Biol Med. 1985;1:87–95. doi: 10.1016/0748-5514(85)90011-x. [DOI] [PubMed] [Google Scholar]
- Goldman R, Claycamp GH, Sweetland MA, Sedlov AV, Tyurin VA, Kisin ER, Tyurina YY, Ritov VB, Wenger SL, Grant SG, Kagan VE. Myeloperoxidase-catalyzed redox-cycling of phenol promotes lipid peroxidation and thiol oxidation in HL-60 cells. Free Radic Biol Med. 1999;27:1050–63. doi: 10.1016/s0891-5849(99)00140-9. [DOI] [PubMed] [Google Scholar]
- Goss JR, Taffe KM, Kochanek PM, DeKosky ST. The antioxidant enzymes glutathione peroxidase and catalase increase following traumatic brain injury in the rat. Exp Neurol. 1997;146:291–4. doi: 10.1006/exnr.1997.6515. [DOI] [PubMed] [Google Scholar]
- Gray B, Carmichael AJ. Kinetics of superoxide scavenging by dismutase enzymes and manganese mimics determined by electron spin resonance. Biochem J. 1992;281(Pt 3):795–802. doi: 10.1042/bj2810795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieb P. Neuroprotective properties of citicoline: facts, doubts and unresolved issues. CNS Drugs. 2014;28:185–93. doi: 10.1007/s40263-014-0144-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisham MB. Myoglobin-catalyzed hydrogen peroxide dependent arachidonic acid peroxidation. J Free Radic Biol Med. 1985;1:227–32. doi: 10.1016/0748-5514(85)90122-9. [DOI] [PubMed] [Google Scholar]
- Gueraud F, Atalay M, Bresgen N, Cipak A, Eckl PM, Huc L, Jouanin I, Siems W, Uchida K. Chemistry and biochemistry of lipid peroxidation products. Free Radic Res. 2010;44:1098–124. doi: 10.3109/10715762.2010.498477. [DOI] [PubMed] [Google Scholar]
- Hakan T, Toklu HZ, Biber N, Ozevren H, Solakoglu S, Demirturk P, Aker FV. Effect of COX-2 inhibitor meloxicam against traumatic brain injury-induced biochemical, histopathological changes and blood-brain barrier permeability. Neurol Res. 2010;32:629–35. doi: 10.1179/016164109X12464612122731. [DOI] [PubMed] [Google Scholar]
- Hall E. The mouse head injury model: utility in the discovery of acute cerebroprotective agents. CRC Press; Boca Raton, FL: 1995. pp. 213–223. [Google Scholar]
- Hall ED. Free radicals and CNS injury. Crit Care Clin. 1989;5:793–805. [PubMed] [Google Scholar]
- Hall ED. The role of oxygen radicals in traumatic injury: clinical implications. J Emerg Med. 1993;11(Suppl 1):31–6. [PubMed] [Google Scholar]
- Hall ED, Andrus PK, Smith SL, Oostveen JA, Scherch HM, Lutzke BS, Raub TJ, Sawada GA, Palmer JR, Banitt LS, Tustin JS, Belonga KL, Ayer DE, Bundy GL. Neuroprotective efficacy of microvascularly-localized versus brain-penetrating antioxidants. Acta Neurochir Suppl. 1996;66:107–13. doi: 10.1007/978-3-7091-9465-2_19. [DOI] [PubMed] [Google Scholar]
- Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B. Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med. 1991;91:14S–22S. doi: 10.1016/0002-9343(91)90279-7. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–23. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr. 1993;57:715S–724S. doi: 10.1093/ajcn/57.5.715S. discussion 724S–725S. [DOI] [PubMed] [Google Scholar]
- Hammarstrom S. Leukotrienes. Annu Rev Biochem. 1983;52:355–77. doi: 10.1146/annurev.bi.52.070183.002035. [DOI] [PubMed] [Google Scholar]
- Hammond VJ, O'Donnell VB. Esterified eicosanoids: generation, characterization and function. Biochim Biophys Acta. 2012;1818:2403–12. doi: 10.1016/j.bbamem.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri RJ, Ghajar JB, Pomerantz KB, Hajjar DP, Giannuzzi RF, Tomich E, Andrews DW, Patterson RH., Jr Human glial cell production of lipoxygenase-generated eicosanoids: a potential role in the pathophysiology of vascular changes following traumatic brain injury. J Trauma. 1989;29:1203–10. doi: 10.1097/00005373-198909000-00003. [DOI] [PubMed] [Google Scholar]
- Harrison JL, Rowe RK, O'Hara BF, Adelson PD, Lifshitz J. Acute over-the-counter pharmacological intervention does not adversely affect behavioral outcome following diffuse traumatic brain injury in the mouse. Exp Brain Res. 2014;232:2709–19. doi: 10.1007/s00221-014-3948-3. [DOI] [PubMed] [Google Scholar]
- Harrison JL, Rowe RK, Ellis TW, Yee NS, O’Hara BF, Adelson PD, Lifshitz J. Resolvins AT-D1 and E1 differentially impact functional outcome, post-traumatic sleep, and microglial activation following diffuse brain injury in the mouse. Brain, behavior, and immunity. 2015 doi: 10.1016/j.bbi.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartig W, Michalski D, Seeger G, Voigt C, Donat CK, Dulin J, Kacza J, Meixensberger J, Arendt T, Schuhmann MU. Impact of 5-lipoxygenase inhibitors on the spatiotemporal distribution of inflammatory cells and neuronal COX-2 expression following experimental traumatic brain injury in rats. Brain Res. 2013;1498:69–84. doi: 10.1016/j.brainres.2012.12.022. [DOI] [PubMed] [Google Scholar]
- Hawkins KE, DeMars KM, Singh J, Yang C, Cho HS, Frankowski JC, Dore S, Candelario-Jalil E. Neurovascular protection by post-ischemic intravenous injections of the lipoxin A4 receptor agonist, BML-111, in a rat model of ischemic stroke. J Neurochem. 2014;129:130–42. doi: 10.1111/jnc.12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey RW, Adelson PD, Johnnides MJ, Davis DS, Yu Z, Rose ME, Chang YF, Graham SH. Cyclooxygenase-2 activity following traumatic brain injury in the developing rat. Pediatr Res. 2007;62:271–6. doi: 10.1203/PDR.0b013e3180db2902. [DOI] [PubMed] [Google Scholar]
- Hillered L, Persson L, Bolander HG, Hallstrom A, Ungerstedt U. Increased extracellular levels of ascorbate in the striatum after middle cerebral artery occlusion in the rat monitored by intracerebral microdialysis. Neurosci Lett. 1988;95:286–90. doi: 10.1016/0304-3940(88)90672-6. [DOI] [PubMed] [Google Scholar]
- Hillered L, Nilsson P, Ungerstedt U, Ponten U. Trauma-induced increase of extracellular ascorbate in rat cerebral cortex. Neurosci Lett. 1990;113:328–32. doi: 10.1016/0304-3940(90)90606-a. [DOI] [PubMed] [Google Scholar]
- Hoffer ME, Balaban C, Slade MD, Tsao JW, Hoffer B. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One. 2013;8:e54163. doi: 10.1371/journal.pone.0054163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrocks LA, Demediuk P, Saunders RD, Dugan L, Clendenon NR, Means ED, Anderson DK. The degradation of phospholipids, formation of metabolites of arachidonic acid, and demyelination following experimental spinal cord injury. Cent Nerv Syst Trauma. 1985;2:115–20. doi: 10.1089/cns.1985.2.115. [DOI] [PubMed] [Google Scholar]
- Huang W, Bhavsar A, Ward RE, Hall JC, Priestley JV, Michael-Titus AT. Arachidonyl trifluoromethyl ketone is neuroprotective after spinal cord injury. J Neurotrauma. 2009;26:1429–34. doi: 10.1089/neu.2008.0835. [DOI] [PubMed] [Google Scholar]
- Hurtado O, Cardenas A, Pradillo JM, Morales JR, Ortego F, Sobrino T, Castillo J, Moro MA, Lizasoain I. A chronic treatment with CDP-choline improves functional recovery and increases neuronal plasticity after experimental stroke. Neurobiol Dis. 2007;26:105–11. doi: 10.1016/j.nbd.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Itoh T, Imano M, Nishida S, Tsubaki M, Hashimoto S, Ito A, Satou T. (-)-Epigallocatechin-3-gallate protects against neuronal cell death and improves cerebral function after traumatic brain injury in rats. Neuromolecular Med. 2011;13:300–9. doi: 10.1007/s12017-011-8162-x. [DOI] [PubMed] [Google Scholar]
- Jacotte-Simancas A, Costa-Miserachs D, Coll-Andreu M, Torras-Garcia M, Borlongan CV, Portell-Cortes I. Effects of voluntary physical exercise, citicoline, and combined treatment on object recognition memory, neurogenesis, and neuroprotection after traumatic brain injury in rats. J Neurotrauma. 2015;32:739–51. doi: 10.1089/neu.2014.3502. [DOI] [PubMed] [Google Scholar]
- Jensen K, Ohrstrom J, Cold GE, Astrup J. The effects of indomethacin on intracranial pressure, cerebral blood flow and cerebral metabolism in patients with severe head injury and intracranial hypertension. Acta Neurochir (Wien) 1991;108:116–21. doi: 10.1007/BF01418518. [DOI] [PubMed] [Google Scholar]
- Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, Tyurina YY, Fink B, Manole MD, Puccio AM, Okonkwo DO, Cheng JP, Alexander H, Clark RS, Kochanek PM, Wipf P, Kagan VE, Bayir H. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci. 2012a;15:1407–13. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Tyurina YY, Tang M, Feng W, Stolz DB, Clark RS, Meaney DF, Kochanek PM, Kagan VE, Bayir H. Mitochondrial injury after mechanical stretch of cortical neurons in vitro: biomarkers of apoptosis and selective peroxidation of anionic phospholipids. J Neurotrauma. 2012b;29:776–88. doi: 10.1089/neu.2010.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Baart S, Vikulina AS, Clark RS, Anthonymuthu TS, Tyurin VA, Du L, St Croix CM, Tyurina YY, Lewis J, Skoda EM, Kline AE, Kochanek PM, Wipf P, Kagan VE, Bayir H. Deciphering of mitochondrial cardiolipin oxidative signaling in cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2015;35:319–28. doi: 10.1038/jcbfm.2014.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Arai K, Murata Y, Wang S, Stins MF, Lo EH, van Leyen K. Protecting against cerebrovascular injury: contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke. 2008;39:2538–43. doi: 10.1161/STROKEAHA.108.514927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature chemical biology. 2005a;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005b;1:223–32. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, Greenberger JS, Pitt B, Shvedova AA, Borisenko G. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med. 2009a;46:1439–53. doi: 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Wipf P, Stoyanovsky D, Greenberger JS, Borisenko G, Belikova NA, Yanamala N, Samhan Arias AK, Tungekar MA, Jiang J, Tyurina YY, Ji J, Klein-Seetharaman J, Pitt BR, Shvedova AA, Bayir H. Mitochondrial targeting of electron scavenging antioxidants: Regulation of selective oxidation vs random chain reactions. Adv Drug Deliv Rev. 2009b;61:1375–85. doi: 10.1016/j.addr.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappus H. A survey of chemicals inducing lipid peroxidation in biological systems. Chem Phys Lipids. 1987;45:105–15. doi: 10.1016/0009-3084(87)90062-4. [DOI] [PubMed] [Google Scholar]
- Kapralov AA, Kurnikov IV, Vlasova, Belikova NA, Tyurin VA, Basova LV, Zhao Q, Tyurina YY, Jiang J, Bayir H, Vladimirov YA, Kagan VE. The hierarchy of structural transitions induced in cytochrome c by anionic phospholipids determines its peroxidase activation and selective peroxidation during apoptosis in cells. Biochemistry. 2007;46:14232–44. doi: 10.1021/bi701237b. [DOI] [PubMed] [Google Scholar]
- Kasprzak HA, Wozniak A, Drewa G, Wozniak B. Enhanced lipid peroxidation processes in patients after brain contusion. J Neurotrauma. 2001;18:793–7. doi: 10.1089/089771501316919157. [DOI] [PubMed] [Google Scholar]
- Kawashima H, Kusunose E, Thompson CM, Strobel HW. Protein expression, characterization, and regulation of CYP4F4 and CYP4F5 cloned from rat brain. Arch Biochem Biophys. 1997;347:148–54. doi: 10.1006/abbi.1997.0342. [DOI] [PubMed] [Google Scholar]
- Kellogg EW, 3rd, Fridovich I. Superoxide, hydrogen peroxide, and singlet oxygen in lipid peroxidation by a xanthine oxidase system. J Biol Chem. 1975;250:8812–7. [PubMed] [Google Scholar]
- Khan NA. Infrared studies on initiation of the autoxidation of some fatty acid esters with and without light-sensitized chlorophyll, ultraviolet light and lipoxidase. Biochim Biophys Acta. 1955;16:159–60. doi: 10.1016/0006-3002(55)90195-5. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Levasseur JE, Patterson JL, Jr, Jackson GF, Madge GE, Povlishock JT, Kontos HA. Effect of indomethacin pretreatment on acute mortality in experimental brain injury. J Neurosurg. 1989;71:565–72. doi: 10.3171/jns.1989.71.4.0565. [DOI] [PubMed] [Google Scholar]
- Kissner R, Nauser T, Kurz C, Koppenol W. Peroxynitrous Acid-Where is the Hydroxyl Radical? IUBMB life. 2003;55:567–572. doi: 10.1080/15216540310001628690. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Potentiation of hippocampal synaptic transmission by superoxide requires the oxidative activation of protein kinase C. J Neurosci. 2002;22:674–83. doi: 10.1523/JNEUROSCI.22-03-00674.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontos HA, Wei EP. Superoxide production in experimental brain injury. Journal of neurosurgery. 1986;64:803–807. doi: 10.3171/jns.1986.64.5.0803. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta. 2015;1851:308–30. doi: 10.1016/j.bbalip.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz T, Marklund N, Hillered L, Oliw EH. Effects of the selective cyclooxygenase-2 inhibitor rofecoxib on cell death following traumatic brain injury in the rat. Restor Neurol Neurosci. 2006;24:55–63. [PubMed] [Google Scholar]
- Lai Y, Hickey RW, Chen Y, Bayır H, Sullivan ML, Chu CT, Kochanek PM, Dixon CE, Jenkins LW, Graham SH. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant γ-glutamylcysteinyl ethyl ester. Journal of Cerebral Blood Flow & Metabolism. 2008;28:540–550. doi: 10.1038/sj.jcbfm.9600551. [DOI] [PubMed] [Google Scholar]
- Laneuville O, Breuer DK, Xu N, Huang ZH, Gage DA, Watson JT, Lagarde M, DeWitt DL, Smith WL. Fatty acid substrate specificities of human prostaglandin-endoperoxide H synthase-1 and -2. Formation of 12-hydroxy-(9Z, 13E/Z, 15Z)- octadecatrienoic acids from alpha-linolenic acid. J Biol Chem. 1995;270:19330–6. doi: 10.1074/jbc.270.33.19330. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Maher P, Schubert D, Zivin JA. Baicalein, an antioxidant 12/15-lipoxygenase inhibitor improves clinical rating scores following multiple infarct embolic strokes. Neuroscience. 2007;150:585–91. doi: 10.1016/j.neuroscience.2007.09.033. [DOI] [PubMed] [Google Scholar]
- Lee DW, Andersen JK, Kaur D. Iron dysregulation and neurodegeneration: the molecular connection. Mol Interv. 2006;6:89–97. doi: 10.1124/mi.6.2.6. [DOI] [PubMed] [Google Scholar]
- Lin CJ, Chen TH, Yang LY, Shih CM. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 2014a;5:e1147. doi: 10.1038/cddis.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W-M, Chen M-H, Wang H-C, Lu C-H, Chen P-C, Chen H-L, Tsai N-W, Su Y-J, Li S-H, Kung C-T. Association between peripheral oxidative stress and white matter damage in acute traumatic brain injury. BioMed research international. 2014b;2014 doi: 10.1155/2014/340936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente L, Martín MM, Abreu-González P, Ramos L, Argueso M, Cáceres JJ, Solé-Violán J, Lorenzo JM, Molina I, Jiménez A. Association between serum malondialdehyde levels and mortality in patients with severe brain trauma injury. Journal of neurotrauma. 2015;32:1–6. doi: 10.1089/neu.2014.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacova N, Halat G, Chavko M, Marsala J. Ischemia-reperfusion injury in the spinal cord of rabbits strongly enhances lipid peroxidation and modifies phospholipid profiles. Neurochem Res. 1996;21:869–73. doi: 10.1007/BF02532334. [DOI] [PubMed] [Google Scholar]
- Luo CL, Li QQ, Chen XP, Zhang XM, Li LL, Li BX, Zhao ZQ, Tao LY. Lipoxin A4 attenuates brain damage and downregulates the production of pro-inflammatory cytokines and phosphorylated mitogen-activated protein kinases in a mouse model of traumatic brain injury. Brain Res. 2013;1502:1–10. doi: 10.1016/j.brainres.2013.01.037. [DOI] [PubMed] [Google Scholar]
- MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10:997–1030. doi: 10.1089/ars.2007.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso DJ, Kotzbauer P, Wozniak DF, Sims HF, Jenkins CM, Guan S, Han X, Yang K, Sun G, Malik I, Conyers S, Green KG, Schmidt RE, Gross RW. Genetic ablation of calcium-independent phospholipase A2{gamma} leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. J Biol Chem. 2009;284:35632–44. doi: 10.1074/jbc.M109.055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, Hardy M, Gimenez JM, Chiang N, Serhan CN, Bazan NG. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. 2003;278:43807–17. doi: 10.1074/jbc.M305841200. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Lovell MA. Damage to lipids, proteins, DNA, and RNA in mild cognitive impairment. Arch Neurol. 2007;64:954–6. doi: 10.1001/archneur.64.7.954. [DOI] [PubMed] [Google Scholar]
- Marshall LF, Maas AI, Marshall SB, Bricolo A, Fearnside M, Iannotti F, Klauber MR, Lagarrigue J, Lobato R, Persson L. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. Journal of neurosurgery. 1998;89:519–525. doi: 10.3171/jns.1998.89.4.0519. [DOI] [PubMed] [Google Scholar]
- Maskrey BH, Bermudez-Fajardo A, Morgan AH, Stewart-Jones E, Dioszeghy V, Taylor GW, Baker PR, Coles B, Coffey MJ, Kuhn H, O'Donnell VB. Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. J Biol Chem. 2007;282:20151–63. doi: 10.1074/jbc.M611776200. [DOI] [PubMed] [Google Scholar]
- Mendes Arent A, de Souza LF, Walz R, Dafre AL. Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. Biomed Res Int. 2014;2014:723060. doi: 10.1155/2014/723060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikawa S, Kinouchi H, Kamii H, Gobbel GT, Chen SF, Carlson E, Epstein CJ, Chan PH. Attenuation of acute and chronic damage following traumatic brain injury in copper, zinc-superoxide dismutase transgenic mice. J Neurosurg. 1996;85:885–91. doi: 10.3171/jns.1996.85.5.0885. [DOI] [PubMed] [Google Scholar]
- Miller DM, Wang JA, Buchanan AK, Hall ED. Temporal and spatial dynamics of nrf2-antioxidant response elements mediated gene targets in cortex and hippocampus after controlled cortical impact traumatic brain injury in mice. J Neurotrauma. 2014;31:1194–201. doi: 10.1089/neu.2013.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison B, 3rd, Eberwine JH, Meaney DF, McIntosh TK. Traumatic injury induces differential expression of cell death genes in organotypic brain slice cultures determined by complementary DNA array hybridization. Neuroscience. 2000;96:131–9. doi: 10.1016/s0306-4522(99)00537-0. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Kiwak KJ, Hekimian K, Levine L. Synthesis of compounds with properties of leukotrienes C4 and D4 in gerbil brains after ischemia and reperfusion. Science. 1984;224:886–9. doi: 10.1126/science.6719118. [DOI] [PubMed] [Google Scholar]
- Muizelaar JP. Cerebral ischemia-reperfusion injury after severe head injury and its possible treatment with polyethyleneglycol-superoxide dismutase. Ann Emerg Med. 1993;22:1014–21. doi: 10.1016/s0196-0644(05)82744-1. [DOI] [PubMed] [Google Scholar]
- Munsiff AV, Chander PN, Levine S, Stier CT., Jr The lipoxygenase inhibitor phenidone protects against proteinuria and stroke in stroke-prone spontaneously hypertensive rats. Am J Hypertens. 1992;5:56–63. doi: 10.1093/ajh/5.2.56. [DOI] [PubMed] [Google Scholar]
- Nigam S, Zafiriou MP, Deva R, Ciccoli R, Roux-Van der Merwe R. Structure, biochemistry and biology of hepoxilins: an update. FEBS J. 2007;274:3503–12. doi: 10.1111/j.1742-4658.2007.05910.x. [DOI] [PubMed] [Google Scholar]
- Niki E. Lipid peroxidation: physiological levels and dual biological effects. Free Radic Biol Med. 2009;47:469–84. doi: 10.1016/j.freeradbiomed.2009.05.032. [DOI] [PubMed] [Google Scholar]
- Niki E. Biomarkers of lipid peroxidation in clinical material. Biochimica et Biophysica Acta (BBA)-General Subjects. 2014;1840:809–817. doi: 10.1016/j.bbagen.2013.03.020. [DOI] [PubMed] [Google Scholar]
- Nishi H, Watanabe T, Sakurai H, Yuki S, Ishibashi A. Effect of MCI-186 on brain edema in rats. Stroke. 1989;20:1236–40. doi: 10.1161/01.str.20.9.1236. [DOI] [PubMed] [Google Scholar]
- O'Donnell VB, Murphy RC. New families of bioactive oxidized phospholipids generated by immune cells: identification and signaling actions. Blood. 2012;120:1985–92. doi: 10.1182/blood-2012-04-402826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbal S, Cankurt U, Tugyan K, Pekcetin C, Sisman AR, Gunduz K, Micili SC. The effects of alpha-lipoic acid on immature rats with traumatic brain injury. Biotech Histochem. 2015;90:206–15. doi: 10.3109/10520295.2014.977950. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Readnower RD, Patel SP, Yonutas HM, Pauly JR, Goldstein GA, Rabchevsky AG, Sullivan PG. N-acetylcysteine amide confers neuroprotection, improves bioenergetics and behavioral outcome following TBI. Exp Neurol. 2014;257:106–13. doi: 10.1016/j.expneurol.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolin A, Nardin L, Gaetani P, Rodriguez YBR, Pansarasa O, Marzatico F. Oxidative damage after severe head injury and its relationship to neurological outcome. Neurosurgery. 2002;51:949–54. doi: 10.1097/00006123-200210000-00018. discussion 954–5. [DOI] [PubMed] [Google Scholar]
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol. 2005;202:199–211. doi: 10.1016/j.taap.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Patel V, Rink C, Khanna S, Sen CK. Tocotrienols: the lesser known form of natural vitamin E. Indian J Exp Biol. 2011;49:732–8. [PMC free article] [PubMed] [Google Scholar]
- Pederson TC, Aust SD. The mechanism of liver microsomal lipid peroxidation. Biochim Biophys Acta. 1975;385:232–41. doi: 10.1016/0304-4165(75)90351-7. [DOI] [PubMed] [Google Scholar]
- Polidori MC, Mecocci P, Frei B. Plasma vitamin C levels are decreased and correlated with brain damage in patients with intracranial hemorrhage or head trauma. Stroke. 2001;32:898–902. doi: 10.1161/01.str.32.4.898. [DOI] [PubMed] [Google Scholar]
- Porter NA. Chemistry of lipid peroxidation. Methods Enzymol. 1984;105:273–82. doi: 10.1016/s0076-6879(84)05035-7. [DOI] [PubMed] [Google Scholar]
- Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30:277–90. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- Pratico D, Uryu K, Sung S, Tang S, Trojanowski JQ, Lee VM. Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J. 2002;16:1138–40. doi: 10.1096/fj.02-0012fje. [DOI] [PubMed] [Google Scholar]
- Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol. 1995;268:L699–722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- Razmkon A, Sadidi A, Sherafat-Kazemzadeh E, Mehrafshan A, Jamali M, Malekpour B, Saghafinia M. Administration of vitamin C and vitamin E in severe head injury: a randomized double-blind controlled trial. Clin Neurosurg. 2011;58:133–7. doi: 10.1227/neu.0b013e3182279a8f. [DOI] [PubMed] [Google Scholar]
- Reeder BJ, Svistunenko DA, Wilson MT. Lipid binding to cytoglobin leads to a change in haem co-ordination: a role for cytoglobin in lipid signalling of oxidative stress. Biochem J. 2011;434:483–92. doi: 10.1042/BJ20101136. [DOI] [PubMed] [Google Scholar]
- Repetto MG, Ferrarotti NF, Boveris A. The involvement of transition metal ions on iron-dependent lipid peroxidation. Arch Toxicol. 2010;84:255–62. doi: 10.1007/s00204-009-0487-y. [DOI] [PubMed] [Google Scholar]
- Robak J, Gryglewski RJ. Nitric oxide donors as generators and scavengers of superoxide anions. Pol J Pharmacol. 1993;45:51–8. [PubMed] [Google Scholar]
- Roberts LJ, 2nd, Morrow JD. Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci. 2002;59:808–20. doi: 10.1007/s00018-002-8469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–85. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- Roof RL, Hoffman SW, Stein DG. Progesterone protects against lipid peroxidation following traumatic brain injury in rats. Mol Chem Neuropathol. 1997;31:1–11. doi: 10.1007/BF02815156. [DOI] [PubMed] [Google Scholar]
- Roveri A, Maiorino M, Nisii C, Ursini F. Purification and characterization of phospholipid hydroperoxide glutathione peroxidase from rat testis mitochondrial membranes. Biochim Biophys Acta. 1994;1208:211–21. doi: 10.1016/0167-4838(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Sabel BA, Slavin MD, Stein DG. GM1 ganglioside treatment facilitates behavioral recovery from bilateral brain damage. Science. 1984;225:340–2. doi: 10.1126/science.6740316. [DOI] [PubMed] [Google Scholar]
- Samuelsson B. Prostaglandins, thromboxanes, and leukotrienes: formation and biological roles. Harvey Lect. 1979;75:1–40. [PubMed] [Google Scholar]
- Sapirstein A, Bonventre JV. Phospholipases A2 in ischemic and toxic brain injury. Neurochem Res. 2000;25:745–53. doi: 10.1023/a:1007583708713. [DOI] [PubMed] [Google Scholar]
- Savaskan NE, Borchert A, Bräuer AU, Kuhn H. Role for glutathione peroxidase-4 in brain development and neuronal apoptosis: specific induction of enzyme expression in reactive astrocytes following brain injury. Free Radical Biology and Medicine. 2007;43:191–201. doi: 10.1016/j.freeradbiomed.2007.03.033. [DOI] [PubMed] [Google Scholar]
- Schurks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: meta-analysis of randomised controlled trials. BMJ. 2010;341:c5702. doi: 10.1136/bmj.c5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secades JJ, Frontera G. CDP-choline: pharmacological and clinical review. Methods Find Exp Clin Pharmacol. 1995;17(Suppl B):1–54. [PubMed] [Google Scholar]
- Serbinova EA, Kadiiska MB, Bakalova RA, Koynova GM, Stoyanovsky DA, Karakashev PC, Stoytchev TS, Wolinsky I, Kagan VE. Lipid peroxidation activation and cytochrome P-450 decrease in rat liver endoplasmic reticulum under oxidative stress. Toxicol Lett. 1989;47:119–23. doi: 10.1016/0378-4274(89)90066-0. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci U S A. 1984;81:5335–9. doi: 10.1073/pnas.81.17.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000;192:1197–204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Gotlinger K, Hong S, Arita M. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: an overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat. 2004;73:155–72. doi: 10.1016/j.prostaglandins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevanian A, Hochstein P. Mechanisms and consequences of lipid peroxidation in biological systems. Annu Rev Nutr. 1985;5:365–90. doi: 10.1146/annurev.nu.05.070185.002053. [DOI] [PubMed] [Google Scholar]
- Shishido Y, Furushiro M, Hashimoto S, Yokokura T. Effect of nordihydroguaiaretic acid on behavioral impairment and neuronal cell death after forebrain ischemia. Pharmacol Biochem Behav. 2001;69:469–74. doi: 10.1016/s0091-3057(01)00572-x. [DOI] [PubMed] [Google Scholar]
- Shohami E, Shapira Y, Sidi A, Cotev S. Head injury induces increased prostaglandin synthesis in rat brain. J Cereb Blood Flow Metab. 1987;7:58–63. doi: 10.1038/jcbfm.1987.8. [DOI] [PubMed] [Google Scholar]
- Shohami E, Shapira Y, Yadid G, Reisfeld N, Yedgar S. Brain phospholipase A2 is activated after experimental closed head injury in the rat. Journal of neurochemistry. 1989;53:1541–1546. doi: 10.1111/j.1471-4159.1989.tb08550.x. [DOI] [PubMed] [Google Scholar]
- Shohami E, Glantz L, Nates J, Feuerstein G. The mixed lipoxygenase/cyclooxygenase inhibitor SK&F 105809 reduces cerebral edema after closed head injury in rat. J Basic Clin Physiol Pharmacol. 1992;3:99–107. doi: 10.1515/jbcpp.1992.3.2.99. [DOI] [PubMed] [Google Scholar]
- Skolnick BE, Maas AI, Narayan RK, van der Hoop RG, MacAllister T, Ward JD, Nelson NR, Stocchetti N, Investigators ST. A clinical trial of progesterone for severe traumatic brain injury. N Engl J Med. 2014;371:2467–76. doi: 10.1056/NEJMoa1411090. [DOI] [PubMed] [Google Scholar]
- Sparvero LJ, Amoscato AA, Kochanek PM, Pitt BR, Kagan VE, Bayir H. Mass-spectrometry based oxidative lipidomics and lipid imaging: applications in traumatic brain injury. J Neurochem. 2010;115:1322–36. doi: 10.1111/j.1471-4159.2010.07055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector R, Eells J. Deoxynucleoside and vitamin transport into the central nervous system. Fed Proc. 1984;43:196–200. [PubMed] [Google Scholar]
- Stellwagen E. The reversible unfolding of horse heart ferricytochrome c. Biochemistry. 1968;7:2893–8. doi: 10.1021/bi00848a028. [DOI] [PubMed] [Google Scholar]
- Strauss KI, Barbe MF, Marshall RM, Raghupathi R, Mehta S, Narayan RK. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma. 2000;17:695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebeni J, Winterbourn CC, Carrell RW. Oxidative interactions between haemoglobin and membrane lipid. A liposome model. Biochem J. 1984;220:685–92. doi: 10.1042/bj2200685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazaki Y, Sakai F, Otomo E, Kutsuzawa T, Kameyama M, Omae T, Fujishima M, Sakuma A. Treatment of acute cerebral infarction with a choline precursor in a multicenter double-blind placebo-controlled study. Stroke. 1988;19:211–6. doi: 10.1161/01.str.19.2.211. [DOI] [PubMed] [Google Scholar]
- Thau-Zuchman O, Shohami E, Alexandrovich AG, Trembovler V, Leker RR. The anti-inflammatory drug carprofen improves long-term outcome and induces gliogenesis after traumatic brain injury. J Neurotrauma. 2012;29:375–84. doi: 10.1089/neu.2010.1673. [DOI] [PubMed] [Google Scholar]
- Thiels E, Urban NN, Gonzalez-Burgos GR, Kanterewicz BI, Barrionuevo G, Chu CT, Oury TD, Klann E. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. The Journal of Neuroscience. 2000;20:7631–7639. doi: 10.1523/JNEUROSCI.20-20-07631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CE, Morehouse LA, Aust SD. Ferritin and superoxide-dependent lipid peroxidation. J Biol Chem. 1985;260:3275–80. [PubMed] [Google Scholar]
- Thorogood MC, Armstead WM. Influence of polyethylene glycol superoxide dismutase/catalase on altered opioid-induced pial artery dilation after brain injury. Anesthesiology. 1996;84:614–25. doi: 10.1097/00000542-199603000-00017. [DOI] [PubMed] [Google Scholar]
- Tinoco J. Dietary requirements and functions of alpha-linolenic acid in animals. Prog Lipid Res. 1982;21:1–45. doi: 10.1016/0163-7827(82)90015-7. [DOI] [PubMed] [Google Scholar]
- Toffano G, Savoini G, Aldinio C, Valenti G, Dal Toso R, Leon A, Calza L, Zini I, Agnati LF, Fuxe K. Effects of gangliosides on the functional recovery of damaged brain. Adv Exp Med Biol. 1984;174:475–88. doi: 10.1007/978-1-4684-1200-0_40. [DOI] [PubMed] [Google Scholar]
- Topcu I, Gumuser G, Bayram E, Aras F, Cetin I, Temiz C, Civi M. The effects of lornoxicam on brain edema and blood brain barrier following diffuse traumatic brain injury in rats. Ulus Travma Acil Cerrahi Derg. 2013;19:294–8. doi: 10.5505/tjtes.2013.32458. [DOI] [PubMed] [Google Scholar]
- Tyurin VA, Tyurina YY, Borisenko GG, Sokolova TV, Ritov VB, Quinn PJ, Rose M, Kochanek P, Graham SH, Kagan VE. Oxidative stress following traumatic brain injury in rats: quantitation of biomarkers and detection of free radical intermediates. J Neurochem. 2000;75:2178–89. doi: 10.1046/j.1471-4159.2000.0752178.x. [DOI] [PubMed] [Google Scholar]
- Tyurin VA, Tyurina YY, Ritov VB, Lysytsya A, Amoscato AA, Kochanek PM, Hamilton R, Dekosky ST, Greenberger JS, Bayir H, Kagan VE. Oxidative lipidomics of apoptosis: quantitative assessment of phospholipid hydroperoxides in cells and tissues. Methods Mol Biol. 2010;610:353–74. doi: 10.1007/978-1-60327-029-8_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyurina YY, Kini V, Tyurin VA, Vlasova, Jiang J, Kapralov AA, Belikova NA, Yalowich JC, Kurnikov IV, Kagan VE. Mechanisms of cardiolipin oxidation by cytochrome c: relevance to pro- and antiapoptotic functions of etoposide. Mol Pharmacol. 2006;70:706–17. doi: 10.1124/mol.106.022731. [DOI] [PubMed] [Google Scholar]
- Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthu TS, Kapralova VI, Vikulina AS, Jung MY, Epperly MW, Mohammadyani D, Klein-Seetharaman J, Jackson TC, Kochanek PM, Pitt BR, Greenberger JS, Vladimirov YA, Bayir H, Kagan VE. A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem. 2014;6:542–52. doi: 10.1038/nchem.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyurina YY, Polimova AM, Maciel E, Tyurin VA, Kapralova VI, Winnica DE, Vikulina AS, Domingues MR, McCoy J, Sanders LH, Bayir H, Greenamyre JT, Kagan VE. LC/MS analysis of cardiolipins in substantia nigra and plasma of rotenone-treated rats: Implication for mitochondrial dysfunction in Parkinson's disease. Free Radic Res. 2015;49:681–91. doi: 10.3109/10715762.2015.1005085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leyen K, Kim HY, Lee SR, Jin G, Arai K, Lo EH. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke. 2006;37:3014–8. doi: 10.1161/01.STR.0000249004.25444.a5. [DOI] [PubMed] [Google Scholar]
- van Leyen K, Arai K, Jin G, Kenyon V, Gerstner B, Rosenberg PA, Holman TR, Lo EH. Novel lipoxygenase inhibitors as neuroprotective reagents. J Neurosci Res. 2008;86:904–9. doi: 10.1002/jnr.21543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt C, Donat CK, Hartig W, Forschler A, Skardelly M, Stichtenoth D, Arendt T, Meixensberger J, Schuhmann MU. Effect of leukotriene inhibitors on evolution of experimental brain contusions. Neuropathol Appl Neurobiol. 2012;38:354–66. doi: 10.1111/j.1365-2990.2011.01211.x. [DOI] [PubMed] [Google Scholar]
- Wada M, DeLong CJ, Hong YH, Rieke CJ, Song I, Sidhu RS, Yuan C, Warnock M, Schmaier AH, Yokoyama C, Smyth EM, Wilson SJ, FitzGerald GA, Garavito RM, Sui de X, Regan JW, Smith WL. Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derived substrates and products. J Biol Chem. 2007;282:22254–66. doi: 10.1074/jbc.M703169200. [DOI] [PubMed] [Google Scholar]
- Wagner AK, Bayir H, Ren D, Puccio A, Zafonte RD, Kochanek PM. Relationships between cerebrospinal fluid markers of excitotoxicity, ischemia, and oxidative damage after severe TBI: the impact of gender, age, and hypothermia. J Neurotrauma. 2004;21:125–36. doi: 10.1089/089771504322778596. [DOI] [PubMed] [Google Scholar]
- Wang H-C, Lin Y-J, Shih F-Y, Chang H-W, Tsai N-W, Chang Y-T, Kwan A-L, Lu C-H. The Role of Serial Oxidative Stress Levels in Acute Traumatic Brain Injury and as. 2015 doi: 10.1016/j.wneu.2015.10.010. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhao Y, Bradbury JA, Graves JP, Foley J, Blaisdell JA, Goldstein JA, Zeldin DC. Cloning, expression, and characterization of three new mouse cytochrome p450 enzymes and partial characterization of their fatty acid oxidation activities. Mol Pharmacol. 2004;65:1148–58. doi: 10.1124/mol.65.5.1148. [DOI] [PubMed] [Google Scholar]
- Wei L, Zhang Y, Yang C, Wang Q, Zhuang Z, Sun Z. Neuroprotective effects of ebselen in traumatic brain injury model: involvement of nitric oxide and p38 mitogen-activated protein kinase signalling pathway. Clin Exp Pharmacol Physiol. 2014;41:134–8. doi: 10.1111/1440-1681.12186. [DOI] [PubMed] [Google Scholar]
- Williams KI, Higgs GA. Eicosanoids and inflammation. J Pathol. 1988;156:101–10. doi: 10.1002/path.1711560204. [DOI] [PubMed] [Google Scholar]
- Wills ED. Mechanisms of lipid peroxide formation in animal tissues. Biochem J. 1966;99:667–76. doi: 10.1042/bj0990667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witting LA. Lipid peroxidation in vivo. J Am Oil Chem Soc. 1965;42:908–13. doi: 10.1007/BF02632443. [DOI] [PubMed] [Google Scholar]
- Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M, Goldstein FC, Caveney AF, Howlett-Smith H, Bengelink EM. Very early administration of progesterone for acute traumatic brain injury. New England Journal of Medicine. 2014;371:2457–2466. doi: 10.1056/NEJMoa1404304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Ying Z, Schubert D, Gomez-Pinilla F. Brain and spinal cord interaction: a dietary curcumin derivative counteracts locomotor and cognitive deficits after brain trauma. Neurorehabil Neural Repair. 2011;25:332–42. doi: 10.1177/1545968310397706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Ye XH, Guo PP, Xu SP, Wang J, Yuan SY, Yao SL, Shang Y. Neuroprotective effect of lipoxin A4 methyl ester in a rat model of permanent focal cerebral ischemia. J Mol Neurosci. 2010;42:226–34. doi: 10.1007/s12031-010-9355-8. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang YP, Guo P, Ye XH, Wang J, Yuan SY, Yao SL, Shang Y. A lipoxin A4 analog ameliorates blood-brain barrier dysfunction and reduces MMP-9 expression in a rat model of focal cerebral ischemia-reperfusion injury. J Mol Neurosci. 2012;46:483–91. doi: 10.1007/s12031-011-9620-5. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Peterson PL, Lee CP. Effect of N-acetylcysteine on mitochondrial function following traumatic brain injury in rats. J Neurotrauma. 1999;16:1067–82. doi: 10.1089/neu.1999.16.1067. [DOI] [PubMed] [Google Scholar]
- Yagami T, Yamamoto Y, Koma H. The role of secretory phospholipase A(2) in the central nervous system and neurological diseases. Mol Neurobiol. 2014;49:863–76. doi: 10.1007/s12035-013-8565-9. [DOI] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, Prolla TA. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radical Biology and Medicine. 2003;34:496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- Ye XH, Wu Y, Guo PP, Wang J, Yuan SY, Shang Y, Yao SL. Lipoxin A4 analogue protects brain and reduces inflammation in a rat model of focal cerebral ischemia reperfusion. Brain Res. 2010;1323:174–83. doi: 10.1016/j.brainres.2010.01.079. [DOI] [PubMed] [Google Scholar]
- Yermakova A, O'Banion MK. Cyclooxygenases in the central nervous system: implications for treatment of neurological disorders. Curr Pharm Des. 2000;6:1755–76. doi: 10.2174/1381612003398672. [DOI] [PubMed] [Google Scholar]
- Yokoyama T, Date C, Kokubo Y, Yoshiike N, Matsumura Y, Tanaka H. Serum vitamin C concentration was inversely associated with subsequent 20-year incidence of stroke in a Japanese rural community. The Shibata study. Stroke. 2000;31:2287–94. doi: 10.1161/01.str.31.10.2287. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Busto R, Abe K, Santiso M, Ginsberg MD. Compression-induced brain edema in rats: effect of dietary vitamin E on membrane damage in the brain. Neurology. 1985;35:126–30. doi: 10.1212/wnl.35.1.126. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Itoh N, Hayakawa M, Habuchi Y, Saito Y, Tsukamoto Y, Cynshi O, Jishage K, Arai H, Niki E. The role of alpha-tocopherol in motor hypofunction with aging in alpha-tocopherol transfer protein knockout mice as assessed by oxidative stress biomarkers. J Nutr Biochem. 2010;21:66–76. doi: 10.1016/j.jnutbio.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Young B, Runge JW, Waxman KS, Harrington T, Wilberger J, Muizelaar JP, Boddy A, Kupiec JW. Effects of pegorgotein on neurologic outcome of patients with severe head injury. A multicenter, randomized controlled trial. JAMA. 1996;276:538–43. [PubMed] [Google Scholar]
- Yuan C, Sidhu RS, Kuklev DV, Kado Y, Wada M, Song I, Smith WL. Cyclooxygenase Allosterism, Fatty Acid-mediated Cross-talk between Monomers of Cyclooxygenase Homodimers. J Biol Chem. 2009;284:10046–55. doi: 10.1074/jbc.M808634200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf HK. Understanding the brain and its development: A chemical approach. World Scientific; 1992. [Google Scholar]
- Zafonte R, Friedewald WT, Lee SM, Levin B, Diaz-Arrastia R, Ansel B, Eisenberg H, Timmons SD, Temkin N, Novack T, Ricker J, Merchant R, Jallo J. The citicoline brain injury treatment (COBRIT) trial: design and methods. J Neurotrauma. 2009;26:2207–16. doi: 10.1089/neu.2009.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang WP, Hu H, Wang ML, Sheng WW, Yao HT, Ding W, Chen Z, Wei EQ. Expression patterns of 5-lipoxygenase in human brain with traumatic injury and astrocytoma. Neuropathology. 2006;26:99–106. doi: 10.1111/j.1440-1789.2006.00658.x. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Fang SH, Ye YL, Chu LS, Zhang WP, Wang ML, Wei EQ. Caffeic acid ameliorates early and delayed brain injuries after focal cerebral ischemia in rats. Acta Pharmacol Sin. 2006;27:1103–10. doi: 10.1111/j.1745-7254.2006.00406.x. [DOI] [PubMed] [Google Scholar]
- Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X, Chen F, Wang CS, Feng H, Lin JK. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. J Neuroinflammation. 2014;11:59. doi: 10.1186/1742-2094-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]