

Abstract
Histone deacetylases (HDACs), originally described as histone modifiers, have more recently been demonstrated to target a variety of other proteins unrelated to the chromatin environment. In this context, our present work demonstrates that the pharmacological or genetic abrogation of HDAC6 in primary melanoma samples and cell lines, down‐regulates the expression of PD‐L1, an important co‐stimulatory molecule expressed in cancer cells, which activates the inhibitory regulatory pathway PD‐1 in T‐cells. Our data suggests that this novel mechanism of PD‐L1 regulation is mainly mediated by the influence of HDAC6 over the recruitment and activation of STAT3. Additionally, we observed that selective HDAC6 inhibitors impairs tumor growth and reduce the in vivo expression of several inhibitory check‐point molecules and other regulatory pathways involved in immunosurveillance. Most importantly, these results provide a key pre‐clinical rationale and justification to further study isotype selective HDAC6 inhibitors as potential immuno‐modulatory agents in cancer.
Keywords: Histone deacetylases, HDAC6, PD‐L1, STAT3, PP2A, Melanoma, Nexturastat, Tubastatin A
Highlights
The inhibition of HDAC6, induces an in vivo delay in tumor growth and down‐regulates the expression of PD‐L1.
Several key immunological check‐point modulators are regulated by HDAC6.
The specific recruitment of HDAC6 to the PD‐L1 promoter, which is required for the STAT3‐mediated activation of this promoter.
1. Introduction
In spite of recent progress made in understanding the pathobiology, genetics, and immunology of melanoma, the outcome for patients with advanced‐stage disease remains poor with a median survival ranging from 8 to 16 months and an overall survival (OS) at 5 years of less than 10% (Nikolaou and Stratigos, 2014). However, new findings provide optimism for patients with metastatic melanoma. This is due to improved clinical outcomes observed in patients receiving targeted therapies aiming to block negative immuno‐modulatory pathways such as cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4), program death receptor‐1 (PD‐1) and program death receptor ligand‐1 (PD‐L1) receptors (Hodi et al., 2010; Topalian et al., 2012), ultimately, augmenting T‐cell anti‐tumor activity. This work has paved a novel role in the development of rational combinatorial approaches aimed at augmenting the efficacy of immuno‐modulatory drugs and antibodies targeting immunological checkpoints. An emerging awareness is currently addressing the role of epigenetic modifiers in the regulation of immuno‐modulatory pathways. Among these, histone deacetylases (HDACs) are attractive targets due to the availability of a broad spectrum of inhibitors targeting their enzymatic activity (HDACi). However, despite the well documented effects of HDACi in the control of the cell cycle and apoptosis, their participation in the regulation of immune‐related pathways is still not completely understood. Additionally, the reported immunological outcomes when using these drugs are heterogeneous, and in many cases contradictory when using different HDAC inhibitors (Woan et al., 2012; Tomasi et al., 2006). This lack of understanding and the observed disparities in HDAC inhibition can be attributed, at least in part, to the non‐specific action of pan‐HDACi targeting all 11 zinc‐dependent HDACs, or a subset thereof, and the intrinsic variations in the expression of these enzymes among different cell types in both physiological and pathological conditions. Therefore, the generation of selective HDACi and mechanistic insight into their role in the immune response against cancer cells are highly desirable goals, and has the potential to augment anti‐tumor immunity.
HDACs, originally described as histone modifiers, have recently been demonstrated to modify a variety of other proteins involved in diverse cellular processes unrelated to the chromatin environment. This includes deacetylation of multiple non‐histone targets, such as members of oncogenic‐ and immune‐related pathways (Woan et al., 2012; Villagra et al., 2010). In this regard, a considerable number of reports have analyzed the role of unspecific inhibition of HDACs through pan‐HDACi in cancer as well as processes of immune regulation. However, the contribution of single HDACs is still poorly understood and only a few reports identified the role of specific HDACs in these cellular processes. In this context, we recently reported that the genetic and pharmacological inhibition of a single HDAC, HDAC6, resulted in decreased proliferation of melanoma cells in both in vitro and in in vivo models (Woan et al., 2015). In addition to this effect on survival, HDAC6 was found to be a modulator of the expression of specific tumor associated antigens, MHC class I and co‐stimulatory molecules in melanoma (Woan et al., 2015). Furthermore, HDAC6 seems to be an important regulator of the STAT3 pathways (Cheng et al., 2014a), which is commonly altered in melanoma and other malignancies (Yu et al., 2009). Here, we report that HDAC6 is also involved in the regulation of the co‐inhibitory molecule Program Death Receptor Ligand 1 (PD‐L1). This protein is one of the natural ligands for the PD‐1 receptor present on T‐cells, which suppresses T‐cell activation, proliferation, and induces T‐cell anergy and apoptosis (Taube et al., 2014). During the last few years, a number of important studies demonstrated that PD‐L1 is present on cancer cells (Tomasi et al., 2006; Pardoll, 2012), and its over‐expression is often associated with poor prognosis in several malignancies, including melanoma (Hino et al., 2010), ovarian (Hamanishi et al., 2007), gastric (Wu et al., 2006) and breast cancer (Ghebeh et al., 2006), among many others.
The already described participation of HDAC6 as regulator of immune‐related pathways in melanoma and its new role as PD‐L1 regulator opens the possibility of using its selective inhibition as a potential immuno‐modulatory option in ongoing therapies aiming to ameliorate negative pathways affecting the T‐cell response against cancer.
2. Materials and methods
2.1. Mice
Experiments involving mice were performed in accordance with approved protocols by the IACUC at the University of South Florida. C57BL/6 mice were obtained form the National Institutes of Health (Fredrick, Maryland, USA). For in vivo tumor studies, mice were subcutaneously injected into the shaved flank with 1.3 × 105 B16‐F10 melanoma cells suspended in 100 μL Hank's buffered salt solution (HBSS).
2.2. Patient samples
Patient‐derived resected melanoma specimens were obtained from Dr. Sarnaik's Lab at Moffitt Cancer Center through a University of South Florida Institutional Review Board‐approved regulatory protocol. The cells were extracted directly from melanoma tumor and cultured in RPMI 1640 supplemented with l‐glutamine, 10% FBS, 100 IU/mL Penicillin, 100 μg/mL Streptomycin, 1% sodium pyruvate, 1% non‐essential amino acid, 0.05 mM of 2‐mercaptoethanol and 1% gentamycin. The cells were grown under humidified conditions at 37 °C and 5% CO2.
2.3. Cells
B16‐F10‐luc murine melanoma cell line was obtained from ATCC and cultured in RPMI 1640 supplemented with 10% FBS, 100 IU/mL Penicillin, and 100 μg/mL Streptomycin. SM1 cell line was obtained from Dr. Antoni Ribas's Lab at University of California Los Angeles. Human melanoma cell lines were obtained from Dr. Smalley's Lab at Moffitt Cancer Center. Cells were cultured in RPMI 1640 media, supplemented with 10% FBS, penicillin/streptomycin (50 U/ml), l‐glutamine (2 mM), and 2‐mercaptoethanol (50 μM) (complete media), and grown under humidified conditions at 37 °C and 5% CO2.
2.4. HDACi
MGCD0103 and LBH589, were purchased from Selleck Chemicals. The HDAC6 selective inhibitors Tubastatin A and Nexturastat B were synthesized by Dr. Alan Kozikowski (University of Illinois, Chicago, IL). All HDACi were reconstituted in DMSO at greater than 10 mM and stored in aliquots at −80 °C. For in vitro use, stocks were diluted in complete medium immediately before use. For in vivo studies, Nexturastat B was dissolved in 5% DMSO plus 95% Hank's buffered salt solution (HBSS) 1X.
2.5. Immunoblotting
The cells were lysed in a buffer containing 280 mM NaCl, 50 mM Tris HCL PH 8.0, 0.5% Igepal, 5 mM MgCl2, 10% glycerol and 1X protease inhibitor (Roche), phosphatase inhibitor (Santa Cruz Biotechnology). Lysates were sonicated on ice for 8 min (2 cycles of 30 s on, 30 s rest) and then mixed with 6x gel loading buffer and boiled for 5 min. Samples were then resolved on 10% or 4–15% gradient gels and transferred to nitrocellulose membranes. Membranes were blocked with 5% milk‐PBS‐Tween. Bands were detected by scanning blots with an LI‐COR Odyssey imaging system using both 700 and 800 channels. The antibodies used for immunoblotting included anti‐acetyl‐α‐Tubulin (SC‐23950) and anti‐α‐Tubulin (SC‐32293), which were purchased from Santa Cruz Biotechnology. Anti‐HDAC6 (C0226) was from Assay Biotech. Anti‐GAPDH (68795) was from Sigma Aldrich. Anti‐STAT3 (12640), anti P‐STAT3 Y‐705 (9138), anti P‐STAT3 S727 (9136), and anti Acetil‐STAT3 (2523) were purchased from Cell signaling. Anti PD‐L1 (PA5‐28115) was obtained from Thermo Scientific. Anti‐FLAG (F1804) antibody was from Sigma.
2.6. Flow cytometry
For surface marker analysis, wild type melanoma cells were treated with Tubastatin A, Nexturastat B, or DMSO for 24 h, and NT and HDAC6KD cell lines were maintained in growth medium. All the cell lines after the treatment were incubated for an extra 24 h with either IL6 (30 ng/ml) or IFN gamma (100 ng/ml). Cells were rendered into single cell suspension and stained with phycoerythryn (PE) conjugated antibodies against PD‐L1 (CD274). Conjugated antibodies were purchased from BDBioscience, human PD‐L1 (557924) and murine PD‐L1 (558091). After staining for 30 min at 4 °C, cells were washed three times and then resuspended in buffer containing DAPI (50 ng/mL) for viability. At least 10,000 events were collected using an LSR II (BD) and subsequently analyzed using FlowJo software.2.2.7.
2.7. Generation of stable knockdown clones
shRNA lentiviral transduction particles for murine HDAC6 (NM010413, TRCN0000008415), for human HDAC6 (NM00604, TRC0000004839) and non‐target shRNA (SHC002V) were obtained from Sigma Aldrich (St. Louis, MO). Transductions were performed according to the manufacturer's instructions. Melanoma cells were grown in antibiotic‐free medium and transduced with shRNA particles in the presence of hexadimethrine bromide. After 72 h, puromycin was added to the culture media and cells were cultured until 50% confluence and analyzed for HDAC6 expression. Monoclonal populations were generated by serial dilutions of polyclonal populations. Multiple colonies were selected and tested to ensure the reproducibility of effects from knocking down HDAC6 and not an effect of individual clones.
2.8. Protein over‐expression experiments
Over‐expression of HDAC6‐flag and STAT3C‐flag and respective control vectors were performed in WM164 human melanoma cell line. Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. The cells were transfected with plasmids for 24 h, followed by 24 h stimulation with the respective cytokine cells expressing each protein variant were treated for their further analysis; lysed and immunoblotted or the cells were collected for flow cytometer analysis.
2.9. Reagents and plasmids
The recombinant cytokines human IL‐16 (570804), human IFNgamma (570204), mouse IL‐6 (575704), mouse IFNgamma (575304) were purchased from Biolegend. Lipofectamine 2000 was purchased from Invitrogen. The over‐expression plasmid HDAC6‐flag were kindly provided by Dr. Zhang's lab at University of South Florida, and the STAT3C‐flag plasmid were obtained from Addgene (24983).
2.10. Quantitative real‐time RT‐PCR
Melanoma cells were plated at 2 × 106 cells per 35 mm well and cultured under conditions detailed above. The total RNA was extracted from cells using TRIzol® as per manufacturer's instructions (Invitrogen, NY). Briefly, cells were completely homogenized in TRIzol® reagent by pipetting and incubation for 5 min at room temperature. Samples were then processed immediately or stored at −80 °C. RNA was extracted by chloroform and RNA precipitated from the aqueous phase using isopropyl alcohol. The RNA pellet was then washed with 75% ethanol, air dried, and suspended in DEPC‐treated water. RNA was quantified spectrophotometrically using a Nanodrop. 260/280 and 260/230 ratios were routinely over 1.8. The cDNA was produced using iScript cDNA synthesis kit (Bio‐Rad, Hercules, CA). Target mRNA was quantified using MyIQ single color real time PCR detection system (Bio‐Rad) and iQ SYBR green Supermix (Bio‐Rad, Hercules, CA) using previously assayed parameters (Cheng et al., 2014, 2014). Primers used CDKN1A, IL‐10, FOS and PD‐L1 for qRT‐PCR were purchased from Qiagen and the cycling conditions were used as per manufacturer's instructions. Single product amplification was confirmed by melting curve analysis and primer efficiency was near 100% in all the experiments performed. Quantification is expressed in arbitrary units and target mRNA levels were normalized to GAPDH expression using the method of Pfaffl (Pfaffl, 2001).
2.11. In vivo studies
Mice received 1.3 × 105/mouse B16 tumor cells B16‐F10‐Luc subcutaneously in a shaved rear flank. Once the tumor was barely palpable Nexturastat B was injected daily intraperitoneally at a dose of 25 mg/kg. Tumor growth was monitored every 2 days in individually tagged mice by measuring the longest diameter (length) and the orthogonal diameter (width) with calipers. Mice were euthanized when the tumor volume reached 4000 mm3 (length × width × width/2). Results are presented as the mean tumor size (volume in mm3) and standard deviation for every treatment group at various time points until the termination of the experiment.
Chromatin Immunoprecipitation (ChIP) assays. ChIP studies were performed as previously described (Cheng et al., 2014a). All steps were carried out at 4 °C using WM164 melanoma cells: wild type, non target, KDHDAC6 or KDSTAT3. The DNA was recovered using Qiagen (51304) Quiaquick™ columns.
The antibodies used for CHIP experiments were obtained as follows. Anti‐HDAC6 (07‐732), anti‐STAT3 (06‐596), anti‐Acetyl‐Histone3 (06‐599) were purchased from Millipore. The anti‐RNA Polymerase II CDT (ab817) was obtained from Abcam. The 14 different sets of primers used for the PD‐L1 promoter analysis are described in Table 1.
Table 1.
Primers used for chromatin immunoprecipitation.
| Primer name | Sequence 5′ 3′ | Location in PD‐L1 promoter |
|---|---|---|
| A‐FW | TGTGGATTTGCTTTAATCTTC | UTR |
| A‐RV | AGGTATCTAGTGTTGGTGTCCT | |
| B‐FW | CATTTCTATACACAGCTTTATTCC | UTR |
| B‐RV | CAAGGCAGCAAATCCA | |
| C‐FW | TGTTTCACTTTCTGTTTCATTT | −193/−82 |
| C‐RV | GTTGGACTTTCCTGACCTT | |
| D‐FW | TCAGATGTTGGCTTGTTGT | −312/−192 |
| D‐RV | CATGTCAGTCCAGTTTTCTTG | |
| E‐FW | GCTGCTGACTTTTTATATGTTG | −469/−312 |
| E‐RV | ACGCACCTTGATTTTACCT | |
| F‐FW | AAAGATGTAGCTCGGGATG | −783/−632 |
| F‐RV | GTGTGTGTGTGTATGGGTGT | |
| G‐FW | ACACCATCGTCTGTCATCTT | −959/−832 |
| G‐RV | CATCCCGAGCTACATCTTT | |
| H‐FW | CAACGAAGAGTCCAATTTCT | −1153/−956 |
| H‐RV | TTCTCGAACTCCTTGACCT | |
| I‐FW | CATGCTCCTGCCAAATC | −1267/−1123 |
| I‐RV | TTGTTTGCCTTTCCTTCTT | |
| J‐FW | ACAGTCACCAAAATTGCTCT | −1728/−1588 |
| J‐RV | CTGACACTGCCTTGATTTG | |
| K‐FW | CAAAAACAAAATACCCATCC | −1931/−1804 |
| K‐RV | TCAGAGCCATCTACCACTAAC | |
| L‐FW | TTGTATGGGAAAATGAATGG | −2035/−1861 |
| L‐RV | GATGGGAATTGAGGGTATTT | |
| M‐FW | CCCACTCATTAACCATCTGT | −2180/−2010 |
| M‐RV | GTGAGGATTCGTGTTTTTGT | |
| N‐FW | CATGAATAGGAAGTGGTGGT | −2449/−2271 |
| N‐RV | GCAAAACAGATGGTTAATGAG |
All samples and inputs were quantified using MyIQ single color real time PCR detection system (Bio‐Rad) and iQ SYBR green Supermix (Bio‐Rad). Single product amplification was confirmed by melting curve analysis and primer efficiency was near or close to 100% in all experiments performed. Quantification is expressed in arbitrary units indicating fold over non‐stimulation condition, and target sequence levels were normalized to the input signal using the method of Pfaffl (Pfaffl, 2001). All ChIP experiments were repeated twice starting from the crosslinking, and final quantitative real time PCR was done in triplicates.
2.12. Statistical analysis
All experiments were repeated at least two times unless indicated otherwise. Unpaired t‐tests were performed using Microsoft Excel Software (Microsoft, Redmond, WA) with significance at p < 0.05.
3. Results
3.1. HDAC6 modulates the activation of the STAT3 pathway
We recently reported the important role of HDAC6 in the regulation of the STAT3 pathway in antigen presenting cells (APC) (Cheng et al., 2014a). Although, the exact regulatory mechanism is not completely understood, it was observed that the pharmacological inhibition and genetic abrogation of HDAC6, impairs the phosphorylation of STAT3 and subsequently inactivates STAT3 target genes, including number of cytokines and immune‐related genes (Cheng et al., 2014a). Since, the deregulation of STAT3 has been described to be important in the pathogenesis of melanoma (Messina et al., 2008), we interrogated whether the effect of HDAC6 in the activation of STAT3 could be recapitulated in melanoma tumor cells. Using specific HDAC6 shRNA lentiviral constructs we created stable monoclonal cell lines lacking HDAC6 (HDAC6KD) in the human melanoma cell lines WM164, WM983A and WM793. Next, we evaluated the activation of the STAT3 pathway in these knock‐down cells in parallel to their respective homologous control cells transduced with non‐target shRNA (NT). As shown in Figure 1A, the absence of HDAC6 impaired the proper phosphorylation of the STAT3‐Tyr705 after IL‐6 stimulation, suggesting that the role of HDAC6 in the activation of STAT3 was not restricted to APCs, and may be a global regulatory mechanism modulating the activity of this pathway. In parallel, we evaluated the phosphorylation of STAT3‐Ser727, which has been described to be essential for enhanced activation of STAT3 (Sakaguchi et al., 2012), and an important post‐translational modification in the melanocytic lineage (Sakaguchi et al., 2012). Similarly, the phosphorylation of this residue was diminished in the absence of HDAC6. However, this occurred to a lesser extent when compared to Tyr705 (Figure 1A).
Figure 1.
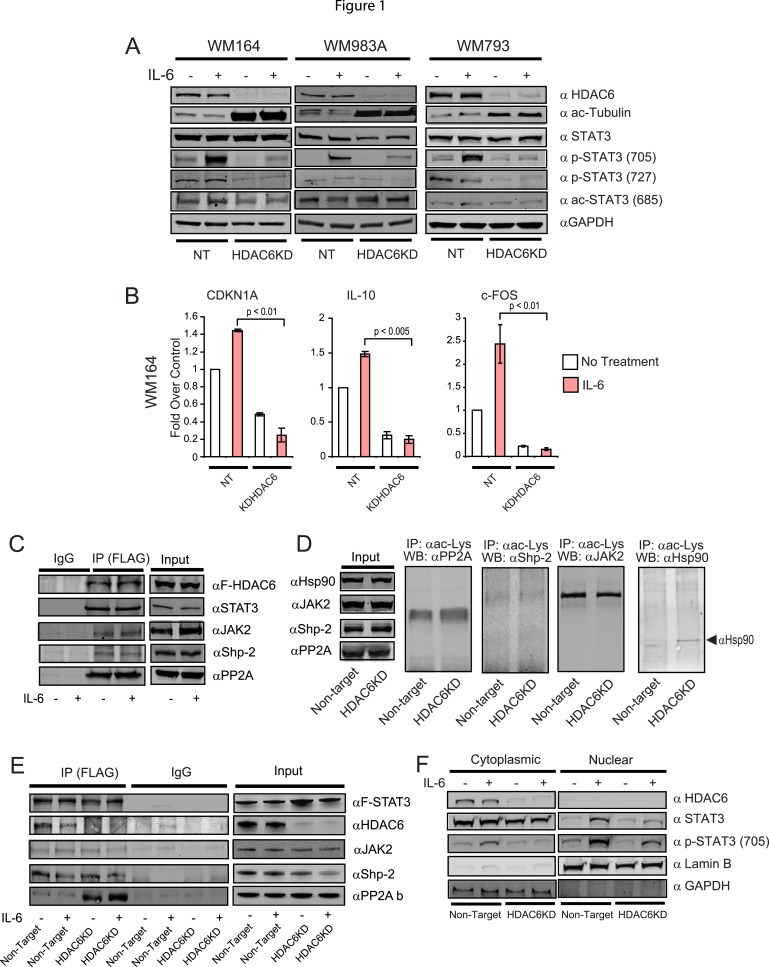
The absence of HDAC6 impairs the STAT3 activation in melanoma cells. (A) NT and HDAC6KD WM164, WM983A and WM793 melanoma cells were treated with IL‐6 (30 ng/mL) or left untreated. Then, the presence of HDAC6, acetylated tubulin, STAT3, P‐STAT3Y705, P‐STAT3‐S727, acetylated STAT3 and GAPDH was evaluated by immunoblot. (B) Total RNA was isolated from NT and HDAC6KD WM164 melanoma treated with IL‐6 (30 ng/mL) or left untreated. Next, the expression of CDKN1A, IL‐10 and FOS was analyzed by quantitative real‐time RT‐PCR (qRT‐PCR). The results are expressed as a percent over control cells, and data normalized by GAPDH expression. This experiment was performed three times with similar results. Error bars represent standard deviation from triplicates. (C) HDAC6‐flag plasmid was transfected in WM164 cells. Cellular extracts were subjected to immunoprecipitation against flag. The immunoprecipitated fraction was then assayed for the presence of Flag, STAT3, JAK2, Shp‐2 and PP2A using specific antibodies. (D) Total acetylated proteins were immunoprecipitated from NT and HDAC6KD WM164 cells. The immunoprecipitated fraction was then assayed for the presence of PP2A, Shp‐2Jak2 and Hsp90. (E) A construct carrying Flag‐STAT3 was transfected in WM164 cells. Then, cellular extracts were subjected to immunoprecipitation against Flag. Next, the immunoprecipitated fraction was assayed for the presence of Flag, HDAC6, JAk2, Shp‐2 and PP2A using specific antibodies. (F) Nuclear and cytoplasmic fractions were isolated from NT and HDAC6KD WM164 cells and immunoblotted against HDAC6, STAT3, p‐STAT3 Y705, Lamin B, GAPDH.
Another post‐translational modification reported as important in the regulation of STAT3 is the acetylation of Lys685 (Ray et al., 2008; Z‐lGuan et al., 2005). Therefore, we tested the possible participation of HDAC6 in this process by analyzing the acetylation status of this particular residue. However, we did not find major changes when comparing HDAC6KD against their NT control cell lines (Figure 1A, anti ac‐STAT3). To further study the consequences of the inactivation of the STAT3 pathway in the absence of HDAC6, we evaluated the expression of well‐characterized STAT3 target genes (Cheng et al., 2014a) in melanoma cell lines. As shown in Figure 1B, the expression of CDKN1A, IL‐10 and c‐FOS was significantly reduced in the HDAC6KD cell lines when compared with their respective NT controls. Importantly, these differences were more evident in the IL‐6 stimulated condition, suggesting that the effect of HDAC6 over the expression of these genes is mainly due to the inactivation of the STAT3 pathway.
3.2. Enhanced interaction of STAT3 and PP2A in the absence of HDAC6
We previously reported the interaction of HDAC6 with STAT3 in macrophages (Cheng et al., 2014a). Extending this previous observation, and using a similar experimental approach, we detected that this interaction occurred also in melanoma cells (Figure 1C, αSTAT3). However, our results indicate that despite this interaction, HDAC6 does not alter the acetylation status of STAT3 (Figure 1A, αac‐STAT3). Then, we explored other potential molecular mechanisms that might explain the participation of HDAC6 in the phosphorylation of STAT3. Our attention was directed to the identification of a possible interaction of HDAC6 with JAK2, Shp‐2 and PP2A, proteins directly involved in the homeostasis of phospho‐STAT3 (Murray, 2007; Yu et al., 2014). As shown in Figure 1C, we were able to detect a physical interaction of HDAC6 with these three candidates. Therefore, we next ventured to evaluate them as potential enzymatic targets for HDAC6. Since, there are no available antibodies for the acetylated variants of these proteins, our experimental approach consisted of the immunoprecipitation of all acetylated proteins from NT (control) and HDAC6KD cells and then evaluating for these specific targets in the immunoprecipitated fraction. As shown in Figure 1D, we did not find any changes in the acetylation of PP2A, Shp‐2 and JAK2 proteins in the absence of HDAC6. In order to verify this technique, we evaluated the acetylation of Hsp90, a known target for HDAC6 (Boyault et al., 2007), which was found to be hyperacetylated in the absence of HDAC6 (Figure 1D).
Another proposed regulatory mechanism affecting the activity of STAT3 is its ability to interact with PP2A. In this regard, it has been shown that histone deacetylase 3 (HDAC3) functions as a scaffold protein to enhance the interaction of STAT3 and PP2A, and subsequently down‐regulates the expression of STAT3 target genes (Togi et al., 2009). Considering this antecedent, and in order to evaluate the potential participation of HDAC6 in the stability of the protein complex STAT3‐PP2A, we performed a co‐immunoprecipitation of STAT3 in the presence or absence of HDAC6. The analysis of the immunoprecipitated fraction suggested that HDAC6 was not essential for the interaction of STAT3 with Jak2 and Shp‐2 (Figure 1E). However, we observed an enhanced interaction of STAT3 with PP2A in the absence of HDAC6 (Figure 1E, αPP2A), suggesting that the effect of HDAC6 in the phosphorylation of STAT3 could be mediated by this mechanism.
It has been demonstrated that STAT3 needs to be phosphorylated in order to allow its dimerization and subsequent translocation to the nucleus (Yu et al., 2014). Since our results indicate that HDAC6 is necessary for STAT3 phosphorylation, we hypothesized that the intracellular localization of STAT3 was also affected in the absence of HDAC6. To further explore this possibility, we analyzed nuclear and cytosolic extracts from NT and HDAC6KD melanoma cells previously stimulated with IL‐6. As expected, the presence of STAT3 in the nucleus increased after IL‐6 treatment in NT cells. However, in HDAC6KD cells we found minimal differences between the stimulated and non‐stimulated conditions (Figure 1F, nuclear panel).
3.3. HDAC6 is necessary for the cytokine‐mediated up‐regulation of PD‐L1 expression
STAT3 regulates critical functions in mammalian cells, such as proliferation, survival, differentiation, and immune response, among others (Yu et al., 2014). Constitutive active STAT3 has been observed in human melanoma cell lines and primary tumors, but not in matched normal skin specimens from the same patients (Niu et al., 2002a). In fact, it has been shown that hyperactivity of STAT3 deregulates the expression of several important immune‐related pathways, including those involved in the regulation of pro‐ and anti‐inflammatory pathways and anti‐tumor responses (Woan et al., 2012; Villagra et al., 2010; Yu et al., 2009). It has been reported that STAT3 is a potent activator of the co‐stimulatory molecule PD‐L1 in antigen presenting cells (APCs) (Wolfle et al., 2011) and in melanoma (Jiang et al., 2013). In order to validate these early observations we evaluated the expression of PD‐L1 in STAT3 knock‐down (STAT3KD) in the same parental melanoma cell lines that were used to generate the HDAC6KD monoclonal cell lines. As expected, the expression of PD‐L1 in NT control cells was enhanced upon IL‐6 stimulation. However, its expression was importantly diminished in the STAT3KD cells after IL‐6 stimulation (Figure 2A). Analysis of the PD‐L1 mRNA by qRT‐PCR confirmed that our observations were due to transcriptional regulation and discarded any potential post‐translational regulatory mechanism affecting the presence of the PD‐L1 (Figure 2B).
Figure 2.
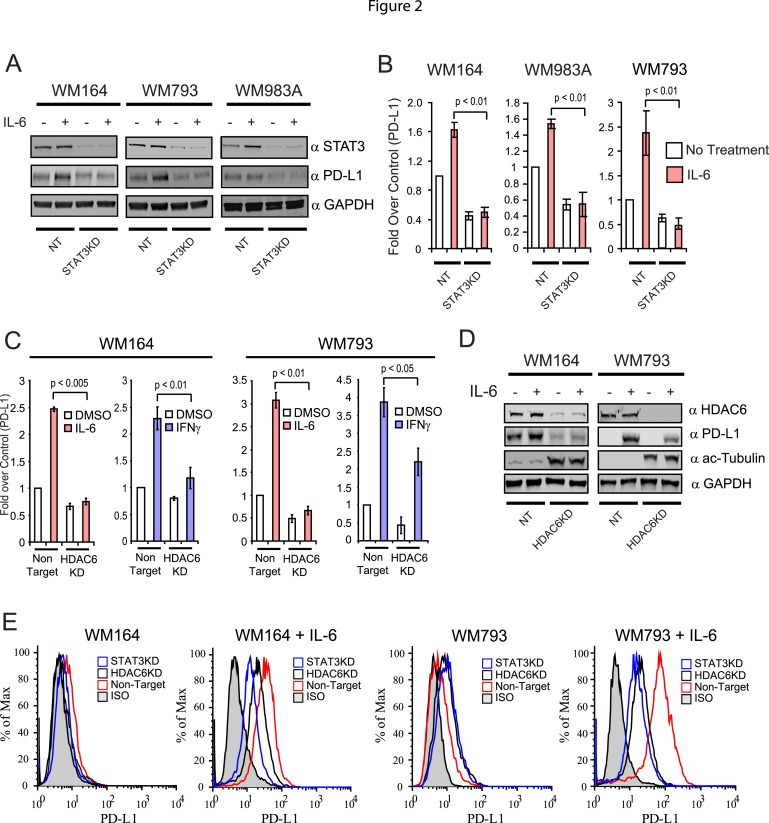
HDAC6 modulates the expression of PD‐L1 in melanoma cells. (A) NT and STAT3KD WM164, WM983A and WM793 melanoma cells were treated with IL‐6 (30 ng/mL) or left untreated. The presence of STAT3, PD‐L1 and GAPDH was evaluated by immunoblot. (B) Total RNA was isolated from NT and STAT3KD WM164 melanoma treated with IL‐6 (30 ng/mL) or left untreated. Then, the expression of PD‐L1 was analyzed by quantitative qRT‐PCR. The results are expressed as a percent over control cells, and data normalized by GAPDH expression. This experiment was performed three times with similar results. Error bars represent standard deviation from triplicates. (C) Total RNA was isolated from NT and HDAC6KD WM164 and WM793 melanoma cells treated with IL‐6 (30 ng/mL), IFNγ (100 ng/mL) or untreated. The expression of PD‐L1 was analyzed by quantitative qRT‐PCR. These results are expressed as a percent over control cells, and data normalized by GAPDH expression. This experiment was performed three times with similar results. Error bars represent standard deviation from triplicates. (D) Immunoblotting analysis of PD‐L1, acetylated tubulin and GAPDH proteins in NT and HDAC6KD WM164 and WM793 melanoma cells under stimulation of IL‐6 (30 ng/mL). (E) Expression of PD‐L1 was measured by flow cytometry in NT, HDAC6KD and STAT3KD melanoma cell lines with or without stimulation of IL‐6 (30 ng/mL).
Our results also suggest that HDAC6 is absolutely necessary for the phosphorylation of STAT3. Consequently, we expected that HDAC6 might also indirectly control the expression of PD‐L1 by modulating the activation of STAT3. In order to test this hypothesis, we evaluated the expression of PD‐L1 mRNA in HDAC6KD melanoma cells stimulated with either IL‐6 or IFNγ. As shown in Figure 2C, after cytokine stimulation, the expression of PD‐L1 increased between 2.5 and 4 times over the non‐stimulated condition assigned as value 1.0 on each chart. While the absence of HDAC6 slightly diminished the expression of PD‐L1 in the non‐cytokine treatment, we observed an important abrogation in its production after the stimulation with both cytokines. However, this effect was more pronounced in the IL‐6 treatment when compared to the IFNγ stimulation, which has been reported previously to be a potent activator of PD‐L1 expression in professional antigen presenting cells (APCs) (Loke and Allison, 2003). To further study whether the diminished expression of PD‐L1 mRNA was also affecting the protein levels of this co‐stimulatory molecule, we evaluated the total amount of PD‐L1, which was found to be also substantially diminished in HDAC6KD cell lines after stimulation with IL‐6 (Figure 2D). Likewise, changes in PD‐L1 protein were similar when we evaluated the expression of surface PD‐L1 on HDAC6KD melanoma cells by flow cytometry (Figure 2E, HDAC6KD). In parallel, we evaluated the presence of surface PD‐L1 in STAT3KD melanoma cells, demonstrating a similar decrease of its presence when compared to the HDAC6KD cells (Figure 2E, STAT3KD).
3.4. The recruitment of STAT3 to the PD‐L1 promoter precedes the transcriptional activation of PD‐L1
The initial work describing the binding of STAT3 to the PD‐L1 promoter in APCs only evaluated the recruitment of STAT3 at the −200 bp PD‐L1 proximal promoter region (Wolfle et al., 2011). After performing a manual search of consensus binding sites for STAT3 in the PD‐L1 promoter (Ehret et al., 2001), we identified several other potential binding sites along the first 2.5 kbp of the promoter region (−578, −1113, −1812, −1929, −2182 and −2429 bp) (Figure 3A). This analysis suggested that STAT3 could be a regulator of the expression of PD‐L1 through multiple response elements on its promoter. Additionally, the regulation of PD‐L1 in melanoma could obey different molecular mechanisms in comparison to those encountered in APCs, such as differential presence/activation of cellular pathways and/or intrinsic variations in the chromatin accessibility. Therefore, we decided to evaluate the recruitment of STAT3 to all these potential binding sites at the PD‐L1 promoter in the human melanoma cell line WM164.
Figure 3.
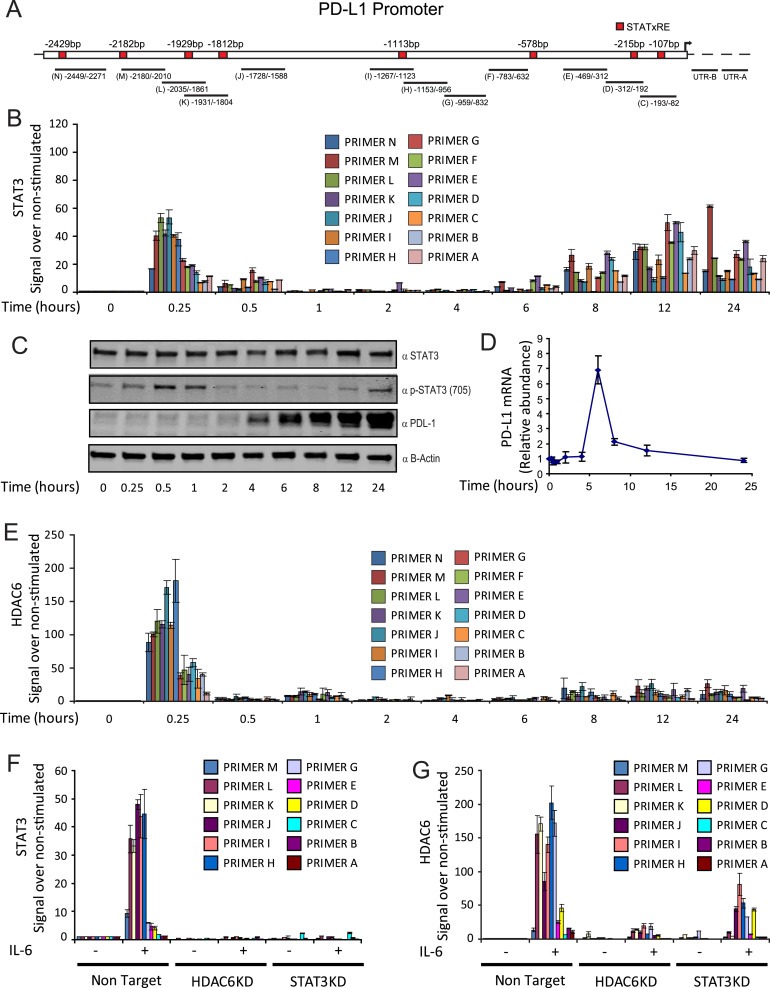
HDAC6 and STAT3 are recruited to the PD‐L1 promoter. (A) Schematic diagram of the PD‐L1 promoter showing the potential STAT3 binding sites and primers used in the ChiP assays. ChiP analysis of WM164 wild type melanoma cell line stimulated with IL‐6 (30 ng/mL) and collected at baseline (time 0) or at 0.25, 0.5, 1, 2, 4, 6, 8, 12 and 24 h after stimulation, assessed with HDAC6 (B) and STAT3 (E) antibodies, followed by quantitative real‐time PCR analysis of the 14 different primers described above (detailed in Table 1). (C) WM164 cells were treated with IL‐6 (30 ng/mL) and then evaluated by immunoblotting for the presence of STAT3, pSTAT3, PD‐L1 and b‐Actin. (D) Following the same conditions and time points from the previous experiment, RNA was isolated and the expression of PD‐L1 analyzed by qRT‐PCR. (E) ChiP analysis of WM164 cells following the same conditions and time points described in experiment (B). NT, HDAC6KD, STAT3KD WM164 cells with or without stimulation of IL‐6 were subjected to ChIP and the presence of STAT3 (F) and HDAC6 (G) was evaluated after 15 min of IL‐6 stimulation. The values obtained from all ChiP experiments were analyzed using the PfaffI method (Pfaffl, 2001) and are presented relative to input before immunoprecipitation. Data presented is from one representative experiment out of two independent experiments with similar results (error bars, s.d. of triplicates).
Of note, the kinetics for the recruitment of STAT3 to the PD‐L1 promoter has not been explored previously. Therefore, we performed chromatin immunoprecipitation (ChIP) of STAT3 at several time points after IL‐6 stimulation, starting at 15 min up to 24 h. As shown in Figure 3B, we observed a peak in STAT3 recruitment at 15 min, followed by a decline in its detection at 4 h, reaching similar levels to the non‐stimulated condition (time zero). Interestingly, we found a new recruitment event starting at 6 h, and a sustained slight increase up to 24 h post‐stimulation. In a parallel experiment, we determined that the phosphorylation of STAT3 occurs shortly after IL‐6 treatment, starting at 15 min and having a maximal peak at 1 h, suggesting that STAT3 is recruited to the PD‐L1 promoter as soon is phosphorylated and subsequently activated (Figure 3C, anti‐STAT3). We also measured the PD‐L1 protein production at the previously mentioned time points, detecting its expression right after the activation peak of STAT3 at 4 h post‐stimulation with IL‐6 (Figure 3C, anti‐PD‐L1). This pattern in the PD‐L1 production kinetics was further explored by the analysis of the PD‐L1 mRNA, which demonstrated a peak at around 6 h (Figure 3D). Additionally, we observed that the recruitment of activated Pol II (Supplementary Figure 1A) and hyperacetylation of histone 3 (ac‐H3) (Supplementary Figure 1B) occurred after the recruitment of STAT3 at 30 min and 2 h, respectively. These findings suggest that the recruitment of STAT3 is an early event necessary for the proper transcriptional activation of PD‐L1 expression. Another well characterized stimulator of PD‐L1 production is IFNγ (Supplementary Figure 1C). However, we observed that this stimulus occurred after 12 h post‐stimulation, supporting an indirect mechanism mediated by this cytokine (Supplementary Figure 1D).
3.5. The recruitment of STAT3 to the PD‐L1 promoter is impaired in the absence of HDAC6
We demonstrated previously that the interaction of HDAC6 with STAT3 in APCs is necessary for the STAT3 recruitment to the IL‐10 promoter (Cheng et al., 2014a). Therefore, we hypothesized that HDAC6 might be also recruited to the PD‐L1 promoter in melanoma. Following the same conditions described for STAT3 in Figure 3B, we performed ChIP for HDAC6, confirming that this deacetylase was recruited to the PD‐L1 promoter at the same time as STAT3 (Figure 3E). Remarkably, the pattern of recruitment along the 2500 bp of the promoter was similar to STAT3, as we observed an enhanced recruitment between the −956 bp to −2429 bp regions.
In order to further study, the relationship and interdependence of STAT3 and HDAC6, we evaluated their recruitment to the PD‐L1 promoter in the absence of each other. Briefly, as in the preceding experiments, we used the human melanoma cell line WM164, which was subjected to ChIP against STAT3 at 15 min post‐stimulation with IL‐6. As shown in Figure 3F, STAT3 was recruited to the PD‐L1 promoter in NT control cells following the same pattern observed in Figure 2B. However, STAT3 was not detected at the PD‐L1 promoter in the absence of HDAC6, implying that HDAC6 is required for its recruitment. Conversely, the same experiment was repeated in STAT3KD cells, but this time we evaluated the presence of HDAC6 at the PD‐L1 promoter. As shown in Figure 3G, the recruitment of HDAC6 was not completely impaired in the absence of STAT3, suggesting that HDAC6 could be a partner for another transcription factor involved in the regulation of PD‐L1.
3.6. PD‐L1 expression is rescued upon constitutive STAT3 activation in HDAC6KD melanoma cells
Next, we wanted to know whether the effect of HDAC6 over the expression of PD‐L1 was a consequence of its regulatory role over the STAT3 activation or mediated by another cellular event. In order to answer this question, we over‐expressed a constitutively active variant of STAT3 (STAT3c) which activates its target genes in the absence of cytokine stimulation and independently of the activation status of the upstream STAT3 pathway (Niu et al., 2002b). As expected, in the absence of either STAT3 or HDAC6, we observed a decreased presence of PD‐L1 post IL‐6 stimulation (Figure 4A). However, the presence of PD‐L1 was rescued after the over‐expression of STAT3C in all tested conditions, NT, HDAC6KD and STAT3KD (Figure 4B), demonstrating that STAT3 is a downstream target in the inhibitory effect observed in the absence of HDAC6. Similar results were also achieved after IFNγ stimulation (Supplementary Figure 1E and F).
Figure 4.
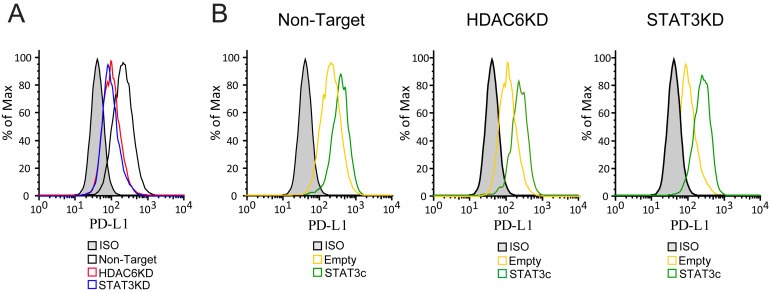
Constitutive active STAT3 rescue PD‐L1 expression in the absence of HDAC6. NT, HDAC6KD and STAT3KD WM164 melanoma cells were transfected with either STAT3C‐flag plasmid (B) or empty vector (A) and then stimulated with IL‐6 (30 ng/mL) or left untreated. Then, PD‐L1 was evaluated by flow cytometry.
3.7. HDAC6 selective inhibitors recapitulate the effect of HDAC6 knock‐down over the production of PD‐L1
Acknowledging the requirement of HDAC6 for the proper activation of the STAT3 pathway and PD‐L1 expression in melanoma cells, we analyzed whether the enzymatic activity of this deacetylase is responsible for the observed outcome. In order to test this hypothesis, we used the selective HDAC6 inhibitor Tubastatin A in melanoma cells. First, we evaluated the acetylation status of tubulin, which has been described as a quantifiable target to evaluate HDAC6 activity (Hubbert et al., 2002). As expected, we observed a strong induction of acetyl‐tubulin in all human and mouse melanoma cell lines tested (Figure 5A, ac‐Tubulin), confirming the pharmacological inhibition of HDAC6 under these conditions. The subsequent evaluation of STAT3 phosphorylation and PD‐L1 production revealed that the enzymatic inhibition of HDAC6 mirrored the results previously found with the HDAC6 knock‐down (Figure 5A, pSTAT3‐Y705 and PD‐L1). A reduced presence of PD‐L1 on the surface of melanoma cells was also observed by flow cytometry, confirming the previous results (Figure 5B). In order to determine if the effect mediated by HDAC6 inhibitors was also occurring with other non‐selective inhibitors we evaluated the expression of PD‐L1 in melanoma cells treated with the pan‐HDACi LBH589 and the class I HDAC inhibitor MGCD0103. As shown in the Supplementary Figure 2A and 2B, both inhibitors enhanced the expression of PD‐L1 in melanoma cells, suggesting that these inhibitors might regulate PD‐L1 expression through other molecular mechanisms. It has been reported that class I HDACs selectively deacetylate STAT3‐Lys685, inactivating the STAT3 pathway. Therefore, we hypothesized that the enzymatic inhibition of these HDACs could enhance PD‐L1 production by increasing the acetylation status of STAT3. In order to test this possibility, we evaluated the activation of STAT3 after treatment with LBH589 and MGCD0103 in WM164 melanoma cells. As expected, both HDACi increased the production of PD‐L1 in a dose dependent fashion (Supplementary Figure 2C and 2D, αPD‐L1). Additionally, we observed that the acetylation status of STAT3 increased after treatment with HDACi, having a pronounced effect at a high drug concentration (Supplementary Figure 2C and 2D, αac‐STAT3 685).
Figure 5.
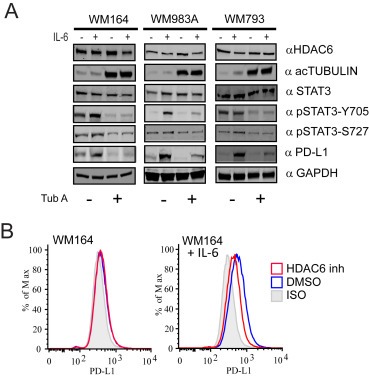
Selective HDAC6 inhibitors down‐regulate PD‐L1 expression in vitro. (A) WM164, WM983A and WM793 human melanoma cell lines were incubated with the HDAC6 inhibitor Tubastatin A (3 μM) for 24 h, followed for IL‐6 stimulation (30 ng/mL). Then, the cells were lyzed and immunoblotted using the specific antibodies listed in figure. (B) WM164 cells were treated with Tubastatin A (3 μM), with or without stimulation of IL‐6 and then the expression of PD‐L1 was analyzed by flow cytometry.
3.8. HDAC6 inhibition reduces tumor growth and PD‐L1 production in vivo
Given the potent effect of HDAC6 disruption over the production of PD‐L1 in vitro, we asked whether this finding would translate in vivo. As previously reported by our group (Cheng et al., 2014a), we found an important delay in B16‐F10 mouse melanoma tumor growth in mice treated with the HDAC6 inhibitors Nexturastat A (Figure 6A) or Tubastatin A (Figure 6B). Additionally, and as we hypothesized, the drug treatment impaired the phosphorylation of STAT3 and PD‐L1 production in all tumors isolated at the end point (Figure 6C), closely resembling the abrogation induced after the in vitro genetic or pharmacological inhibition of HDAC6 in B16‐F10 cells. In further analysis we treated cells obtained from melanoma patient biopsies. As shown in Figure 6D, the treatment with the selective HDAC6i Nexturastat diminished the STAT3 phosphorylation and the production of PD‐L1, demonstrating that this effect was not restricted to cell lines or mouse models.
Figure 6.
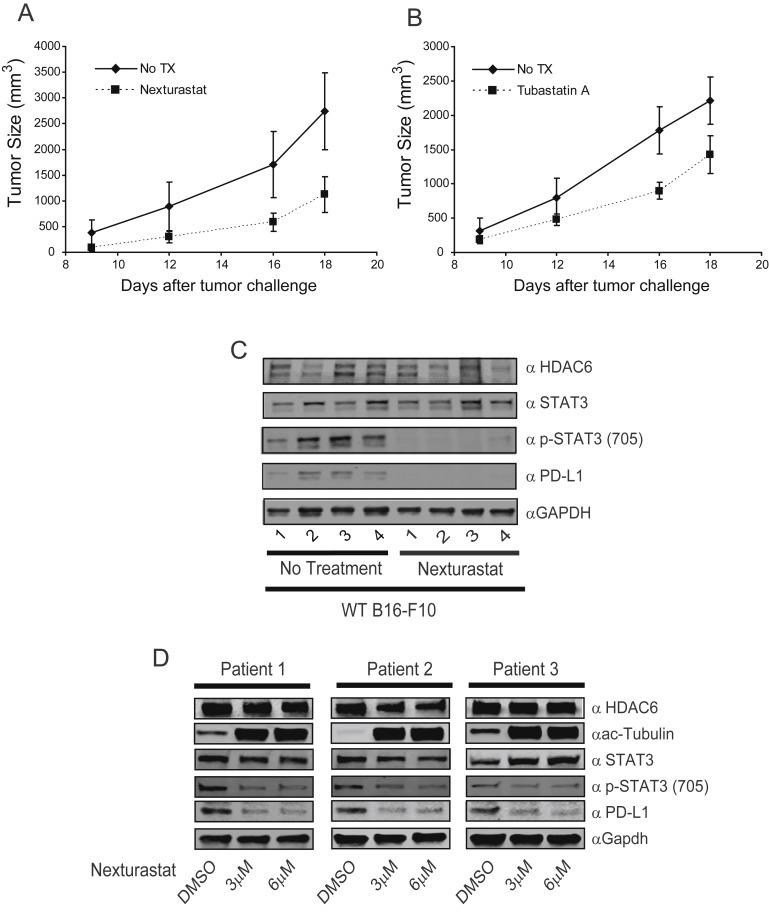
Selective HDAC6 inhibitors down‐regulate PD‐L1 expression in vivo. In vivo tumor growth of C57BL/6 mice injected subcutaneously with B16‐F10‐luc WT cells. Mice were treated by intraperitoneal injection daily with the HDAC6 inhibitors Nexturastat 25 mg/kg (A) and Tubastatin A 25 mg/kg (B). (C) Tumors were collected at the end point from C57BL mice either treated with Nexturastat or non‐treated, and the presence of HDAC6, STAT3, pSTAT3, PD‐L1 and GAPDH was evaluated by immunoblot. (D) Cells from melanoma patient's biopsies were treated or untreated with Nexturastat (3 μM and 6 μM) and then the presence of HDAC6, STAT3, pSTAT3, PD‐L1 and GAPDH was evaluated by immunoblot.
3.9. The HDAC6 abrogation controls multiple immune‐related pathways and check‐point molecules
In addition to the effect of HDAC6 inhibition over the expression of PD‐L1, we previously observed that the genetic and pharmacological inhibition of HDAC6 induced the expression of MHC class I and several tumor associated antigens such as gp100, MART1, TYRP1 and TYRP2 (Woan et al., 2015). Therefore, we hypothesized that the overall effect of the HDAC6 manipulation could be the consequence of multiple pathways affecting tumor growth, immune surveillance and anti‐tumor immune responses.
To investigate the participation of HDAC6 in the regulation of other anti‐tumor response mechanisms, we evaluated the expression of several inhibitory check‐point molecules and other regulatory tumor receptors in the absence of HDAC6. As shown in Figure 7A, the expression of total PD‐L2, B7‐H4 (inhibitory check‐point molecule VTCN1) and TRAIL‐R1 were importantly diminished in the WM164 HDAC6KD cells, while B7‐H3 (inhibitory check‐point molecule CD276), Galectin‐9 and TRAIL‐R2 were just slightly modified. In a parallel flow cytometry experiment we confirmed that the presence of PD‐L2, B7‐H4 and TRAIL‐R1 in the cell surface was also affected (Figure 7B). In further experiments, we assessed the expression of the aforementioned surface proteins in primary melanoma cells isolated from patient biopsies. As expected, the expression of PD‐L2, B7‐H3 and TRAIL‐R1 was diminished, while the other proteins were modified in a lesser magnitude, with the exception of B7‐H4 that was strongly down‐regulated in the patient 3 (Figure 7C).
Figure 7.
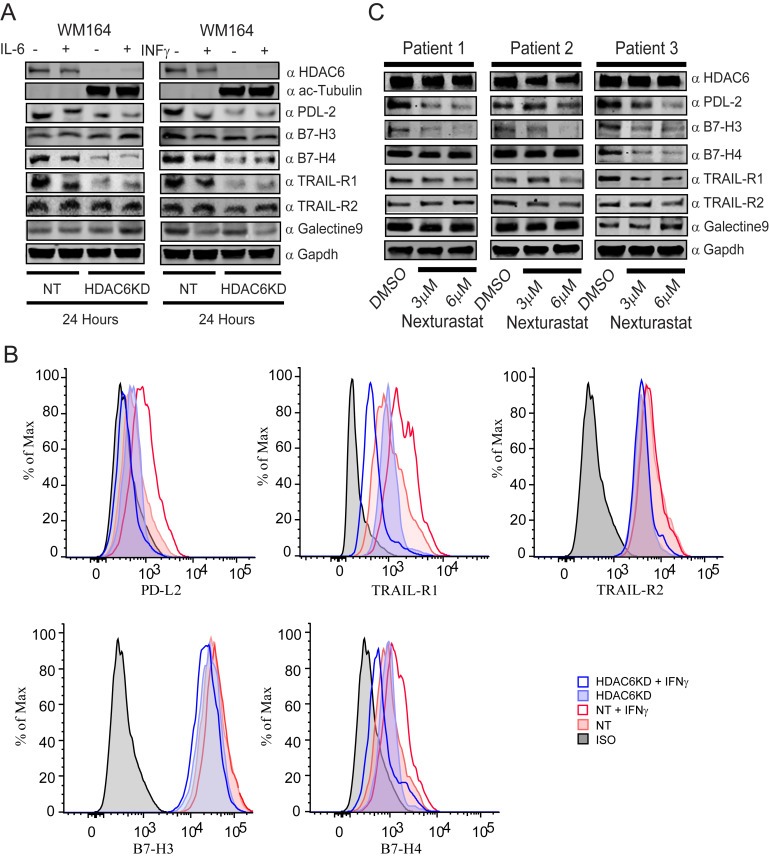
Manipulation of HDAC6 modulates the expression of membrane co‐stimulatory molecules and other regulatory proteins. NT and HDAC6KD WM164 human melanoma cells were stimulated with IFNγ (100 ng/ml) or left untreated. Next, the presence of PD‐L2, TRAIL‐R1, TRAIL‐R2, B7‐H3 and B7‐H4 were measured by western blot (A) and flow cytometry (B). (C) Cells from melanoma patient's biopsies were treated or untreated with Nexturastat (3 μM and 6 μM) and then the presence of HDAC6, PD‐L2, B7‐H3, B7‐H4, TRAIL‐R1, TRAIL‐R2, Galectin 9 and GAPDH was evaluated by immunoblot.
4. Discussion
Recent clinical trials using PD‐1 blocking antibodies have demonstrated promising results in several malignancies. However, not all patients respond equally to this treatment, and the reported objective response rate (ORR) is 26% for patients diagnosed with advanced melanoma (Robert et al., 2014), and 40% for patients diagnosed with wild type BRAF melanoma (Robert et al., 2015). Additionally, a slight improvement in the ORR has been found when using combined checkpoint blockade agents, as reported for the combination of anti PD‐1 and anti‐CTLA‐4 blocking antibodies, where the ORR increased to 40% (Callahan et al., 2014). As a result, there is a special interest to identify new potential therapeutic options and/or adjuvants targeting multiple cellular processes, thereby improving immunotherapeutic options in melanoma treatment. Some HDACs have captured special attention due to their recently assigned roles as modulators of oncogenesis and the immune response (Woan et al., 2012; Villagra et al., 2010; Kroesen et al., 2014). Among them, the genetic and/or pharmacological inhibition of HDAC6 has been reported to inhibit tumor growth in vitro and in vivo and control the expression of tumor associated antigens and MHC class I in melanoma (Woan et al., 2015). Additionally, HDAC6 is an essential regulator in the activation of the pro‐oncogenic STAT3 pathway, and we previously demonstrated that selective inhibitors for this deacetylase effectively down‐regulate the expression of STAT3 target genes, such as IL‐10 (Cheng et al., 2014a). These findings, along with the important known role of STAT3 hyperactivation in melanoma (Yu et al., 2009), encouraged us to further study the potential involvement of HDAC6 in the regulation of STAT3 target genes in this particular cancer. Our initial characterization focused in proving that HDAC6 was also modulating STAT3 Tyr‐705 phosphorylation in melanoma, which was evaluated across several human cell lines (Figure 1A). Additionally, we found that the abrogation of HDAC6 impaired the proper phosphorylation of STAT3 Ser‐727, which was not found to be affected in the original work reporting HDAC6 as a modulator of STAT3 activation in APCs (Cheng et al., 2014a). Although, it was initially suggested that Ser‐727 phosphorylation is a secondary event after Tyr705 phosphorylation, and only required for the maximal transcriptional activity of STAT3 (Wen et al., 1995), it is largely accepted now that this specific phosphorylation is playing a more complex role in melanoma tumorigenesis (Sakaguchi et al., 2012). Acetylation of STAT3 is also a regulator of the STAT3 activity (Z‐lGuan et al., 2005). In this regard, previous reports did not find major changes in the acetylation status of STAT3 in the absence or inhibition of HDAC6 in macrophages (Cheng et al., 2014a), and similar results were obtained in the present work (Figure 1A, anti ac‐STAT3), suggesting that HDAC6 might be controlling the phosphorylation of STAT3 by indirect means. A potential alternative regulatory mechanism is the previously reported action of additional proteins affecting the interaction of STAT3 with the phosphatase PP2A (Togi et al., 2009). Our data clearly demonstrated that a possible explanation for the impaired phosphorylation of STAT3 in the absence of HDAC6 could be originated by the enhanced interaction of PP2A with STAT3, which in turn could facilitate the dephosphorylation of STAT3‐mediated by PP2A.
The significance of these findings is in line with several other reports positioning STAT3 as a tolerogenic pathway influencing both professional APCs and tumor cells to inhibit T‐cell function and evade immune recognition (Kortylewski et al., 2005). In addition to STAT3, several other cellular pathways have been reported to be involved in the regulation of PD‐L1, including those activated by IL‐6, IL‐10, IL‐4, GM‐CSF, TLRs, interferons and TNFα (Loke and Allison, 2003; Francisco et al., 2010; Yamazaki et al., 2002). Among them, IFNγ has been reported as a potent stimulator of PD‐L1 production, a process believed to be mediated by the STAT1 pathway (Loke and Allison, 2003). Since most of the aforementioned observations have been made in immune cells, we evaluated whether PD‐L1 was also up‐regulated by IFNγ in melanoma cells. As shown in Figure 2C, the stimulation of human melanoma cells with IFNγ was closely comparable with the values achieved using IL‐6. In spite of that observation, both stimuli were able to induce the expression of PD‐L1 and we observed that the negative effect of HDAC6KD over PD‐L1 was more pronounced after IL‐6 stimulation, suggesting that HDAC6 was mainly regulating the STAT3 pathway and probably to a lesser magnitude STAT1, possibly by heterodimerization of STAT3 and STAT1 as demonstrated previously (Haan et al., 2005; Delgoffe and Vignali, 2013).
It has been reported that HDAC6 is recruited to regulatory sequences in gene promoters such as MYC (Toropainen et al., 2010), glucocorticoid receptor (Govindan, 2010) and estrogen receptor α‐inducible genes (Palijan et al., 2009). However, there is no evidence suggesting that HDAC6 directly affects the acetylation status of chromatin. In fact, the deacetylation of histones by HDAC6 has been demonstrated only by in vitro assays (Todd et al., 2010). The transcriptional regulatory effects observed for HDAC6 could be mediated by other regulatory factors recruited along with this deacetylase to specific DNA sequences. This hypothesis suggests that HDAC6 may be a regulator of the activation status of these transcription factors, perhaps by modulating their acetylation and/or modifying other essential regulators of these transcription factors. We have previously observed that HDAC6 and STAT3 are recruited to the same region of the IL‐10 promoter, and the recruitment of HDAC6 is impaired when cells are treated with the STAT3 inhibitor CPA‐7 (Cheng et al., 2014b). Moreover, the recruitment of STAT3 to the IL‐10 promoter diminishes considerably in HDAC6KD cells, suggesting that the down‐regulation of PD‐L1 expression in the absence or inhibition of HDAC6 could be part of a global regulatory mechanism affecting STAT3 target genes.
As mentioned before, the recruitment of STAT3 to the PD‐L1 promoter has been shown previously in APCs. However, this work only evaluated a very discrete region around the first 200 bp region of the PD‐L1 promoter. Our search for consensus potential STAT3 binding sites identified several regions along the first 2.5 kbp of the PD‐L1 promoter, which we demonstrated by analyzing the recruitment of this transcription factor to all the potential STAT3 binding sites using ChIP. Interestingly, these results indicate that STAT3 and HDAC6 were preferentially recruited to the far region comprised between −956 bp and −2429 bp, suggesting that this distal region could be responsible for the recruitment of STAT3. Needless to say, future experiments aiming towards the identification of single binding sequences are necessary for a more detailed characterization of the STAT3 recruitment to the PD‐L1 promoter.
In addition to the role of HDAC6 in the modulation of immune‐related pathways, it has also been reported that HDAC6KD melanoma cells have decreased proliferation when compared to their respective controls, and this effect is mainly mediated by G1 cell cycle arrest with minimal effects in apoptotic signaling (Woan et al., 2015). As mentioned before, selective HDAC6 inhibitors are currently available, making this deacetylase a very attractive target to pursue using small molecules as potential anticancer drugs. In this context, selective HDAC6 inhibitors, alone or in combination with other agents, are currently under evaluation in clinical trials, including the ongoing Phase 2 multiple myeloma clinical trial using the partially selective HDAC6 inhibitor ACY1215, which has shown important anti‐tumor activity in pre‐clinical studies (Santo et al., 2012). The in vivo results presented in this work suggest that HDAC6 inhibitors are also endowed with anti‐tumor activity in melanoma, and this effect could be the result of the modulation of multiple regulatory mechanisms involved in tumor survival and immune recognition, including the down‐regulation of PD‐L1, PD‐L2, B7‐H4 and TRAIL‐R1. In overall, our results provide a rational framework to consider the use of selective HDAC6 inhibitors as anti‐tumor agents in melanoma.
Disclosures
The authors have no financial conflict of interest.
Supporting information
The following is the supplementary data related to this article:
Figure S1. ChiP analysis of WM164 wild type melanoma cell line stimulated with IL‐6 (30 ng/mL) and collected at baseline (time 0) or at 0.25, 0.5, 1, 2, 4, 6, 8, 12 and 24 h after stimulation, assessed with PS5‐CTD Pol II (A) and acetyl H3 (B) antibodies, followed by quantitative real‐time PCR analysis of the 7 different primers described above (detailed in Table 1). (C) WM164 cells were treated with IFNγ, and then subjected to immunoblot to evaluate HDAC6, PD‐L1, ac‐Tubulin and GAPDH. (D) WM164 cells were treated with IFNγ, then total RNA was isolated and the expression of PD‐L1 analyzed by qRT‐PCR. NT, HDAC6KD and STAT3KD WM164 melanoma cells were transfected with either STAT3C‐flag plasmid (E) or empty vector (F) and then stimulated with IFNγ (100 ng/mL) or left untreated. Then, PD‐L1 was evaluated by flow cytometry.
Figure S2. WM164 cells were treated with LBH589 10 nM or 20 nM (A and C) or MGCD0103 1 μM or 2 μM (B and D) for 24 h and then stimulated with IL‐6 or left untreated. Then, the presence of HDAC6, STAT3, P‐STAT3 Y705, P‐STAT3 S727, acetylated STAT3, PD‐L1 and GAPDH was assessed by flow cytometry (A and B) and immunoblot (C and D).
Figure S3. (A) NT and HDAC6KD B16‐F10 murine melanoma cells were stimulated with IL‐6 (30 ng/ml) or left untreated. Next, the presence of HDAC6, acetylated tubulin, STAT3, P‐STAT3Y705, P‐STAT3‐S727, acetylated STAT3 and GAPDH was assessed by immunoblot. (B) B16‐F10‐luc melanoma cell line were treated with the HDAC6 inhibitor Nexturastat for 24 h and then stimulated with IL‐6 (30 ng/mL) or left untreated. The presence of HDAC6, acetylated tubulin, STAT3, P‐STAT3Y705, P‐STAT3‐S727, acetylated STAT3 and GAPDH was assessed by immunoblot.
Figure S4. NT and HDAC6KD WM164 human melanoma cells were stimulated with IL‐6 (30 ng/ml) or left untreated. Next, the presence of PD‐L2, TRAIL‐R1, TRAIL‐R2, B7‐H3 and B7‐H4 were measured by flow cytometry.
Acknowledgments
Funded by NIH R21 CA184612‐01 and Melanoma Research Foundation CDA Grant Award (AV, ID288760).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.12.012.
Lienlaf M., Perez-Villarroel P., Knox T., Pabon M., Sahakian E., Powers J., Woan K.V., Lee C., Cheng F., Deng S., Smalley K.S.M., Montecino M., Kozikowski A., Pinilla-Ibarz J., Sarnaik A., Seto E., Weber J., Sotomayor E.M., Villagra A., (2016), Essential role of HDAC6 in the regulation of PD‐L1 in melanoma, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.12.012.
References
- Boyault, C. , Zhang, Y. , Fritah, S. , Caron, C. , Gilquin, B. , Kwon, S.H. , 2007. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 21, 2172–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, M.K. , Postow, M.A. , Wolchok, J.D. , 2014. CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front. Oncol. 4, 385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F. , Lienlaf, M. , Wang, H.W. , Perez-Villarroel, P. , Lee, C. , Woan, K. , 2014. A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J. Immunol. 193, 2850–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F. , Lienlaf, M. , Perez-Villarroel, P. , Wang, H.W. , Lee, C. , Woan, K. , 2014. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol. Immunol. 60, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe, G.M. , Vignali, D.A. , 2013. STAT heterodimers in immunity: a mixed message or a unique signal?. Jak-Stat. 2, e23060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret, G.B. , Reichenbach, P. , Schindler, U. , Horvath, C.M. , Fritz, S. , Nabholz, M. , 2001. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J. Biol. Chem. 276, 6675–6688. [DOI] [PubMed] [Google Scholar]
- Francisco, L.M. , Sage, P.T. , Sharpe, A.H. , 2010. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghebeh, H. , Mohammed, S. , Al-Omair, A. , Qattan, A. , Lehe, C. , Al-Qudaihi, G. , 2006. The B7-H1 (PD-L1) T lymphocyte-inhibitory molecule is expressed in breast cancer patients with infiltrating ductal carcinoma: correlation with important high-risk prognostic factors. Neoplasia. 8, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindan, M.V. , 2010. Recruitment of cAMP-response element-binding protein and histone deacetylase has opposite effects on glucocorticoid receptor gene transcription. J. Biol. Chem. 285, 4489–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haan, S. , Keller, J.F. , Behrmann, I. , Heinrich, P.C. , Haan, C. , 2005. Multiple reasons for an inefficient STAT1 response upon IL-6-type cytokine stimulation. Cell Signal. 17, 1542–1550. [DOI] [PubMed] [Google Scholar]
- Hamanishi, J. , Mandai, M. , Iwasaki, M. , Okazaki, T. , Tanaka, Y. , Yamaguchi, K. , 2007. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. U. S. A. 104, 3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino, R. , Kabashima, K. , Kato, Y. , Yagi, H. , Nakamura, M. , Honjo, T. , 2010. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 116, 1757–1766. [DOI] [PubMed] [Google Scholar]
- Hodi, F.S. , O'Day, S.J. , McDermott, D.F. , Weber, R.W. , Sosman, J.A. , Haanen, J.B. , 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert, C. , Guardiola, A. , Shao, R. , Kawaguchi, Y. , Ito, A. , Nixon, A. , 2002. HDAC6 is a microtubule-associated deacetylase. Nature. 417, 455–458. [DOI] [PubMed] [Google Scholar]
- Jiang, X. , Zhou, J. , Giobbie-Hurder, A. , Wargo, J. , Hodi, F.S. , 2013. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin. Cancer Res. 19, 598–609. [DOI] [PubMed] [Google Scholar]
- Kortylewski, M. , Jove, R. , Yu, H. , 2005. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 24, 315–327. [DOI] [PubMed] [Google Scholar]
- Kroesen, M. , Gielen, P. , Brok, I.C. , Armandari, I. , Hoogerbrugge, P.M. , Adema, G.J. , 2014. HDAC inhibitors and immunotherapy; a double edged sword?. Oncotarget. 5, 6558–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loke, P. , Allison, J.P. , 2003. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. U. S. A. 100, 5336–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina, J.L. , Yu, H. , Riker, A.I. , Munster, P.N. , Jove, R.L. , Daud, A.I. , 2008. Activated stat-3 in melanoma. Cancer Control J. Moffitt Cancer Cent. 15, 196–201. [DOI] [PubMed] [Google Scholar]
- Murray, P.J. , 2007. The JAK-STAT signaling pathway: input and output integration. J. Immunol. 178, 2623–2629. [DOI] [PubMed] [Google Scholar]
- Nikolaou, V. , Stratigos, A.J. , 2014. Emerging trends in the epidemiology of melanoma. Br. J. Dermatol. 170, 11–19. [DOI] [PubMed] [Google Scholar]
- Niu, G. , Bowman, T. , Huang, M. , Shivers, S. , Reintgen, D. , Daud, A. , 2002. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene. 21, 7001–7010. [DOI] [PubMed] [Google Scholar]
- Niu, G. , Wright, K.L. , Huang, M. , Song, L. , Haura, E. , Turkson, J. , 2002. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 21, 8 [DOI] [PubMed] [Google Scholar]
- Palijan, A. , Fernandes, I. , Bastien, Y. , Tang, L. , Verway, M. , Kourelis, M. , 2009. Function of histone deacetylase 6 as a cofactor of nuclear receptor coregulator LCoR. J. Biol. Chem. 284, 30264–30274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll, D.M. , 2012. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl, M.W. , 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucl. Acids Res. 29, e45- [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, S. , Lee, C. , Hou, T. , Boldogh, I. , Brasier, A.R. , 2008. Requirement of histone deacetylase1 (HDAC1) in signal transducer and activator of transcription 3 (STAT3) nucleocytoplasmic distribution. Nucleic Acids Res. 36, 4510–4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert, C. , Ribas, A. , Wolchok, J.D. , Hodi, F.S. , Hamid, O. , Kefford, R. , 2014. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 384, 1109–1117. [DOI] [PubMed] [Google Scholar]
- Robert, C. , Long, G.V. , Brady, B. , Dutriaux, C. , Maio, M. , Mortier, L. , 2015. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 372, 320–330. [DOI] [PubMed] [Google Scholar]
- Sakaguchi, M. , Oka, M. , Iwasaki, T. , Fukami, Y. , Nishigori, C. , 2012. Role and regulation of STAT3 phosphorylation at Ser727 in melanocytes and melanoma cells. J. Invest Dermatol. 132, 1877–1885. [DOI] [PubMed] [Google Scholar]
- Santo, L. , Hideshima, T. , Kung, A.L. , Tseng, J.C. , Tamang, D. , Yang, M. , 2012. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 119, 2579–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube, J.M. , Klein, A. , Brahmer, J.R. , Xu, H. , Pan, X. , Kim, J.H. , 2014. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 20, 5064–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd, P.K. , Oh, S.Y. , Krans, A. , Pandey, U.B. , Di Prospero, N.A. , Min, K.-T. , 2010. Histone deacetylases suppress CGG repeat–induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. Plos Genet. 6, e1001240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togi, S. , Kamitani, S. , Kawakami, S. , Ikeda, O. , Muromoto, R. , Nanbo, A. , 2009. HDAC3 influences phosphorylation of STAT3 at serine 727 by interacting with PP2A. Biochem. Biophys. Res. Commun. 379, 616–620. [DOI] [PubMed] [Google Scholar]
- Tomasi, T.B. , Magner, W.J. , Khan, A.N. , 2006. Epigenetic regulation of immune escape genes in cancer. Cancer Immunol. Immunother. – CII. 55, 1159–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian, S.L. , Hodi, F.S. , Brahmer, J.R. , Gettinger, S.N. , Smith, D.C. , McDermott, D.F. , 2012. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toropainen, S. , Väisänen, S. , Heikkinen, S. , Carlberg, C. , 2010. The down-regulation of the human MYC gene by the nuclear hormone 1[alpha],25-dihydroxyvitamin D3 is associated with cycling of corepressors and histone deacetylases. J. Mol. Biol. 400, 284–294. [DOI] [PubMed] [Google Scholar]
- Villagra, A. , Sotomayor, E.M. , Seto, E. , 2010. Histone deacetylases and the immunological network: implications in cancer and inflammation. Oncogene. 29, 157–173. [DOI] [PubMed] [Google Scholar]
- Wen, Z. , Zhong, Z. , Darnell, J.E. , 1995. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 82, 241–250. [DOI] [PubMed] [Google Scholar]
- Woan, K.V. , Sahakian, E. , Sotomayor, E.M. , Seto, E. , Villagra, A. , 2012. Modulation of antigen-presenting cells by HDAC inhibitors: implications in autoimmunity and cancer. Immunol. Cell Biol. 90, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woan, K.V. , Lienlaf, M. , Perez-Villaroel, P. , Lee, C. , Cheng, F. , Knox, T. , 2015. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: enhanced antitumor immunity and impaired cell proliferation. Mol. Oncol. 9, 1447–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfle, S.J. , Strebovsky, J. , Bartz, H. , Sahr, A. , Arnold, C. , Kaiser, C. , 2011. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur. J. Immunol. 41, 413–424. [DOI] [PubMed] [Google Scholar]
- Wu, C. , Zhu, Y. , Jiang, J. , Zhao, J. , Zhang, X.G. , Xu, N. , 2006. Immunohistochemical localization of programmed death-1 ligand-1 (PD-L1) in gastric carcinoma and its clinical significance. Acta Histochem. 108, 19–24. [DOI] [PubMed] [Google Scholar]
- Yamazaki, T. , Akiba, H. , Iwai, H. , Matsuda, H. , Aoki, M. , Tanno, Y. , 2002. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 169, 5538–5545. [DOI] [PubMed] [Google Scholar]
- Yu, H. , Pardoll, D. , Jove, R. , 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 9, 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. , Lee, H. , Herrmann, A. , Buettner, R. , Jove, R. , 2014. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer. 14, 736–746. [DOI] [PubMed] [Google Scholar]
- Z-l, Yuan , Guan, Y.-j. , Chatterjee, D. , Chin, Y.E. , 2005. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 307, 269–273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Figure S1. ChiP analysis of WM164 wild type melanoma cell line stimulated with IL‐6 (30 ng/mL) and collected at baseline (time 0) or at 0.25, 0.5, 1, 2, 4, 6, 8, 12 and 24 h after stimulation, assessed with PS5‐CTD Pol II (A) and acetyl H3 (B) antibodies, followed by quantitative real‐time PCR analysis of the 7 different primers described above (detailed in Table 1). (C) WM164 cells were treated with IFNγ, and then subjected to immunoblot to evaluate HDAC6, PD‐L1, ac‐Tubulin and GAPDH. (D) WM164 cells were treated with IFNγ, then total RNA was isolated and the expression of PD‐L1 analyzed by qRT‐PCR. NT, HDAC6KD and STAT3KD WM164 melanoma cells were transfected with either STAT3C‐flag plasmid (E) or empty vector (F) and then stimulated with IFNγ (100 ng/mL) or left untreated. Then, PD‐L1 was evaluated by flow cytometry.
Figure S2. WM164 cells were treated with LBH589 10 nM or 20 nM (A and C) or MGCD0103 1 μM or 2 μM (B and D) for 24 h and then stimulated with IL‐6 or left untreated. Then, the presence of HDAC6, STAT3, P‐STAT3 Y705, P‐STAT3 S727, acetylated STAT3, PD‐L1 and GAPDH was assessed by flow cytometry (A and B) and immunoblot (C and D).
Figure S3. (A) NT and HDAC6KD B16‐F10 murine melanoma cells were stimulated with IL‐6 (30 ng/ml) or left untreated. Next, the presence of HDAC6, acetylated tubulin, STAT3, P‐STAT3Y705, P‐STAT3‐S727, acetylated STAT3 and GAPDH was assessed by immunoblot. (B) B16‐F10‐luc melanoma cell line were treated with the HDAC6 inhibitor Nexturastat for 24 h and then stimulated with IL‐6 (30 ng/mL) or left untreated. The presence of HDAC6, acetylated tubulin, STAT3, P‐STAT3Y705, P‐STAT3‐S727, acetylated STAT3 and GAPDH was assessed by immunoblot.
Figure S4. NT and HDAC6KD WM164 human melanoma cells were stimulated with IL‐6 (30 ng/ml) or left untreated. Next, the presence of PD‐L2, TRAIL‐R1, TRAIL‐R2, B7‐H3 and B7‐H4 were measured by flow cytometry.