

Abstract
Despite significant efforts to translate nanotechnology for cancer application, lack of identification of biodistribution/accumulation of these nanovehicles in vivo remains a substantial barrier for successful implementation of theranostic nanoparticles in the clinic. The purpose of the study was to develop a tumor targeted- theranostic nanovehicle for pancreatic cancer detectable by multispectral optoacoustic tomography (MSOT). To improve the tumor specificity of our mesoporous silica nanoparticle (MSN), we utilized a dual targeting strategy: 1) an elevated tumor receptor, urokinase plasminogen activator receptor (UPAR), and 2) the acidic tumor microenvironment. The tumor specificity of the MSN particle was improved with the addition of both chitosan, targeting acidic pH, and urokinase plasminogen activator (UPA), targeting UPAR. Drug release assays confirmed pH responsive release of gemcitabine in vitro. The UPAR specific binding of MSN-UPA nanoparticles was confirmed by reduction in fluorescence signal following MSN-UPA nanoparticle treatment in UPAR positive cells blocked with a UPAR-blocking antibody. Based upon indocyanine green encapsulation within the nanoparticles, UPA ligand targeted MSNs demonstrated increased intensity compared to untargeted MSNs at both pH 7.4 (7X) and 6.5 (20X); however the signal was much more pronounced at a pH of 6.5 using tissue phantoms (p<0.05). In vivo, MSN-UPA particles demonstrated orthotopic pancreatic tumor specific accumulation compared to liver or kidney as identified using multispectral optoacoustic tomography (p<0.05) and confirmed by ex vivo analysis. By tracking in vivo nanoparticle biodistribution with MSOT, it was shown that pH responsive, ligand targeted MSNs preferentially bind to pancreatic tumors for payload delivery.
Keywords: pancreatic cancer, nanomedicine multispectral optoacoustic tomography, mesoporous silica, theranostics
1. Introduction
Clinical outcomes are significantly worse in patients diagnosed with pancreatic cancer compared to other solid organ cancers [1, 2]. The majority (91%) of patients present with advanced disease, limiting the probability of long term survival with only 2% of patients alive 5 years from diagnosis [2]. The difficulties in both diagnosing and treating pancreatic cancer stem largely from constraints in current diagnostic imaging modalities, advanced clinical stage at presentation, and inadequate delivery of chemotherapeutic agents. Optoacoustic-based nanomedicine is an emerging modality that addresses some of these current shortcomings and may have significant clinical implications for the early diagnosis of pancreatic cancer and targeted drug delivery.
Multispectral optoacoustic tomography (MSOT) is an imaging tool that detects acoustic signals generated from the thermoelastic expansion of particles absorbing electromagnetic energy [3]. Because optoacoustic imaging is dependent on the detection of acoustic signals, which unlike photons do not scatter in tissues, sub-millimeter resolution can be obtained at clinically relevant depths [3-6]. Nanoparticles can be modified to act as a contrast agent to enhance MSOT imaging and also double as a targeted drug delivery vehicle. Mesoporous silica nanoparticles are ideal for this type of system because they are physically customizable, nontoxic, and safely carry drug in vivo [7-8]. The diameter and pore size can be manipulated during MSN creation. Both parameters must be considered for nanoparticle applications in drug delivery systems for two main reasons: if the particles are too large they are unable to pass through the vasculature to reach the desired target and pore size is important to control to provide optimal drug loading.
In vivo identification of nanoparticle accumulation and biodistribution is one of the most important aspects for translation of nanotechnology into the clinic [10-11]. Unfortunately, the majority of studies evaluating nanoparticle performance in the context of cancer generally inject the cancer cells subcutaneously [7-11]. These subcutaneous tumors are highly vascularized, tumors lack the proper tumor microenvironment, and often have little in common with cancer in human patients. While some studies have attempted to determine biodistribution of nanomaterials in vivo using NIR-fluorescent imaging and other conventional methods, these studies have largely been unsuccessful in locating accumulation of nanomaterials in orthotopic abdominal tumors or genetically modified organisms without sacrificing the animal [9-10]. Multispectral optoacoustic tomography offers a solution to this problem, allowing one to view nanoparticle biodistribution in real-time at depths required to detect abdominal tumors [5-6]. This imaging technique utilizes optical excitation of chromophores to induce vibration and produce a distinctive spectrum. Reconstruction of the various acoustic spectra allows for quantitative analysis of specific chromophore distribution [4-6]. The ability to track nanoparticles in this way is a critical step in the translation of nanoparticle based therapies into clinical environments, especially in the context of abdominal tumors.
Two characteristics of pancreatic cancer that can be exploited for the purposes of targeted therapy are the acidic tumor environment and overexpressed extracellular receptors [12-15]. It has been established that the pH in the local environment of pancreatic tumors is below physiologic levels at 6.5 [6,12,14]. This degree of acidity is attributable to the disorganized vasculature in tumors creating a hypoxic environment resulting in the cancer cells utilizing anaerobic metabolism resulting in the formation of acid metabolites [12]. The difference in pH between normal physiologic tissues and the tumor environment can be employed for the purposes of precise drug delivery to pancreatic tumors. Chitosan is a polysaccharide that is insoluble in neutral and basic pH, but in acidic environments it is protonated to a water soluble form [16]. Therefore, coating drug containing mesoporous silica nanoparticles with chitosan creates a pH responsive delivery vehicle.
As mentioned, another unique feature of pancreatic cancer cells is overexpression of urokinase plasminogen activator receptors. Urokinase plasminogen activator is a serine protease that likely plays a role in the development of pancreatic cancer invasion and subsequent metastases [17]. Normal pancreatic cells express very low levels of the UPAR; therefore, conjugation of UPA to a nanoparticle increases the specificity for pancreatic cancer cells [18]. Targeting the acidic tumor environment as well as UPA receptors should greatly increase the specificity of a nanoparticle for pancreatic cancer cells resulting improved diagnostic accuracy and drug delivery while sparing normal tissues.
Gemcitabine is a standard first-line chemotherapy used in pancreatic cancer treatment that has been shown to improve overall survival in patients with both resectable and unresectable disease [2, 19]. Its mechanism of action involves the disruption of cellular DNA synthesis resulting in cell death, but the side effects associated with gemcitabine often results in treatment breaks and even cessation of potentially curative therapy in patients with pancreatic cancer [1,19]. Targeted theranostic nanomedicine promises to deliver effective treatment to pancreatic cancer and avoid high off-target accumulation. The aim of this study was to use UPA ligand targeted, pH responsive mesoporous silica nanoparticles loaded with gemcitabine and Indocyanine Green, a MSOT contrast agent, as a theranostic nanodelivery system for pancreatic cancer.
2. Materials and Methods
2.1. Materials
Cetrimonium bromide (CTAB) (≥99% for molecular biology), tetraethyl orthosilicate (TEOS), (3-glycidoxypropyl) methyldiethoxysilane (GPTMS), (3-aminopropyl) triethoxysilane (APTES), Indocyanine Green (ICG), sodium phosphate monobasic, sodium phosphate dibasic, Whatman Qualitative Filter Paper (grade-1), Rhodamine, DAPI, sodium orthovanadate, Agar, ammonium nitrate, chitosan polymer, acetic acid, sodium hydroxide (NaOH, 5M), and ethanol (99.5%) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received. NuPage 4-12% Bis Tris gels, Nitrocellulose iBlot Membranes, RPMI-1640 medium, Dulbecco's Modified Eagle's Medium (DMEM), and L-glutamine was purchased from Life Technologies (New York, NY, USA). Rabbit IR-Dye secondary antibody, mouse IR-Dye secondary antibody, and Odyssey blocking buffer were purchased from LI-COR (Lincoln, NE, USA). Fetal Bovine Serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA, USA). FITC-Phalloidin was purchased from Cytoskeleton (Denver, CO, USA). Urokinase plasminogen activator ligand (UPA) was purchased from Prospec (Ness-Ziona, Israel). Urokinase plasminogen activator receptor antibody and B-actin antibodies were purchased from Pierce (Rockford, IL, USA). While S2VP10 and S2CP9 cell lines were gifts from Michael Hollingsworth (University of Nebraska), PANC-1, MiaPaCa-2, and ASPC1 cell lines were purchased from ATCC (Manassas, VA, USA). Athymic nude mice and 2920X alfalfa-free rodent pellets were purchased from Harlan Laboratories (Indianapolis, IN, USA).
2.2. Particle Synthesis
MSNs were synthesized using the sol-gel method [20-21]. The surfactant cetrimonium bromide (CTAB, 1.0 g) was added to a solution of deionized (DI) water (500 mL) and NaOH (4.0 mL, 5 M), then stirred vigorously for 4 hours at 80 °C. Next, the silica monomer TEOS (5.0 mL) was added dropwise to the solution and stirred for an additional 4 hours. The resulting white precipitate was collected via centrifugation (1000 G, 25 minutes) and rinsed with ethanol three times. The sample was then dried under vacuum (45 °C) overnight. The resultant dry solid was then dissolved in a solution of ethanol (95%) and ammonium nitrate (10 mg/mL) and refluxed at 80°C for 6 hours. Refluxing removed all CTAB from the solution, resulting in the hollow, mesoporous silica structure of the MSNs. The precipitated MSNs were collected via centrifugation and dried under vacuum (45 °C, 24 hours) as described above.
Chitosan was added to MSN surfaces to allow for pH responsive retention and release of a payload. To conjugate the MSNs with the polymer chitosan, a stock solution of chitosan was first prepared. Preparation consisted of placing chitosan (2.0 g) in an aqueous solution of acetic acid (5 wt. %, 200 mL) and stirring at room temperature for 24 hours. In a separate flask, the previously synthesized MSNs (0.1 g) were dispersed in ethanol (10 mL) using sonication for 15 minutes. The pH of the mixture was lowered to 3.5 – 4.5 by the drop wise addition of acetic acid. (3-glycidoxypropyl) methyldiethoxysilane (GPTMS, 0.1 g) was added to the dispersion and stirred at room temperature for 3 hours. Finally, the stock solution of chitosan was added to the MSN mixture (20 mL) and stirred at room temperature for 24 hours. Chitosan conjugated MSNs (C-MSN) were collected via centrifugation (5000 G) and rinsed using DI water and ethanol three times. The sample was then dried under vacuum overnight.
2.3. Characterization
Characterization of the synthesized MSNs was performed with transmission electron microscopy (TEM) and zeta potential analysis. Transmission electron microscopy allowed for qualitative analysis of the morphological characteristics of the MSNs, such as their average size, shape, and structure. For analysis, the sample (10 μL) was placed on a 200-mesh formvar coated copper grid and wicked dry using filter paper before viewing under a Tecnai-F20 transmission electron microscope (FEI Co., Eindhoven, Netherlands).
Electrophoretic mobility (μE) of the samples was measured at 25° C with Zetasizer Nano-ZS (Malvern Instruments, Ltd., Malvern, United Kingdom). The zeta-potential of the samples was calculated from μE using the following Smoluchowski's equation: Where ζ is the zeta-potential, ε the permittivity of solvent, and η the viscosity of solvent. (See Supplemental Figure 1).
2.4. Western Blot Analysis
Cell culture media was created using RPMI in autoclaved phosphate buffer (PBS) (25mM) at pH 6.5, 6.8, and 7.4. Powdered DMEM (13.6 g) was dissolved in PBS (1 L) at each specified pH and subsequently filtered through sterile, grade-1 Whatman Filter Paper. The filtered solution was mixed to include 1% L-glutamine and 10% FBS. Verification of pH was performed using a Denver Instrument Ultrabasic pH meter (Bohemia, NY, USA) and titrated to appropriate pH using autoclaved NaOH (1M) or HCI (1M). Media created in this way was used for all procedures involving cell lines described in this study.
Cell lysates from lines of S2VP10, S2CP9, PANC-1, MiaPaCa-2, and ASPC1 were cultured for blotting at 37 °C in a 5% C02 environment. At 70% confluence, cells of all lines were moved to culture media of pH 7.4 or pH 6.5. The cells were lysed and the whole cell lysates were collected with sodium orthovanadate. Protein electrophoresis was performed with NuPage 4-12% Bis Tris gels and then transferred to iBIot nitrocellulose membranes using the iBIot Dry Transfer System from Invitrogen (Life Technologies, New York, NY, USA). The membrane was blocked with 3 mL Li-Cor blocking buffer (Li-Cor) for 30 minutes, then incubated with rabbit anti-UPAR (Abcam, Cambridge, England, UK) at a concentration of 1:5—and mouse anti-β-actin antibody at 1:5000. The membranes were then incubated overnight at 4 °C and subsequently washed three times using Tris-buffered saline, tween 20 (TBST; 50 mmol/L Tris, 150 mmol/L NaCl, and 0.05% Tween 20), secondary antibodies donkey anti-mouse IRDye 680RD, and donkey anti-rabbit IRDye 800CW (Li-Cor) at concentrations of 1:2000 in blocking buffer for 1 hour at 25 °C. Membranes were then scanned and analyzed using the Li-Cor Odyssey Infrared Imaging System (Li-Cor). Dosimetry was performed using Odyssey software, in which intensity values for UPAR band were divided by the β-actin band to normalize relative signal abundance.
2.5. Ligand Conjugation
In preparation for UPA conjugation, APTES was added to functionalize the surfaces of the MSNs. First, 1.42 g of APTES was dissolved in 3 mL of ethanol and in a separate flask, 700 mg of chitosan coated MSNs were dissolved in 2.5 mL of ethanol. The two solutions were then mixed and stirred at room temperature for 24 hours. The sample was later dried under vacuum as previously described.
The APTES/UPA ligand crosslinkage was created using n-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) system. EDC (2 mM, 1 mL), NHS (2 mM, 1 mL), and 10 μL of UPA stock were mixed into solution (final concentration of UPA is 200 nM). The mixture was stirred at room temperature for 24 hours and then 1.6 mg of functionalized chitosan coated MSNs were added to the ligand solution. The resulting solution was stirred for an additional 24 hours at room temperature to complete the conjugation.
2.6. Particle Loading
Addition of the chemotherapeutic drug began by dissolving 8 mg of chitosan coated MSNs in 8 mL of phosphate-buffered saline (PBS, pH 3). Gemcitabine (144 mM, 7mL) was added to the solution and the pH was set at 3. The solution was stirred at room temperature for 24 hours. Once the drug was loaded, it was secured within the particle by raising the solution pH to 8 with NaOH (5 M) and stirring at room temperature for 3 hours. Centrifugation (1000 G, 25 minutes) and re-suspension in PBS (pH 8) was used to purify sample.
Loading of the nanoparticles with ICG involved the addition of ligand loaded chitosan coated MSN (40 mg) to an aqueous solution of ICG (1 mM, 10 ml_). The pH of the solution was adjusted to 3 by dropwise addition of HCI (5 M), and then the resulting solution was stirred for 24 hours at room temperature. To lock the loaded dye in place the pH of the solution was increased to 8 using NaOH (5 M) and stirring for an additional 3 hours at room temperature. Dialysis with autoclaved Millipore water and dialysis tubing was used to remove any non-bound dye. Encapsulation of dye was confirmed using spectrophotometry (Supplemental Figure 2). The final concentration of ICG dye encapsulated into the nanoparticles was 50 μg/mL. Rhodamine dye loading was performed in a similar manner by using an aqueous solution of the dye (1 mM).
2.7. Drug Release Assay
Gemcitabine loaded MSNs (3 mL) were titrated to a pH of 6.5, 6.8, or 7.4 by addition of NaOH or HCI and placed inside dialysis tubing. The samples were then placed in a solution of PBS (15mL) at a matching pH. The samples were stirred continuously throughout the experiment. Readings of the PBS solutions were analyzed with the NanoDrop 2000c/2000 UV-Vis Spectrophotometer (Thermo Scientific, Wilmington, DE, USA). A solution for PBS was used as blank for the spectrophotometer. It was possible to determine the amount of Gemcitabine released from the MSNs by analyzing the dialysis fluid. Samples were read consecutively every hour for 10 hours and absorbance values were plotted to determine the drug release kinetics of Gemcitabine from the MSNs.
2.8. Cell Binding Assays
2.8.1 Cell Uptake of NIR dye
Initial evaluation of cellular uptake of MSNs and MSN-UPA in the presence pH 7.4 and pH 6.5 was based upon cellular accumulation of ICG. In 6-well plates, S2VP10 and S2CP9 cells (5 × 105/well) were grown in media at pHe of 7.4 or 6.5 and treated with UPA ligand targeted or untargeted MSNs containing ICG (50 μg/mL) incubated for 2 hours. Cells were washed 3× in their appropriately buffered PBS solution containing 10% FBS. Following washing, accumulation of ICG was measured using an Odyssey Infrared System (LiCor). Dosimetry was performed using Li-Cor software.
2.8.2 Immunocytochemisty
In order to confirm the UPAR binding of the UPA-targeting of the nanoparticle inside the UPAR high cells, Rhodamine was encapsulated within the nanoparticle to identify cellular localization of nanoparticles via microscopy. S2VP10L (UPAR high) and BT474 (UPAR low) cells were each seeded into 3 wells of a chamber slide at a density of 3×105 cells/well. Cells were grown overnight in RPMI with 10% FBS and 1% L-Glutamine at 37°C with 5% CO2. The cells were then serum-starved for 3 hours by switching to RPMI with 1% BSA (Fisher). Immunocytochemistry buffers were prepared as follows: PBS++ (0.5 mM CaCl2 and MgCl2 in PBS), PBS++++ (97.9 mL PBS++, 2 mL 10% BSA + 90.08 mg dextrose), and citrate buffer (μmL 20X SCC buffer, 19 mL ddH2O) and adjusted to pH 7.4. Following starvation, cells of each line were treated with 30 μL PBS (control), 30 μL of MSN-UPA (containing 50 μg/mL Rhodamine) for 1 h, or 30 μL of 1 μg/mL Anti-UPAR blocking antibody (Abcam) for 1 h followed 30 μL of MSN-UPA (containing 50 μg/mL Rhodamine) for 1 h. Then, the wells were washed 2X with cold PBS++++, 3X with ice cold citrate buffer, and again 2X with cold PBS++++. Cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 5 minutes at room temperature and 15 minutes at 4°C. Samples were treated with Acti-stain 488 phalloidin (Cytoskeleton) to visualize actin filaments following manufactures protocol.The slides were mounted in ProLong with DAPI (Invitrogen, Waltham, MA) and set overnight. Images were taken at 400× magnification with a Zeiss Image Photomicroscope 2 (Carl Zeiss, Oberkochen, Germany) using DAPI, FITC, and Rhodamine filters. The exposure times used were 20 ms DAPI, 200 ms FITC, and 200 ms Rhodamine.
2.8.3 Tissue Phantoms
Synthesized MSNs were loaded with ICG for evaluation within tissue phantoms using Multispectral optoacoustic tomography as in Ref. [6]. Cells from the S2VP10 line, in media at pH 7.4 and 6.5, were treated with UPA ligand targeted and untargeted MSNs. Following treatment, cells were washed with PBS to remove unbound particles. Pelleted cells were added to phantom agarose media and inserted into the phantom models. Tissue phantoms were created as fixed cylinders with 2 cm diameter. Agar (1.3% w/w) and an intralipid 20% emulsion (6% v/v) were added to DI water to create gels with a reduced light scattering coefficient of μ = 10 cm-1. Nanoparticle treated cells were added to a cylindrical opening in the tissue phantoms of 3 mm diameter. Tissue phantom models were used to simulate in vivo studies. MSOT was performed at a single axial position approximately in the middle of the sample added to the tissue phantom with the inVision 256TF (iThera Medical, Munich, Germany). Samples were evaluated using 680, 710, 730, 740, 760, 770, 780, 800, 850, and 900 nm wavelengths. Twenty averages were performed at each wavelength. Water temperature was 34°C. MSOT signal was analyzed through region of interest selection. Tissue phantoms confirmed MSOT signal strength in preparation for in vivo imaging.
2.9 Biodistribution of MSNs in vivo
2.9.1 Orthotopic implantation of pancreatic cancer cells
Female athymic nude mice 4 weeks of age (Harlan Laboratories, Indianapolis, IN) were used for this study in strict adherence to a University of Louisville Institutional Animal Care and Use Committee (IACUC) approved protocol. A diet of 2920× alfalfa-free feed (Harlan Laboratories) was used in order to reduce background signal during MSOT imaging. Tumor cell implantation was performed using a previously described method [6, 22]. Mice were anesthetized with 2.5% isoflurane and betadine was applied to the area of incision. Incision began in the left lower quadrant and extended 1cm cranially. The spleen was located and retracted to reveal the pancreas. S2VP10 (2.0 × 105) and ASPC1 (2.0 × 106) lines were suspended in 30 μL of serum-free DMEM. The cell suspension was injected into the pancreas using a 28-gauge needle. Spleen and pancreas were then reinserted in the abdominal cavity. Sutures (5-0 Prolene Sutures) were used to close fascia and skin incisions.
2.9.2 In Vivo Imaging
The inVision 256TF MSOT (iThera Medical, Munich, Germany) was used for real-time, quantitative analysis of MSN biodistribution. Mice were anesthetized with 1.6% isoflurane and placed in the MSOT animal holder, ventral side up. Baseline MSOT images were taken prior to MSN injection, 0 hours. Targeted (MSN-UPA) and untargeted MSNs loaded with ICG dye (50 μg/mL) were injected intravenously into xenograft mouse models (5 mice per group). At 4 and 24 hours post-injection, MSOT images were acquired. The method of acquisition involved imaging a 1.8 cm portion of each mouse, from liver to kidney, cranially to caudally, in 0.2 mm steps. Multispectral evaluation was conducted at 680, 710, 730, 740, 760, 770, 780, 800, 850, and 900 nm wavelengths, with 20 averages taken at each wavelength. Water temperature within the MSOT instrument was 34°C. Acquisition time was set to 10 μs to minimize movement artifacts. MSN biodistribution was determined by ICG signal intensity.
Raw data reconstruction was performed using backprojection at a resolution of 75 μm using ViewMSOT software version 3.5 (iThera Medical, Munich, Germany). After spectral unmixing, Multispectral processing was performed in ViewMSOT with linear regression. Multispectral processing assumes that all absorbers in an imaging field are accounted for in order to attribute signal levels to each of the absorbers across wavelengths. Reconstruction and unmixing parameters were maintained across all data to preserve comparability among data sets. No fluence correction was applied during spectral unmixing since all the tumors were located at a similar depth in each mouse. Image stacks were imported to ImageJ (National Institute of Health, USA) and analyzed for probe binding using orthogonal views.
2.9.3 Region of interest analysis
Regions of interests were drawn in ViewMSOT around sites of probe binding and reported in MSOT a.u. Deoxy-hemoglobin was used as a comparison to probe binding in the location of the tumor. Regions of interest were consistent across all slices, resulting in a non-uniform elliptical prism volume of interest. Mean pixel intensity was plotted against position to assess probe signal strength within the tumors. In order to minimize variability from out-of-plane artifacts, maximum mean signal per cross-section was used to find the center of signal intensity and quantify probe binding. Values for treatment and control nanoparticles were assessed with SAS 9.3 (Cary, NC, USA) and reported in MSOT a.u.
2.10. Ex Vivo Analysis
To confirm MSOT findings of MSN biodistribution, organs were removed from the imaged mice 4 hours post injection. The liver, spleen, kidney, and pancreas tumors were placed into a petri dish with PBS 7.4 pH at 37 °C. AMI NIR-fluorescence (Spectral Instruments Imaging, Tucson, AZ, USA) was performed within 30 seconds of organ removal.
2.11 Statistical Considerations
Statistical comparisons of in vitro data utilized analysis of variance (ANOVA) with significance found at p<0.05. For evaluation of accumulation and biodistribution of nanoparticles in vivo, peak intensity was determined in each of the three organs and statistically compared using ANOVA with significance at p < 0.05.
3. Results and Discussion
3.1. Nanoparticle Synthesis and Characterization
The objective of this study was to assess the efficacy of a urokinase plasminogen activator ligand targeted, pH responsive mesoporous silica nanoparticle as a theranostic nanodelivery system in pancreatic cancer. Synthesis of the MSNs involved the sol-gel method, UPA and chitosan conjugation, and the loading of drug and contrast, (Figure 1). TEM analysis showed the MSNs to be an average of 115 ±10 nm in diameter, with an average pore size of 3 nm, and to be mesoporous and spherical in structure, (Figure 2).
Figure 1.
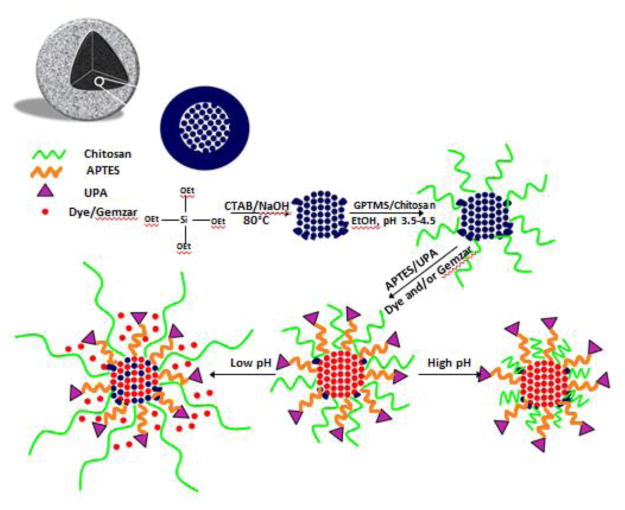
Synthesis of UPA ligand targeted, pH responsive MSNs. Dye and/or gemcitabine were loaded onto the MSNs for release in acidic pancreatic tumor environments. MSN, mesoporous silica nanoparticle; UPA, urokinase plasminogen activator; APTES, (3-aminopropyl)triethoxysilane; GPTMS, (3-glycidoxypropyl) methyldiethoxysilane; ICG, Indocyanine Green; TEM, transmission electron microscopy; UPAR, urokinase plasminogen receptor activator; CTAB, cetrimonium bromide; TEOS, tetraethyl orthosilicate
Figure 2.

Image of a mesoporous silica nanoparticle obtained with transmission electron microscopy (TEM). TEM confirmed a spherical, mesoporous structure. The average diameter of the particles was 115±10 nm with an average pore size of 3 nm.
The MSN nanodelivery system incorporated particle surface modifications to impart pancreatic cancer cell binding specificity and pH responsiveness. The MSNs were first conjugated with chitosan to make the particles pH sensitive. Zeta potential analysis confirmed chitosan conjugation of MSNs (C-MSN -/APTES) by mapping pH dependent particle surface charge. This involves the measurement of the electrical charge existing outside of a chemical species [23]. The surface charge of the MSN decreased with increasing pH, showing a -16 mV potential at pH 6, a -22 mV potential at pH 7, and a -28 mV potential at pH 8, (Supplemental Figure 1A).
The surfaces of the MSNs also needed to be functionalized for ligand attachment. (3-aminopropyl) triethoxysilane (APTES) was used to functionalize the MSN surfaces. The addition of APTES involved the addition of positive surface charge to the MSNs. Conjugation was confirmed through zeta potential analysis, with a 0 mV potential at pH 6, a -7 mV potential at pH 7, and a -8 mV potential at pH 8, (Supplemental Figure 1B). These readings validated the conjugation of the MSN with the chitosan (C-MSN -/APTES) and chitosan and (3-aminopropyl) triethoxysilane (APTES) dual conjugation (C-MSN +/APTES). The decreasing voltage with corresponding increasing pH displayed by the C-MSN -/APTES results from deprotonation of the chitosan compound. The addition of APTES adds positive charge to the particle resulting in a more positive charge for the MSN overall, when compared to the C-MSN -/APTES.
While the addition of chitosan to the MSNs restricted the release of chemotherapeutic payload to acidic environments, a method to actively target the MSNs to the tumor cells required the selection of an extracellular receptor known to be overexpressed in pancreatic cancer. Urokinase plasminogen activator receptor is an extracellular receptors with one of the highest differential expression between malignant and non-malignant pancreatic tissue [24-25]. Therefore, the second key surface modification was the addition of UPA ligand to the MSNs. Before proceeding to ligand conjugation, pancreatic tumor overexpression of urokinase plasminogen activator receptor (UPAR) was confirmed with Western blot analysis. BT474, a UPAR negative breast cancer cell line, was used as a negative control and MCF7, a UPAR positive breast cancer cell line, was used as a positive control for UPAR expression. Pancreatic cancer cell lines S2VP10, S2CP9, PANC-1, MiaPaCa-2, and ASPC1 were blotted to determine relative expression of UPAR in each cell line. Western blot analysis indicated that S2VP10 and S2CP9 cells exhibited the greatest degree of overexpression. S2CP9 cells showed a relative abundance of 3.18 +/- 0.2 and S2VP10 cells showed a relative abundance of 3.93 +/- 0.2 (Figure 3). Although pancreatic cancer cell lines evaluated demonstrated varying levels of UPAR, each cell line expressed UPAR at higher levels than the negative control cells. Additionally, both metastatic pancreatic cancer cell lines, S2VP10 and S2CP9, had higher levels than the positive control breast cancer cells suggesting that the UPA-MSNs could be successful for drug delivery to metastatic lesions.
Figure 3.
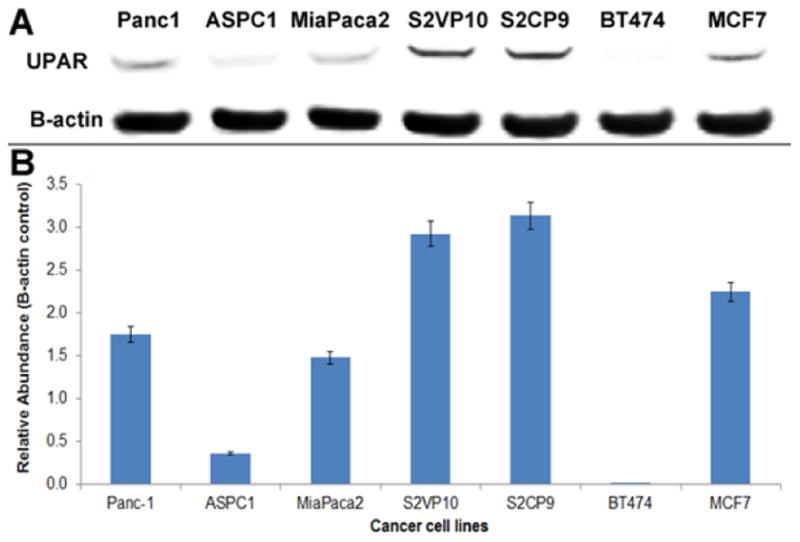
Western blot analysis of cell lines. A) Western Blot analysis of multiple pancreatic cell lines and two breast cancer cell lines was performed to assess the relative expression levels of UPAR. BT474, a UPAR negative breast cancer cell line, was used as a negative control for UPAR expression. MCF7, a UPAR positive breast cancer cell line, was used as a positive control. B) Dosimetry indicates that S2VP10 and S2CP9 cells express UPAR at the highest levels among those cell lines evaluated.
Several methods were utilized to evaluate impact of the pH responsiveness and UPA ligand conjugation of the MSNs. Initial evaluation was conducted based upon cellular uptake of Indocynanine green containing MSN-UPA and control MSNs using Near infrared fluorescence imaging. Cells of the S2VP10 line were plated in pH 7.4 media and pH 6.5 media to mimic normal physiological and pathological tissue pHs, respectively. The data showed that at a pH 7.4 UPA ligand targeted MSNs had 79X higher uptake then control MSNs, and that at a pH 6.5, the UPA ligand targeted MSNs 6750X higher uptake than at pH 7.4 (p<0.05)(Supplemental Figure 3).
To assess the potential of MSN-UPA for UPAR specific binding, fluorescence microscopy was implemented to observe MSN-UPA binding S2VP10 (UPAR high) and BT474 (UPAR low-control) cells. In UPAR high S2VP10 cells, uptake of MSN-UPA particles containing Rhodamine was confirmed in the cytoplasm of the cells (Supplemental Figure 4). Upon prior pre-treatment using a UPAR blocking antibody, the blocked cells exhibited minimal fluorescence, confirming that the fluorescence of the UPA ligand targeted MSNs was attributable to UPAR binding, (Supplemental Figure 4).
Tissue phantoms that approximated the consistency and light scattering properties of organic tissue were created, some with pH values at 7.4 and others at a pH of 6.5. The tissue phantoms were infused with S2VP10 pancreatic cancer cells and then treated with UPA ligand targeted and untargeted MSNs. The phantoms were imaged with MSOT and the observed pattern of cell binding similar to the cell fluorescence pattern previously observed. In the tissue phantoms with a pH 7.4, the UPA ligand targeted MSNs exhibited 7× the binding seen with untargeted MSNs and 20X at a pH of 6.5 (p < 0.05) (Figure 4). These results were consistent with previous cell binding data, which confirmed that MSNs retain their characteristic function in an environment closely mimicking living tissue suggesting that the in vivo model would also be successful.
Figure 4.
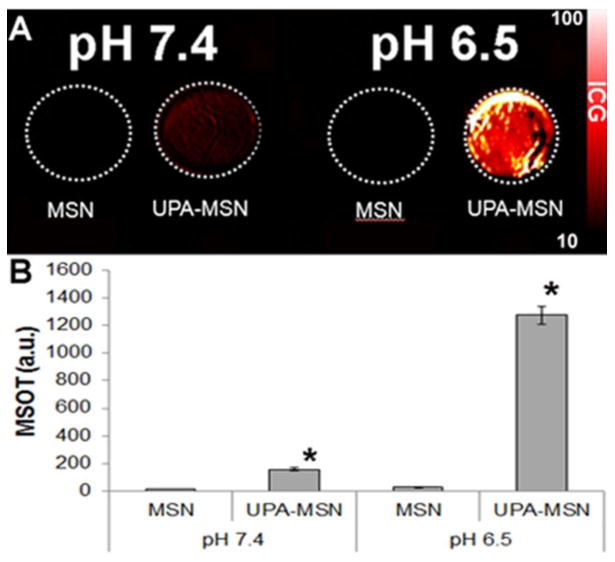
Multispectral optoacoutic tomography (MSOT) of the tissue phantoms infused with S2VP10 pancreatic cancer cells. A) Image of tissue phantoms containing S2VP10 pancreatic cancer cell infused with untargeted and ligand targeted mesoporous silica nanoparticles (MSN) obtained with inVision 256TF MSOT (iThera Medical, Munich, Germany). The signal intensity bar is shown on the right. Ligand targeted MSNs demonstrated increased intensity compared to untargeted MSNs at both pH values; however the was much more pronounced at a pH of 6.5. B) Quantitative analysis of each signal in A. In the tissue phantom with a pH 7.4, the UPA ligand targeted MSNs exhibited 7× the signal seen with untargeted MSNs (p <0.05). At a pH of 6.5 the targeted MSNS demonstrated 20× (p < 0.05) the intensity compared to untargeted MSNs.
3.2. Nanoparticle Loading
Loading the MSNs with gemcitabine involved an acidic phase, where chitosan conformation was relaxed and drug could enter the particle, and a basic phase, where chitosan conformation tightened and prevented release. The drug release characteristics were assessed with dialysis. MSNs loaded with gemcitabine were dialyzed with PBS at three different pH levels. These pHs were chosen because they resemble normal physiological pH (pH 7.4) and cancerous acidity (pH 6.8- 6.5) [6, 12, 26]. As hypothesized, at a pH of 6.5 the MSNs were shown to release 2× the amount of gemcitabine as the MSNs at pH 6.8 and 7.4, with release kinetics leveling off between 2 and 3 hours, confirming preferential drug release in an acidic environment, (Supplemental Figure 5). It can be inferred that these drug release kinetics will result in targeted release of chemotherapeutics in vivo and decreased normal tissue toxicity. This intermediate set of data points also offers a good comparison for determining MSN release kinetic trends as pH decreases.
Indocyanine Green (ICG), which has a characteristic absorbance peak at 800 nm, was used in both in vitro and in vivo assays. The UV-Vis spectrum of the dye alone was compared to the spectrum of MSNs loaded with dye, (Supplemental Figure 2). The spectral comparison confirmed the intact and functional structure of the ICG dye when loaded onto the nanoparticles, with major absorbance peaks at 780 nm and minor peaks at 700 nm. These data also showed that the spectrum of ICG was essentially unaltered by the MSNs; in fact, the MSNs appear to adopt the spectral characteristics of the ICG. Because contrast agents for MSOT depend on the absorption of light by the contrast agent to produce sound waves, the MSNs containing ICG can be expected to appear on MSOT imaging with peak intensity at excitatory wavelengths of 770-790 nm.
3.3 Determining Particle Distribution with Imaging
Before mouse models were injected with the MSNs, it was necessary to create an in vitro pancreatic tumor model to better understand the expected activity of MSNs in vivo. Mouse models were used to assess nanoparticle biodistribution in vivo. Mice were anesthetized with isoflurane and imaged four hours post-treatment. While mice were imaged immediately prior to nanoparticle injection (time 0), four hours post injection and one day post injection, the maximum accumulation of MSN-UPA nanoparticles was observed at four hours 47 ± 8 MSOT a.u. in S2VP10 mice (Figure 5, Supplemental Figure 6 A-B). In S2VP10 mice, untargeted MSNs were observed at the greatest accumulation within the spleen. With MSOT, substantial binding of MSN-UPA particles within the orthotopic S2VP10 mouse tumor was observed with reduced secondary binding in the specimen's liver, spleen, and kidney (Supplemental Figure 6). The liver uptake is thought to represent MSN clearance from the blood stream and spleen uptake is thought to occur via macrophage engulfment. Due to the size of the particle, uptake within the kidney was not expected or observed for either the targeted or untargeted MSNs [27]. Greater clearance can be attributed to increased expression of UPA relative to UPAR by the cancerous cell lines in vivo compared to in vitro levels of expression. It has been shown that, while UPA and UPAR are overexpressed in pancreatic malignancy [24], the ratio of UPA to UPAR expression increases in the presence of multiple biological factors, such as IGF-1 (IGF1-R) and HIF-1 [28-31]. This increased expression of UPA would compete with the MSNs for binding sites on cancerous cells. The pancreatic binding of MSN-UPA particles detected by MSOT is promising and proves the concept of the UPA ligand targeted, pH responsive MSN as a nanodelivery system in vivo.
Figure 5.
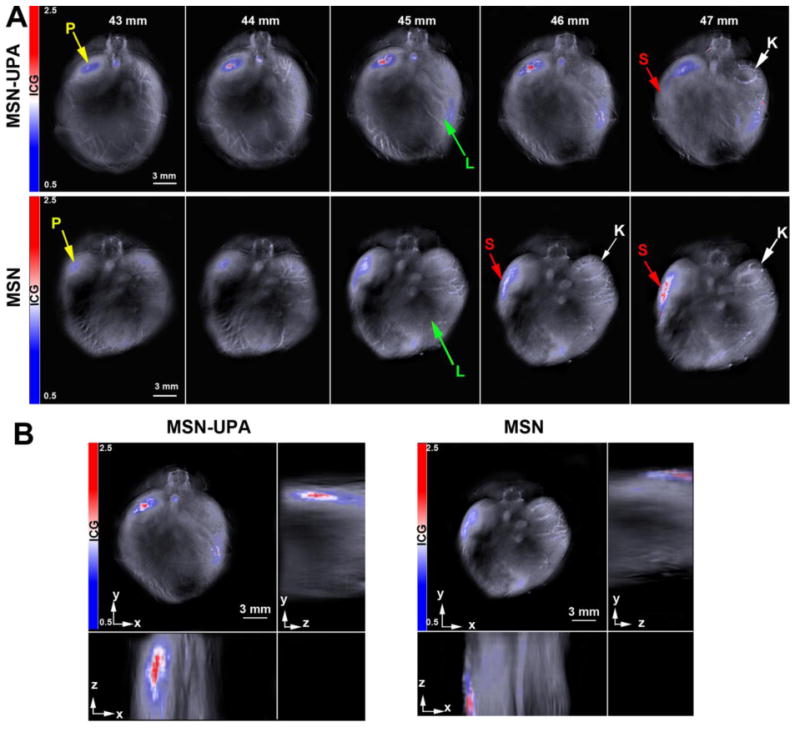
Serial slice images of nanoparticle accumulation. (A) S2VP10 implanted mice were injected with either MSN-UPA or untargeted MSN. The highest signal intensity of the nanoparticles was at 45-46 mm. Organs are identified: Yellow arrow=Pancreas tumor, green arrow=Liver, Red arrow=Spleen, White arrow=Kidney. (B) Orthogonal views of the nanoparticle accumulation through different anatomical planes in the xyz space. Representative locations of organs can be also be viewed in [6].
The biodistribution determined through MSOT was confirmed after imaging, ex vivo. The ex vivo analysis corroborated the initial MSOT readings, confirming primary pancreatic accumulation and secondary splenic accumulation of UPA ligand targeted MSNs, as well as confirming the accuracy of MSOT imaging for determining in vivo nanoparticle biodistribution.
4. Conclusion
The results of this study illustrate the efficacy of UPA ligand targeted, pH responsive MSNs as a theranostic nanodelivery system for use in pancreatic cancer. To further develop and translate nanomedicine for improved cancer detection and treatment, it is vital to utilize clinically relevant models, especially orthotopic or genetically modified mammalian models, and longitudinal evaluations to closely mimic the clinical setting. This study demonstrates the utility of MSOT to monitor nanoparticle accumulation and biodistribution of tumor targeted nanoparticles in an orthotopic pancreatic tumor without the requirement for euthanasia. It also demonstrated the ability of multispectral optoacoustic tomography to obtain high-resolution images in real-time and to quantify nanoparticle biodistributions, in vivo at the depths > 1 cm. Eventual clinical implications for this system may include improved clinical staging of pancreatic cancer with simultaneous treatment. More accurate clinical staging combined with decreasing the time from diagnosis to treatment may lead to an improvement in survival outcomes for patients with pancreatic cancer. Future directions for this study include actual assessment of tumor response to the gemcitabine or other therapeutic agents delivered in this manner.
Supplementary Material
Supplemental Figure 1. Zeta potential analysis confirmed chitosan conjugation of MSNs A) Zeta potential analysis shows the charge distribution of the chitosan conjugated MSNs exhibited pH responsiveness gradually increased in responsiveness as pH decreased from pH 8. B) The addition of APTES was confirmed by the differing surface charge distribution compared to (chitosan coated MSN) C-MSN -/APTES.
Supplemental Figure 2. UV-Vis spectra. A) The UV-Vis absorption spectrum for ICG dye alone. B) The UV-Vis absorption spectrum of MSNs loaded with ICG.
Supplemental Figure 3: Evaluation of acidic pH specificity of ICG dye release from MSNs. S2VP10 cells grown at either pH 7.4 or 6.5 were treated with either UPA-targeted or no-ligand targeted MSNs encapsulated with ICG. Cells were washed with pH specific PBS. Cells were viewed using Odyssey infrared imaging system with dosimetry evaluation. At pH 7.4, UPA ligand targeted MSNs had 79X higher uptake then control MSNs (p<0.05). The UPA ligand targeted MSNs resulted in 6750X higher uptake at pH 6.5 than at pH 7.4 (p<0.05).
Supplemental Figure 4. Fluorescence miscroscopy. A) Untagged MSNs loaded with Rhodamine were used to treat S2VP10 cells and the cells showed minimal Rhodamine fluorescence under microscopy B) Significant rhodamine fluorescence in S2VP10 cells treated with UPA ligand targeted MSNs. C) S2VP10 cells were treated with UPAR blocking antibody to ensure MSN binding and fluorescence was due to UPA ligand binding.
Supplemental Figure 5. Drug release kinetics of Gemcitabine. The MSNs showed 2x the gemcitabine release at pH 6.5 that was observed at pH 6.8 and pH 7.4.
Supplemental Figure 6. ROI analysis on nanoparticle signal in various locations. (A) Nanoparticle accumulation was measured at 4 hours post injection using MSOT. Bar height represents the median value and error bars represent the standard deviation throughout the organ. Peak liposomal accumulation occurred at 4 hours post-injection. Significantly more MSN-UPA nanoparticles accumulated in the S2VP10 tumor versus untargeted MSN nanoparticles, UPAR low ASPC1 tumor, or off-target organs (p<0.05). Significantly lower accumulation of MSN-UPA in the spleen of mice implanted with S2VP10 cells was also observed in comparison to untargeted MSN particles or mice implanted with ASPC1 tumor cells (p<0.05). (B) MSN-UPA nanoparticle accumulation was measured at 0, 4, and 24 hours post injection within S2VP10 mice. Significant accumulation was of MSN-UPA particles was observed in S2VP10 pancreatic tumors at 4 h post injection and remained for 24 h (p<0.05). Accumulation of MSN-UPA within S2VP10 tumors is shown over time.
Acknowledgments
We would like to thank the NCI (R25-CA134283) and the University of Louisville School of Medicine for financial support of this research. This work was presented at the NanoDDS symposium 2015.
Abbreviations
- MSOT
multispectral optoacoustic tomography
- MSN
mesoporous silica nanoparticle
- UPA
urokinase plasminogen activator
- APTES
(3-aminopropyl)triethoxysilane
- GPTMS
(3-glycidoxypropyl) methyldiethoxysilane
- ICG
Indocyanine Green
- TEM
transmission electron microscopy
- UPAR
urokinase plasminogen receptor activator
- CTAB
cetrimonium bromide
- TEOS
tetraethyl orthosilicate
- NHS
n-hydroxysuccinimide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- DI
deionized
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Komar G, Kauhanen S, Liukko K, Seppanen M, Kajander S, Ovaska J, Nuutila P, Minn H. Decreased blood flow with increased metabolic activity: a novel sign of pancreatic tumor aggressiveness. Clin Cancer Res. 2009;15:5511–5517. doi: 10.1158/1078-0432.CCR-09-0414. [DOI] [PubMed] [Google Scholar]
- 3.Ntziachristos V, Razansky D. Molecular imaging by means of multispectral optoacoustic tomography (MSOT) Chem Rev. 2010;110:2783–94. doi: 10.1021/cr9002566. [DOI] [PubMed] [Google Scholar]
- 4.Stoffels I, Morscher S, Helfrich I, Hillen U, Lehy J, Burton NC, Sardella TC, Claussen J, Poeppel TD, Bachmann HS, Roesch A. Metastatic status of sentinel lymph nodes in melanoma determined noninvasively with multispectral optoacoustic imaging. Science Translational Medicine. 2015;7:317ra199-317ra199. doi: 10.1126/scitranslmed.aad1278. [DOI] [PubMed] [Google Scholar]
- 5.Hudson SV, Huang JS, Yin W, Albeituni S, Rush J, Khanal A, Yan J, Ceresa BP, Frieboes HB, McNally LR. Targeted noninvasive imaging of EGFR-expressing orthotopic pancreatic cancer using multispectral optoacoustic tomography. Cancer Res. 2014;74:6271–9. doi: 10.1158/0008-5472.CAN-14-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kimbrough CW, Khanal A, Zeiderman M, Khanal BR, Burton NC, McMasters KM, Vickers SM, Grizzle WE, McNally LR. Targeting Acidity in Pancreatic Adenocarcinoma: Multispectral Optoacoustic Tomography Detects pH-Low Insertion Peptide Probes In Vivo. Clin Cancer Res. 2015;21:4576–4585. doi: 10.1158/1078-0432.CCR-15-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuthati Y, Sung PJ, Weng CF, Mou CY, Lee CH. Functionalization of mesoporous silica nanoparticles for targeting, biocompatibility, combined cancer therapies and theragnosis. J Nanosci Nanotechnol. 2013;13:2399–2430. doi: 10.1166/jnn.2013.7363. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Zhao Q, Han N, Bai L, Li J, Liu J, Che E, Hu L, Zhang Q, Jiang T, Wang S. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomedicine. 2015;11:313–327. doi: 10.1016/j.nano.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 9.Wang YG, Zhou K, Huang G, Hensley C, Huang X, Ma X, Zhao T, Sumer BD, DeBerardinis RJ, Gao J. A nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nature materials. 2014;13:204–212. doi: 10.1038/nmat3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho K, Wang XU, Nie S, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clinical cancer res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 11.Dobrucki LW, Pan D, Smith AM. Multiscale Imaging of Nanoparticle Drug Delivery. Curr Drug Targets. 2015;16:560–70. doi: 10.2174/1389450116666150202163022. [DOI] [PubMed] [Google Scholar]
- 12.Phillips P. Pancreatic stellate cells and fibrosis., in: M.H. Grippo PJ (Ed.) Pancreatic Cancer and Tumor Microenvironment, Transworld Research Network, Trivandrum (India) 2012 [PubMed] [Google Scholar]
- 13.Kong SC, Giannuzzo A, Novak I, Pedersen SF. Corrigendum: Acid-base transport in pancreatic cancer: Molecular mechanisms and clinical potential. Biochem Cell Biol. 2015;93:272. doi: 10.1139/bcb-2015-0026. [DOI] [PubMed] [Google Scholar]
- 14.Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Controlled Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 15.Barth BM, Sharma R, Altınogˇlu EI, Morgan TT, Shanmugavelandy SS, Kaiser JM, McGovern C, Matters GL, Smith JP, Kester M, Adair JH. Bioconjugation of calcium phosphosilicate composite nanoparticles for selective targeting of human breast and pancreatic cancers in vivo. ACS nano. 2010;4:1279–1287. doi: 10.1021/nn901297q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agnihotri SA, Mallikarjuna NN, Aminabhavi TM. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J Control Release. 2004;100:5–28. doi: 10.1016/j.jconrel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Mazar AP, Henkin J, Goldfarb RH. The urokinase plasminogen activator system in cancer: implications for tumor angiogenesis and metastasis. Angiogenesis. 1999;3:15–32. doi: 10.1023/a:1009095825561. [DOI] [PubMed] [Google Scholar]
- 18.Cantero D, Friess H, Deflorin J, Zimmermann A, Brundler MA, Riesle E, Korc M, Buchler MW. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br J Cancer. 1997;75:388–395. doi: 10.1038/bjc.1997.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, Schramm H, Fahlke J, Zuelke C, Burkart C, Gutberlet K, Kettner E, Schmalenberg H, Weigang-Koehler K, Bechstein WO, Niedergethmann M, Schmidt-Wolf I, Roll L, Doerken B, Riess H. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–277. doi: 10.1001/jama.297.3.267. [DOI] [PubMed] [Google Scholar]
- 20.Lodha A, Lodha M, Patel A, Chaudhuri J, Dalal J, Edwards M, Douroumis D. Synthesis of mesoporous silica nanoparticles and drug loading of poorly water soluble drug cyclosporin A. J Pharm Bioallied Sci. 2012;4:S92–94. doi: 10.4103/0975-7406.94153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vallet-Regi M, Balas F, Arcos D. Mesoporous materials for drug delivery. Angew Chem Int Ed Engl. 2007;46:7548–7558. doi: 10.1002/anie.200604488. [DOI] [PubMed] [Google Scholar]
- 22.Khanal A, Ullum C, Kimbrough CW, Garbett NC, Burlison JA, McNally MW, Chuong P, El-Baz AS, Jasinski JB, McNally LR. Tumor targeted mesoporous silica-coated gold nanorods facilitate detection of pancreatic tumors using Multispectral optoacoustic tomography. Nano Research. 2015:1–14. [Google Scholar]
- 23.Gui R, Wang Y, Sun J. Encapsulating magnetic and fluorescent mesoporous silica into thermosensitive chitosan microspheres for cell imaging and controlled drug release in vitro. Colloids Surf B Biointerfaces. 2014;113:1–9. doi: 10.1016/j.colsurfb.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 24.de Bock CE, Wang Y. Clinical significance of urokinase-type plasminogen activator receptor (uPAR) expression in cancer. Medicinal research reviews. 2004;24:13–39. doi: 10.1002/med.10054. [DOI] [PubMed] [Google Scholar]
- 25.Cantero D, Friess H, Deflorin J, Zimmermann A, Bründler MA, Riesle E, Korc M, Büchler MW. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. British journal of cancer. 1997;75:388. doi: 10.1038/bjc.1997.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor invasion: a multidisciplinary study. Cancer research. 2006;66:5216–5223. doi: 10.1158/0008-5472.CAN-05-4193. [DOI] [PubMed] [Google Scholar]
- 27.Kumar R, Roy I, Ohulchanskky TY, Vathy LA, Bergey EJ, Sajjad M, Prasad PN. In vivo biodistribution and clearance studies using multimodal organically modified silica nanoparticles. ACS nano. 2010;4:699–708. doi: 10.1021/nn901146y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang C, Xie D, Cui J, Li Q, Gao Y, Xie K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin Cancer Res. 2014;20:1477–1488. doi: 10.1158/1078-0432.CCR-13-2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin W, Kimbrough CW, Gomez-Gutierrez JG, Burns CT, Chuong P, Grizzle WE, McNally LR. Tumor specific liposomes improve detection of pancreatic adenocarcinoma in vivo using optoacoustic tomography. J nanobiotechnology. 2015;13:90. doi: 10.1186/s12951-015-0139-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semenza GL. Targeting HIF-1 for cancer therapy. Nature reviews cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 31.Keleg S, Büchler P, Ludwig R, Büchler MW, Friess H. Invasion and metastasis in pancreatic cancer. Molecular cancer. 2003;2:14. doi: 10.1186/1476-4598-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Zeta potential analysis confirmed chitosan conjugation of MSNs A) Zeta potential analysis shows the charge distribution of the chitosan conjugated MSNs exhibited pH responsiveness gradually increased in responsiveness as pH decreased from pH 8. B) The addition of APTES was confirmed by the differing surface charge distribution compared to (chitosan coated MSN) C-MSN -/APTES.
Supplemental Figure 2. UV-Vis spectra. A) The UV-Vis absorption spectrum for ICG dye alone. B) The UV-Vis absorption spectrum of MSNs loaded with ICG.
Supplemental Figure 3: Evaluation of acidic pH specificity of ICG dye release from MSNs. S2VP10 cells grown at either pH 7.4 or 6.5 were treated with either UPA-targeted or no-ligand targeted MSNs encapsulated with ICG. Cells were washed with pH specific PBS. Cells were viewed using Odyssey infrared imaging system with dosimetry evaluation. At pH 7.4, UPA ligand targeted MSNs had 79X higher uptake then control MSNs (p<0.05). The UPA ligand targeted MSNs resulted in 6750X higher uptake at pH 6.5 than at pH 7.4 (p<0.05).
Supplemental Figure 4. Fluorescence miscroscopy. A) Untagged MSNs loaded with Rhodamine were used to treat S2VP10 cells and the cells showed minimal Rhodamine fluorescence under microscopy B) Significant rhodamine fluorescence in S2VP10 cells treated with UPA ligand targeted MSNs. C) S2VP10 cells were treated with UPAR blocking antibody to ensure MSN binding and fluorescence was due to UPA ligand binding.
Supplemental Figure 5. Drug release kinetics of Gemcitabine. The MSNs showed 2x the gemcitabine release at pH 6.5 that was observed at pH 6.8 and pH 7.4.
Supplemental Figure 6. ROI analysis on nanoparticle signal in various locations. (A) Nanoparticle accumulation was measured at 4 hours post injection using MSOT. Bar height represents the median value and error bars represent the standard deviation throughout the organ. Peak liposomal accumulation occurred at 4 hours post-injection. Significantly more MSN-UPA nanoparticles accumulated in the S2VP10 tumor versus untargeted MSN nanoparticles, UPAR low ASPC1 tumor, or off-target organs (p<0.05). Significantly lower accumulation of MSN-UPA in the spleen of mice implanted with S2VP10 cells was also observed in comparison to untargeted MSN particles or mice implanted with ASPC1 tumor cells (p<0.05). (B) MSN-UPA nanoparticle accumulation was measured at 0, 4, and 24 hours post injection within S2VP10 mice. Significant accumulation was of MSN-UPA particles was observed in S2VP10 pancreatic tumors at 4 h post injection and remained for 24 h (p<0.05). Accumulation of MSN-UPA within S2VP10 tumors is shown over time.