

Summary
Despite the approval of several anti-angiogenic therapies, clinical results remain unsatisfactory, and transient benefits are followed by rapid tumor recurrence. Here, we demonstrate potent anti-angiogenic efficacy of the multi-kinase inhibitors nintedanib and sunitinib in a mouse model of breast cancer. However, after an initial regression, tumors resume growth in the absence of active tumor angiogenesis. Gene expression profiling of tumor cells reveals metabolic reprogramming toward anaerobic glycolysis. Indeed, combinatorial treatment with a glycolysis inhibitor (3PO) efficiently inhibits tumor growth. Moreover, tumors establish metabolic symbiosis, illustrated by the differential expression of MCT1 and MCT4, monocarboxylate transporters active in lactate exchange in glycolytic tumors. Accordingly, genetic ablation of MCT4 expression overcomes adaptive resistance against anti-angiogenic therapy. Hence, targeting metabolic symbiosis may be an attractive avenue to avoid resistance development to anti-angiogenic therapy in patients.
Graphical Abstract
Highlights
-
•
Tumors can escape anti-angiogenic therapy with multi-kinase inhibitors
-
•
A glycolytic shift underlies resistance against multi-kinase inhibitors
-
•
Metabolic symbiosis between hypoxic and oxygenated cells inspires therapy resistance
-
•
Inhibition of glycolysis or lactate export collapses metabolic symbiosis
Pisarsky et al. examine the role of metabolic symbiosis as a mechanism underlying evasive resistance to anti-angiogenic therapy by the multi-kinase inhibitors nintedanib and sunitinib. Inhibition of glycolysis or genetic ablation of the lactate exporter MCT4 in tumor cells disrupts metabolic symbiosis, overrides therapy resistance, and suppresses tumor growth.
Introduction
An imbalance between pro- and anti-angiogenic factors inducing the formation of new blood vessels from a pre-existing vasculature (angiogenesis) has been described as a hallmark of cancer (Hanahan and Weinberg, 2011). Hence, targeting angiogenesis might plausibly reduce intra-tumoral levels of oxygen and nutrients, resulting in tumor starvation and thus in reduced tumor growth (Folkman, 1971). Anti-angiogenic therapies have been rapidly translated with great expectations from preclinical cancer models to clinical practice (Carmeliet and Jain, 2011, Crawford and Ferrara, 2009, Ferrara and Kerbel, 2005). For example, the identification of vascular endothelial growth factor (VEGF-A) and its receptors as rate-limiting factors for normal and pathological angiogenesis has led to the development of bevacizumab (Avastin), a humanized monoclonal antibody targeting VEGF-A (Ferrara et al., 2004, Ferrara and Kerbel, 2005). Some cancer types, such as colorectal (Hurwitz et al., 2004), renal cell (Motzer et al., 2007), and pancreatic neuroendocrine carcinomas (PNETs) (Raymond et al., 2011), have shown encouraging responses to this therapeutic strategy. However, numerous other cancer types, in particular breast cancer, seem to be poorly responsive to anti-angiogenic regimens. Indeed, metastatic breast cancer patients treated with standard chemotherapy plus bevacizumab benefit from only 1 or 2 months of progression-free survival. The rapid onset of resistance evidently prevents any overall survival benefit (Kerbel, 2009, Miller et al., 2007, Rose, 2011).
These data underline the importance of deciphering the molecular mechanisms underlying intrinsic or adaptive resistance to anti-angiogenic therapy. When blocking the VEGF-A signaling axis in preclinical models, e.g., with bevacizumab, tumors escape by activating alternative pro-angiogenic signaling pathways, including signaling by fibroblast growth factors (FGFs), platelet-derived growth factors (PDGFs), Bv8/prokineticin, and interleukin-17 (IL-17) (Bergers and Hanahan, 2008, Casanovas et al., 2005, Chung et al., 2013, Compagni et al., 2000, Ferrara, 2010). In order to counteract the activation of these alternative pro-angiogenic pathways, several multi-kinase inhibitors, targeting VEGF-dependent and independent pro-angiogenic signaling pathways, are currently in clinical use or in clinical trials. For example, sorafenib, a multi-kinase inhibitor targeting RAF, VEGF receptors (VEGFRs) 1–3, PDGF receptors (PDGFRs) α and β, c-KIT, and FLT-3, is currently used for the treatment of hepatocellular carcinoma. Sunitinib, blocking VEGFR1–3, PDGFRα/β, c-KIT, and FLT-3, is employed for the treatment of renal cancer. Both inhibitors show significant anti-tumor efficacy in preclinical tumor models and in cancer patients; however, they also suffer from resistance development based on thus far unknown mechanisms (Pàez-Ribes et al., 2009, Raymond et al., 2011). Transient benefits are rapidly followed by tumor recurrence, sometimes associated with drug resistance and heightened tumor invasiveness (Bergers and Hanahan, 2008, Ebos and Kerbel, 2011, Pàez-Ribes et al., 2009, Sennino and McDonald, 2012, Singh and Ferrara, 2012).
Nintedanib (BIBF-1120) is an even-wider-spectrum angiokinase inhibitor targeting VEGFR1–3, PDGFα/β, and FGF receptors (FGFRs) 1–4, as well as FLT-3 and SRC family kinases (Hilberg et al., 2008). Nintedanib has recently shown promising results in pre-clinical models of lung cancer, ductal adenocarcinoma of the pancreas, and PNET (Awasthi et al., 2015, Bill et al., 2015, Kutluk Cenik et al., 2013). Furthermore, nintedanib has demonstrated excellent tolerance and potent activity in a phase I clinical trial in early HER2-negative breast cancer (Quintela-Fandino et al., 2014) and in a phase III study in non-small-cell lung cancer (NSCLC), leading to its approval as a second-line treatment in combination with docetaxel for advanced NSCLC (McCormack, 2015, Reck et al., 2014).
We have therefore assessed the effects of nintedanib in mouse models of cancer. We report that tumors treated with nintedanib or sunitinib do not revascularize during the development of therapy resistance. Instead, the cells located in avascular areas escape the lack of oxygen by shifting their metabolism toward a hyperglycolytic state and by producing lactate. Conversely, the cells localized in the vicinity of blood vessels utilize the lactate for oxidative phosphorylation. The data establish metabolic symbiosis (Porporato et al., 2011, Sonveaux et al., 2008) as an alternative route to develop resistance to anti-angiogenic therapy in mouse models of breast cancer and of insulinoma. Notably, interference with glycolysis or disruption of metabolic symbiosis reinstalls nintedanib’s efficacy in repressing tumor growth.
Results
Py2T Tumors Develop Evasive Resistance to Anti-angiogenic Therapy
Nintedanib is a potent angiogenesis inhibitor that represses endothelial cell proliferation and induces their apoptosis (EC50 < 10 nM), yet with limited direct effects on tumor cells (Hilberg et al., 2008). A stable murine breast cancer cell line (Py2T) established from a breast tumor of an MMTV-PyMT transgenic mouse (Waldmeier et al., 2012) displayed an EC50 of 8 μM in vitro, which is above the pharmacologically achievable concentration in mice (Hilberg et al., 2008, Roth et al., 2009; Figure S1A). To study the tumor-suppressive efficacy of nintedanib in vivo, Py2T cells were orthotopically implanted into the mammary fat pad of immune-competent syngeneic FVB/N female mice. When tumors reached a volume of 15–20 mm3, where the angiogenic switch had already taken place (Figure S1B), daily treatment with nintedanib was initiated (50 mg/kg; p.o.). During the first week of treatment (short-term [ST] treatment), tumor volumes as well as tumor weights in nintedanib-treated animals were significantly reduced (Figures 1A and 1B). This nintedanib-responsive phase was associated with decreased cell proliferation and increased apoptosis (Figures 1C–1F). However, after 3 weeks of treatment (long-term [LT] treatment), tumors escaped this therapeutic effect and showed an enhanced tumor growth with increased cell proliferation and reduced apoptosis (Figures 1A and 1C–1F). Apparently, Py2T breast cancer cells escaped nintedanib treatment despite its broad range of inhibitory activities.
Figure 1.

Evasive Resistance to Anti-angiogenic Therapy
Py2T murine breast cancer cells were implanted into the mammary fat pad of FVB/N mice and treated with nintedanib (50 mg/kg daily p.o.) or vehicle control from day 14 after tumor cell implantation.
(A) Primary tumor growth was monitored by assessing tumor volumes over the time of therapy. Values represent mean ± SEM. n = 13 mice per group.
(B) Tumor weights were determined after 7 days of nintedanib short-term (ST) treatment. n = 6–8 mice per group.
(C–F) Cell proliferation (C and D) and the incidence of apoptosis (E and F) were quantified by immunofluorescence staining for phospho-histone 3 (pH3; red) and cleaved caspase-3 (cCasp3; red), respectively, of tumor sections from ST and LT vehicle or nintedanib-treated mice. Representative immunofluorescence microscopy pictures are shown in (D) and (F). DAPI was used to visualize cell nuclei. Values represent the number of pH3-positive (C) and cCasp3-positive (E) cells per area of each microscopic field of view. n = 5–8 mice per group. Mann-Whitney U test. ∗p < 0.05; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. The scale bars represent 50 μm.
See also Figure S1.
Evasive Resistance Is Not Associated with Tumor Revascularization
Next, we investigated whether angiogenesis had been reactivated in LT-treated Py2T tumors, thereby escaping nintedanib treatment. Intriguingly, microvessel density was found decreased both after ST and LT nintedanib regimen, indicating a potent and stable anti-angiogenic effect of nintedanib, even in a phase of drug-refractory exponential tumor growth (Figures 2A, 2B, and S2A). The numbers of blood vessels became more variable following LT nintedanib treatment, potentially indicating an initiation of revascularization. However, immunofluorescence co-staining for CD31 and cleaved caspase 3 (cCasp3) revealed increased apoptosis in endothelial cells after ST and LT nintedanib treatment, demonstrating the sustained anti-angiogenic efficacy of nintedanib even after LT treatment (Figures 2C and 2D). This therapy-resistant tumor growth was not specific for the multi-kinase inhibitor nintedanib; in a head-to-head comparison, Py2T tumors treated with nintedanib and sunitinib displayed comparable tumor growth and reduced microvessel densities after LT treatment (Figures S2B–S2D).
Figure 2.

Lack of Tumor Revascularization during Resistance against Nintedanib Therapy
(A and B) Microvessel densities (A) and CD31-positive area fractions (B) were quantified in Py2T tumors from mice treated for 1 week (ST) or 3 weeks (LT) with vehicle or nintedanib.
(C) Endothelial cell apoptosis (CD31, green; cCasp3, red) is shown on representative immunofluorescence picture of a tumor from a 1-week (ST) nintedanib-treated mouse. DAPI was used to visualize cell nuclei. The scale represents 20 μm.
(D) Quantification of endothelial cell apoptosis by immunofluorescence co-staining for cCasp3 and CD31 in tumors from ST and LT vehicle or nintedanib-treated mice.
(E) Quantification of the percentage of CD31-positive blood vessels that were in contact with NG2-positive perivascular cells in Py2T tumors from ST and LT vehicle or nintedanib-treated mice.
(F) The functionality of blood vessels was assessed by i.v. injection of fluorescein isothiocyanate (FITC)-lectin into Py2T tumor-bearing mice following ST or LT vehicle or nintedanib-treatment. Patent, perfused blood vessels were identified by immunofluorescence staining for CD31 and detection of FITC-lectin and quantified by counting CD31 and lectin double-positive blood vessels.
(G) Hypoxic areas were identified and quantified by immunofluorescence staining for pimonidazole adducts in Py2T tumors from ST and LT vehicle or nintedanib-treated mice.
(H) Representative pictures of the immunofluorescence co-staining for pimonidazole adducts (red) and CD31 (green) on histological sections of tumors from ST and LT vehicle or nintedanib-treated mice. DAPI staining visualizes cell nuclei. The scale bars represent 100 μm.
n = 6–8 mice per group. Mann-Whitney U test. n. s., non-significant; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Figure S2.
Next, we assessed whether Py2T tumors compensate for the lack of blood vessels with increased pericyte coverage. Pericytes promote the maturation and stabilization of blood vessels through PDGFR signaling and thus influence the responsiveness to anti-angiogenic therapy (Hellström et al., 1999). Interestingly, despite its inhibitory activity on PDGFR signaling, nintedanib did not affect the pericyte coverage of blood vessels resisting nintedanib treatment (Figures 2E and S2E). Nintedanib also did not affect the functionality of the remaining blood vessels as determined by the injection of fluorescence-labeled lectin (Figures 2F and S2F). Consistent with decreased tumor perfusion, pimonidazole staining revealed a significant increase in tumor hypoxia not only in the ST-treated, nintedanib-responsive tumors but also in the LT-treated, nintedanib-resistant tumors (Figures 2G and 2H). These data demonstrate a potent anti-angiogenic activity of nintedanib and suggest a mechanism of therapy resistance by which tumors escape anti-angiogenic therapy in the absence of any revascularization.
Tumor Cells Become Hyperglycolytic to Survive Hypoxia
To investigate the molecular mechanisms underlying the resistance against nintedanib treatment, we isolated by flow cytometry endothelial and tumor cells from nintedanib-treated and untreated tumors at different time points of resistance development. To facilitate the isolation of tumor cells, Py2T cells were transduced with a retroviral construct expressing a truncated, non-functional form of murine CD8α (Misteli et al., 2010). A CD45−CD8+ population could only be identified in Py2T-CD8α+ tumors and not in wild-type Py2T tumors (Figure S3A). After ST (1 week) and LT (3 weeks) treatment with nintedanib, CD45−CD8α+ tumor cells and CD45−CD8α−CD31+podoplanin− endothelial cells were sorted by flow cytometry (Figures S3B–S3D). Changes in gene expression were assessed by DNA oligonucleotide microarray analysis. Surprisingly, endothelial cell gene expression profiles between ST and LT nintedanib-treated tumors did not markedly differ, mainly reflecting endothelial cells undergoing apoptosis (data not shown).
In contrast, gene expression analysis of isolated tumor cells revealed a marked difference between untreated and treated groups. The genes resulting from the comparison between LT nintedanib-treated and untreated tumor cells were subjected to Kyoto Encyclopedia of Genes and Genomes (KEGG)-pathway analysis, which showed an enrichment of metabolic pathways, in particular glycolysis (Figure 3A). Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) also showed an enrichment of glycolysis gene expression, especially when comparing the gene expression profiles of LT versus untreated tumor cells, yet also when comparing ST versus untreated tumor cells (Figure 3B). Glycolysis gene enrichment also became evident when the gene expression profiles associated with a core set of glycolytic enzymes were visualized using a heatmap. Indeed, hierarchical clustering correctly interrelated the three different treatment conditions (Figure 3C). qRT-PCR analysis confirmed the upregulated expression of most of the glycolytic enzymes upon ST and LT nintedanib treatment, whereas the expression of genes implicated in mitochondrial biogenesis and oxidative phosphorylation was unaffected (Figures 3D and 3E).
Figure 3.

Tumor Cells Become Hyperglycolytic during Nintedanib Treatment
(A) Differential gene expression between flow-cytometry-isolated LT nintedanib and vehicle-treated tumor cells was assessed by Affymetrix microarray analysis. The list of differentially expressed genes was subjected to KEGG pathway analysis.
(B) Gene set enrichment analysis (GSEA) between gene expression profiles of either ST or LT nintedanib and vehicle-treated tumor cells. Shown are the normalized enrichment score (NES) and the false discovery rate (FDR) q value.
(C) A set of core glycolysis enzymes was used to perform hierarchical clustering of gene expression profiles derived from LT and ST nintedanib and vehicle-treated controls.
(D and E) Expression of different glycolysis and mitochondrial-activity-related transcripts in ST (D) and LT (E) nintedanib-treated tumors analyzed by qRT-PCR is shown. Data are normalized to vehicle-treated tumors. Shown are mean ± SEM. n = 4 mice per group. Mann-Whitney U test. n.s., non-significant; ∗p < 0.05; ∗∗p < 0.01.
See also Figure S3.
Because nintedanib-treated tumors exhibited enhanced hypoxia compared to size-matched, vehicle-treated tumors (Figures 2G and 2H), we hypothesized that hypoxia could be a determinant of tumor cell heterogeneity and a direct inducer of the glycolytic shift. As expected, when compared with normoxic cultures, Py2T cells cultured for 3 days in hypoxic conditions (1% O2) exhibited a significantly increased expression of nine out of ten glycolysis-related transcripts analyzed (Figure S3E).
Together, the data suggest a metabolic adaptation to anti-angiogenic therapy, in which hypoxic tumor cells shift to a hyperglycolytic state to survive and proliferate at reduced oxygen and nutrient supply.
Therapy Resistance Establishes Metabolic Symbiosis
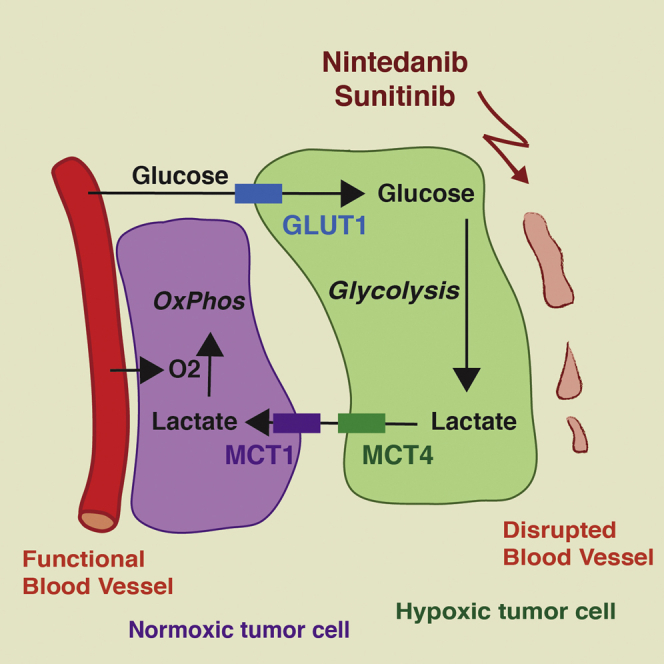
Considering the highly glycolytic phenotype of nintedanib-treated tumor cells, we analyzed lactate production in Py2T tumors. Total lactate production was not increased in nintedanib-treated tumors compared to vehicle-treated tumors (Figure S4A), possibly explained by a fast metabolic utilization of lactate. The alternation between highly hypoxic and normoxic areas in nintedanib-treated tumors (Figure 2H), together with comparable levels of lactate between nintedanib and vehicle-treated tumors, suggested the establishment of lactate-based metabolic symbiosis (Sonveaux et al., 2008). In such symbiosis, hypoxic glycolytic cells use glucose to produce high levels of lactate that is rapidly exported through monocarboxylate transporter 4 (MCT4), mainly a lactate exporter. Oxidative cells located in perfused areas express MCT1, mainly a lactate importer, allowing them to take up lactate and directly fuel their Krebs cycle. These cells do not rely on glycolysis, and glucose can bypass them and diffuse to hypoxic areas, where it is taken up by glycolytic cells expressing high levels of hypoxia-induced glucose transporter 1 (Glut1) to produce lactate.
We assessed the establishment of metabolic symbiosis during the development of resistance against nintedanib-mediated anti-angiogenic therapy in the Py2T transplantation model of breast cancer. Immunofluorescence staining for MCT1 and MCT4 demonstrated a diffuse baseline expression of MCT1 that remained unchanged during nintedanib treatment, whereas MCT4 was highly expressed in non-vascularized areas of LT nintedanib-treated tumors and to a lesser extent in ST-treated tumors (Figures 4A and S4B–S4D). Similar results were observed in sunitinib-treated tumors (Figure S4E). To assess the generality of our findings, we analyzed microvessel densities and MCT4 expression in tumors of Rip1Tag2 transgenic mice that have been treated with nintedanib (Bill et al., 2015). The Rip1Tag2 transgenic mouse model of PNET is highly sensitive to anti-angiogenic therapies and has been instrumental for compound testing and subsequent successful translation to the treatment of patients with PNETs (Tuveson and Hanahan, 2011). With Rip1Tag2 mice, nintedanib treatment was initiated at 10 weeks of age, which prolonged median survival from 24 days in control-treated animals to 55 days in nintedanib-treated animals. Comparable to the Py2T breast cancer model, Rip1Tag2 mice also developed resistance to nintedanib therapy and did not display any revascularization in therapy-refractory tumors (Figure S4F), and MCT4 expression was also only found in tumors after prolonged nintedanib treatment (Figure S4G).
Figure 4.

Tumors Establish Metabolic Symbiosis to Overcome Nintedanib Treatment
(A) Representative pictures of combinatorial immunofluorescence staining for MCT1, MCT4, and CD31 on histological sections of tumors from mice treated with either vehicle or nintedanib (50 mg/kg/day) are shown, as indicated. DAPI was used to visualize cell nuclei. The scale bars represent 100 μm.
(B) Quantification of the closest distance separating blood vessels from MCT4+ areas by immunofluorescence co-staining for MCT4 and CD31 on Py2T tumors from ST and LT vehicle or nintedanib-treated mice. Note that, in ST vehicle-treated tumors, MCT4 was not significantly expressed and thus the distance to blood vessels could not be determined.
(C and D) Quantification of the Glut1+ area fraction (C) and the MCT4+ area fraction within Glut1+ areas (D) by immunofluorescence co-staining for MCT4 and Glut1 on Py2T tumors from ST and LT vehicle or nintedanib-treated mice.
(E) Representative microphotographs of immunofluorescence co-staining for MCT4 and Glut1 on histological sections of tumors from ST and LT vehicle or nintedanib-treated mice. DAPI is used to visualize cell nuclei. The scale bars represent 100 μm.
(F) Quantification of the hypoxic (pimonidazole+) area fraction within Glut1+ areas by immunofluorescence co-staining for pimonidazole and Glut1 on Py2T tumors from ST and LT vehicle or nintedanib-treated mice.
(G) Representative microphotographs of immunofluorescence co-staining for pimonidazole and Glut1 on histological sections of tumors from ST and LT vehicle or nintedanib-treated mice. DAPI was used to visualize cell nuclei. The scale bars represent 100 μm.
(H) Quantification of the MCT4+ area fraction within pimonidazole+ areas by immunofluorescence co-staining for MCT4 and pimonidazole on Py2T tumors from ST and LT vehicle or nintedanib-treated mice.
(I) Representative microphotographs of immunofluorescence co-staining for MCT4 and pimonidazole on histological sections of tumors from ST and LT vehicle or nintedanib-treated mice. DAPI is used to visualize cell nuclei. The scale bars represent 100 μm.
(J) Representative microphotographs of immunofluorescence co-staining for PGC1α and CD31 on histological sections of tumors from ST and LT vehicle or nintedanib-treated mice. DAPI is used to visualize cell nuclei. The scale bars represent 50 μm.
n = 4 mice per group. Mann-Whitney U test. ∗p < 0.05; ∗∗p < 0.01. See also Figure S4.
To further assess the establishment of metabolic symbiosis in nintedanib therapy-resistant tumors, we assessed by immunofluorescence microscopy analysis the expression and localization of markers for hypoxia (pimonidazole), glucose uptake (Glut1), lactate export (MCT4), mitochondrial biogenesis, and oxidative phosphorylation (PGC1α and COX IV; LeBleu et al., 2014, Wu et al., 1999). Notably, the mean shortest distance between MCT4-expressing cells and the nearest blood vessel was increased in LT tumors, although not with statistical significance (Figure 4B), indicating the expression of MCT4 in hypoxic areas. Indeed, the expression of hypoxia-induced Glut1 correlated with the expression of hypoxia-induced MCT4 and with the hypoxia-marker pimonidazole in the hypoxic areas of nintedanib LT tumors (Figures 4C–4G, S4H, and S4I). The expression of MCT4 co-localized with pimonidazole as well (Figures 4H, 4I, and S4J). On the other hand, the expression of PGC1α and COX IV did not specifically localize with vascularized or non-vascularized areas yet increased in ST and LT nintedanib-treated tumors (Figures S4K and S4N). Curiously, the co-expression of MCT4 with PGC1α and COX IV was decreased and unchanged, respectively, in ST nintedanib-treated tumors, yet it was unchanged with PGC1α and increased with COX IV comparing LT vehicle and nintedanib-treated tumors (Figures S4L, S4M, S4O, and S4P). These results suggest a first wave of tumor hypoxia and glycolysis followed by a homeostasis of metabolic symbiosis between anaerobic glycolysis and aerobic oxidative phosphorylation during prolonged anti-angiogenic therapy.
Targeting Glycolysis or Metabolic Symbiosis Delays Resistance Development
The small molecule 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) inhibits the glycolytic activator phosphofructokinase-2/fructose-2,6-bisphosphatase 3 (PFKFB3) in endothelial cells (Schoors et al., 2014). Its combined activity as a glycolysis and endothelial cell inhibitor made it a prime compound to overcome glycolysis-induced resistance to anti-angiogenic therapy. Whereas single treatment with nintedanib significantly repressed tumor growth in Py2T-transplanted mice, single treatment with 3PO only marginally delayed it (Figures 5A and 5B). Notably, the combined treatment with nintedanib and 3PO showed an additive effect on tumor growth inhibition. This combined effect was not mediated by an additive anti-angiogenic effect, because the microvessel densities between the nintedanib single and the nintedanib plus 3PO combination treatments were not significantly altered (Figure 5C). Consistent with its ability to normalize blood vessels, single treatment with 3PO significantly increased pericyte coverage and thus vessel functionality, possibly explaining the limited repression of tumor growth despite the significant decrease in microvessel density (Figure 5D; Schoors et al., 2014). This effect was abrogated upon combined 3PO and nintedanib treatment.
Figure 5.

Targeting Glycolysis or Metabolic Symbiosis in Combination with Nintedanib Treatment Delays Tumor Growth
(A and B) Primary tumor growth over time (A) and tumor weights at the experimental endpoint (B) of mice treated with either vehicle or nintedanib (50 mg/kg/day) in combination with 3PO (70 mg/kg/day) or solvent are shown. 3PO treatment was initiated 8 days after the initiation of nintedanib treatment and then continued as combinatorial treatment (LT treatment). In (A), data are displayed as mean tumor volumes ± SEM.
(C) Quantification of microvessel densities by immunofluorescence staining for CD31 on histological tumor sections from LT nintedanib and 3PO-treated mice. Values represent the number of counts per each area of microscopic field of view, and means are displayed. n = 6–8 mice per group.
(D) Pericyte coverage was assessed by immunofluorescence staining for CD31 and NG2 on histological tumor sections from LT nintedanib and 3PO-treated mice. Values represent the percentage of NG2+ blood vessels, and means are displayed. n = 4–5 mice per group.
(E and F) Primary tumor growth over time (E) and tumor weights at the experimental endpoint (F) of mice injected with Py2T wild-type (WT) or Py2T CRISPR MCT4 no. 1 and no. 2 cells and treated with either vehicle control or nintedanib (50 mg/kg/day) are shown. Nintedanib treatment was initiated 19 days after tumor cell injection, once the tumors were palpable. Mice injected with CRISPR MCT4 no. 1 cells presented a delayed tumor onset and were therefore treated once the tumors became palpable (days 27–38). In (E), data are displayed as mean tumor volumes ± SEM. n = 4–7 mice per group.
Mann-Whitney U test. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Figure S5.
To determine the early effects of 3PO treatment on nintedanib-treated tumors, Py2T-transplanted mice were first treated with nintedanib for 8 days and then subjected to treatment with 3PO and nintedanib for subsequent 5 days. Whereas nintedanib significantly repressed tumor growth upon ST treatment, 3PO treatment did not add further tumor repression (Figures S5A and S5B). However, the extent of tumor hypoxia and the rate of tumor cell apoptosis specifically in the hypoxic tumor areas significantly increased upon combined nintedanib/3PO treatment (Figures S5C–S5E). Collectively, these results suggest that the inhibition of glycolysis is one avenue of overcoming resistance to anti-angiogenic therapy with multi-kinase inhibitors.
To determine whether the inhibition of metabolic symbiosis could overcome the development of resistance against anti-angiogenic therapy, we generated Py2T cell lines that were devoid of MCT4 by CRISPR/Cas9-mediated knockout of the Slc16a3 gene (MCT4 is known as solute carrier 16 a3 [Slc16a3]). Two stable cell clones (CRISPR MCT4 no. 1 and no. 2), which showed targeted recombination in the Slc16a3 gene and did not express MCT4 protein anymore, were used for further experimentation (Figure S5F). The loss of MCT4 expression in these clones significantly repressed tumor growth as compared to wild-type cells under treatment, with nintedanib treatment leading to an additive effect in repressing tumor growth kinetics and final tumor weights (Figures 5E and 5F). These results were confirmed by short hairpin RNA (shRNA)-mediated ablation of MCT4 expression (shMCT4) in Py2T cells (Figure S5G). The loss of MCT4 expression in shMCT4 cell lines significantly retarded tumor growth kinetics and final tumor weights under treatment with nintedanib as compared to shCtrl cells (Figures S5G–S5I). However, after a first delay, shMCT4 tumors resumed growth. Immunofluorescence staining for CD31 did not reveal any increase in microvessel density in nintedanib-treated shMCT4 tumors, excluding an escape route by revascularization (Figure S5J). Instead, we observed an increase of MCT4 expression both at the protein and mRNA level in nintedanib-treated shMCT4 tumors (Figures S5K and S5L), suggesting that cells with poor shRNA-mediated knockdown efficiency developed a selective growth advantage and elicited tumor recurrence.
Hypoxia-Induced Glycolysis Is Reverted by 3PO and Loss of MCT4
The results presented above beg the question whether, in Py2T tumor cells, hypoxia-induced glycolysis is directly affected by treatment with nintedanib and 3PO or the loss of MCT4 expression. Thus, we performed extracellular flux analysis by “Seahorse” methodology to determine the oxygen consumption rate (OCR) as a measure of oxidative phosphorylation and the extracellular acidification rate (ECAR) as a measure of glycolysis. As expected, under hypoxic conditions, Py2T cells exhibited increased ECAR (glycolysis) and decreased OCR (oxidative phosphorylation) as compared to normoxic conditions (Figures 6A and 6B). When directly quantified, hypoxic cells had reduced ATP-coupled OCR, increased ECAR, unchanged glycolytic capacity, and decreased glycolytic reserve as compared to cells cultured under normoxia (Figures 6C–6F). To determine any effects of therapeutic treatments on the rates of glycolysis and oxidative phosphorylation, the ratios between ECAR and OCR were determined in wild-type or MCT4 knockout Py2T cells cultured under normoxia or hypoxia and treated with solvent, nintedanib, or 3PO. These experiments revealed that nintedanib did not affect the ratio between ECAR and OCR (Figure 6G), whereas 3PO reduced this ratio, i.e., it decreased glycolysis and increased oxidative phosphorylation under hypoxic, but not normoxic, conditions (Figure 6H). The genetic ablation of MCT4 expression also reduced ECAR/OCR only under hypoxic growth conditions (Figure 6I), which also resulted into increased tumor cell apoptosis and cell-cycle arrest (Figures 6J and 6K).
Figure 6.

Glycolysis Induced by Hypoxia Can Be Reverted by Treatment with 3PO or the Loss of MCT4
(A and B) Shown are the measurements of representative oxygen consumption rates (A; OCRs) and extracellular acidification rates (B; ECARs) of Py2T cells cultured in normoxic or hypoxic conditions. n = 5.
(C–F) Quantification of ATP-coupled respiration (C), glycolysis (D), glycolytic capacity (E), and glycolytic reserve (F) of Py2T cells cultured under normoxic or hypoxic conditions. See Supplemental Experimental Procedures for details. Data are displayed as mean ± SD. n = 5 (glycolytic reserve: n = 4). Statistical significance was calculated using two-way ANOVA test.
(G and H) ECAR/OCR ratio of Py2T cells cultured under normoxic or hypoxic conditions and treated with DMSO, 0.5 μM or 1 μM nintedanib (G), or 5 μM 3PO (H). Data are displayed as mean ± SD. n = 4. Two-way ANOVA test.
(I) ECAR/OCR ratio of Py2T WT cells or Py2T CRISPR MCT4 no. 1 and no. 2 cells cultured under normoxic or hypoxic conditions. Data are displayed as mean ± SD. n = 4. Two-way ANOVA test.
(J) The percentages of apoptotic Py2T WT cells or Py2T CRISPR MCT4 no. 1 and no. 2 cells cultured under normoxic or hypoxic conditions were assessed using flow cytometry analysis of annexin-V-expressing cells. Data are displayed as mean ± SD. n = 3. One-way ANOVA test.
(K) Cell-cycle analysis for Py2T WT cells or Py2T CRISPR MCT4 clones no. 1 and no. 2 cultured under normoxic or hypoxic conditions was performed using EdU staining. Data are displayed as mean ± SD. n = 3. Two-way ANOVA test.
∗p < 0.05; ∗∗p < 0.01.
Taken together, the data show that anti-angiogenic resistance can occur via the establishment of metabolic symbiosis and that interfering with metabolic symbiosis can overcome resistance to anti-angiogenic therapy with multi-kinase inhibitors.
Discussion
In this and in the accompanying reports by Allen et al. (2016); this issue of Cell Reports and Jiménez-Valerio et al. (2016); this issue of Cell Reports, we report the intriguing finding that a glycolytic shift underlies the development of resistance to anti-angiogenic therapy with multi-kinase inhibitors. Notably, in response to the efficient repression of tumor angiogenesis, tumors compartmentalize into hypoxic regions at a distance from blood perfusion and into normoxic regions in the vicinity of mature and functional blood vessels. The hypoxic tumor cells exhibit high glucose uptake by the hypoxia-induced expression of Glut1, and they efficiently generate and export lactate by the hypoxia-induced expression of the lactate exporter MCT4. Conversely, the normoxic tumor cells take up the lactate produced by the hypoxic tumor cells and oxygen from nearby blood vessels and fuel both into oxidative phosphorylation (Figure 7). Such aspect of metabolic intra-tumoral heterogeneity is portrayed by the concept of metabolic symbiosis (Sonveaux et al., 2008).
Figure 7.

Targeting Metabolic Symbiosis Overcomes Resistance to Anti-angiogenic Therapy
Anti-angiogenic therapy induces hypoxia and reduces the supply of nutrients. As a result, tumor cells shift their metabolism toward a hyperglycolytic state and establish metabolic symbiosis: tumor cells in hypoxic areas upregulate glycolysis, increase lactate production, and export lactate via MCT4. On the other hand, lactate is taken up by tumor cells in more-oxygenated regions of the tumor and is directly fueling the citric acid cycle and thus oxidative phosphorylation. As a consequence, tumor cells in normoxic tumor regions reduce glucose consumption, which increases its diffusion distance. Ablating MCT4 expression (MCT4 KO or shMCT4) or inhibition of glycolysis (3PO) disrupts this homeostatic interplay and decreases tumor growth.
Here, we have analyzed the efficacy of the angiokinase inhibitors nintedanib and sunitinib in a preclinical mouse model of breast cancer and in the Rip1Tag2 transgenic mouse model of pancreatic neuroendocrine cancer. Treatment of Py2T tumor-bearing mice and of Rip1Tag2 mice with the angiogenesis inhibitors has led to a significant therapeutic response, characterized by increased tumor and endothelial cell apoptosis, decreased tumor cell proliferation, and reduced tumor size. However, despite the potent anti-angiogenic efficacies, the treated tumors rapidly escape therapy. Evasive resistance to anti-angiogenic therapy has previously been reported to rely partially on the redundancy of pro-angiogenic growth factors leading to tumor revascularization (Bergers and Hanahan, 2008, Chung et al., 2013, Ferrara, 2010). Intriguingly, the nintedanib- and sunitinib-resistant tumors do not show any evidence of revascularization. Rather, with the reduction in tumor perfusion, hypoxia is increased in resistant tumors and microarray gene expression analysis reveals a metabolic shift to glycolysis in the resistant tumor cells. Indeed, glycolysis and glucose-transport-related genes are well known targets of hypoxia-induced cellular adaptations (Harris, 2002), and glycolysis induction has been recently described in response to VEGF inhibitors (Curtarello et al., 2015, Kumar et al., 2013).
The tumor cells’ shift to glycolysis as a mechanism underlying resistance against anti-angiogenic therapy offers the opportunity of defeating therapy-resistance by interfering with glycolysis. Indeed, in this and in the accompanying reports (Allen et al., 2016, Jiménez-Valerio et al., 2016), combination therapy involving angiokinase inhibitors with 3PO (our work), a glycolytic flux inhibitor (Clem et al., 2008, Schoors et al., 2014), or with rapamycin, an mTOR and glycolysis inhibitor (presented in the accompanying papers by Allen et al. [2016] and Jiménez-Valerio et al. [2016]), surmounts resistance to treatment. However, combination treatment of nintedanib with 2-deoxyglucose, a competitive inhibitor of the production of glucose-6-phosphate from glucose (Wick et al., 1955), did not delay tumor growth, most likely due to the fact that we have been unable to supply the very high concentrations of 2-deoxyglucose in tumors that would be pharmacologically active (data not shown). Dichloroacetate (DCA), a drug inhibiting pyruvate dehydrogenase kinase and thus promoting glucose oxidation over glycolysis by increasing the pyruvate flux into mitochondria (Michelakis et al., 2010), also has not shown any effect on tumor growth (data not shown). Hence, the pharmacological targeting of glycolysis in the context of anti-angiogenic therapy may be more complex than anticipated.
Along these lines, despite a clear hypoxia-response pattern to nintedanib therapy, high-throughput metabolomic analysis of tumor lysates from treated mice has failed to show any significant differences in central carbon metabolism between nintedanib LT and untreated tumors (data not shown). However, this snapshot analysis ex vivo may be obscured by the concomitant presence of cells using hypoxia/glycolysis or oxidative phosphorylation within the same tumor, thus averaging out the metabolites specific for the distinct cellular subpopulations. In a comparable way, metabolic flow analysis with labeled substrates of glycolysis and oxidative phosphorylation is hampered by the lack of a technique to directly measure the metabolites of localized cell subpopulations within a tumor. We have thus used established markers for angiogenesis, hypoxia, metabolite transport, and mitochondrial function to visualize the distinct metabolic compartments. Moreover, we have analyzed the hypoxia-induced metabolic shift between glycolysis and oxidative phosphorylation in cultured tumor cells by Seahorse technology and have found that inhibition of glycolysis by 3PO as well as the genetic ablation of MCT4 expression repress hypoxia-induced glycolysis and induce cell-cycle arrest and apoptosis.
Regions with higher oxygen partial pressure metabolize lactate produced in hypoxic areas and thus increase the diffusion capacity of oxygen and glucose. Indeed, increased expression of MCT4 has been correlated with poor prognosis in melanoma and breast cancer (Doyen et al., 2014, Ho et al., 2012). Accordingly, the genetic ablation of MCT4 expression in Py2T tumors treated with nintedanib show significantly delayed tumor growth. Our data therefore suggest that (1) despite the broad range of activities of the multi-kinase inhibitors nintedanib and sunitinib, tumors can still escape treatment; (2) nintedanib and sunitinib resistance does not occur via tumor revascularization but is induced by a metabolic shift toward glycolysis and the establishment of metabolic symbiosis; and (3) nintedanib and sunitinib treatment should be used in combination with glycolysis/metabolic symbiosis inhibitors for LT efficacy (Figure 7). Along these lines, it has been recently reported that the genetic disruption of MCT1 or MCT4 represses breast tumor growth (Morais-Santos et al., 2015) and sensitizes glycolytic tumor cells to treatment with phenformin, an inhibitor of mitochondrial complex I (Marchiq and Pouysségur, 2016). However, complicating things, a recent investigation of metabolic changes in tumors after cessation of sunitinib or sorafenib therapy has revealed a metabolic shift to lipid synthesis and blockade of lipidogenesis has inhibited tumor regrowth (Sounni et al., 2014).
In conclusion, the data presented here and in the accompanying reports underscore the variety of evasive responses to anti-angiogenic and likely to other targeted therapies. The establishment of metabolic symbiosis adds not only another level of complexity but also a number of druggable targets to the design of combinatorial therapies. The results also emphasize the importance of intra-tumoral heterogeneity as therapy response, in particular with regard to oxygen and nutrient availability. Such heterogeneity likely masks critical adaptation mechanisms when performing cross-sectional analysis without spatial resolution.
Experimental Procedures
Mice
FVB/N mice were kept and bred under specific pathogen-free (SPF) conditions. The generation and characterization of Rip1Tag2 transgenic mice has been described elsewhere (Hanahan, 1985). All experiments were performed following the rules and legislations of the Cantonal Veterinary Office and the Swiss Federal Veterinary Office (SFVO) under license numbers 1878, 1907, and 1908.
Cell Lines and Orthotopic Tumor Cell Transplantation
Py2T murine breast cancer cells were cultured as previously described (Waldmeier et al., 2012). 5 × 105 cells were orthotopically injected into the mammary gland number 9 of 7- to 11-week-old female FVB/N mice under isoflurane/oxygen anesthesia. Tumor length (l) and width (w) were assessed three times per week using a vernier caliper, and tumor volume (V) was calculated using the formula V = 0.543 × l × w2.
Therapy Studies, RNA Isolation, qRT-PCR, Immunofluorescence Microscopy Analysis, Flow Cytometry, Microarray Analysis, and Bioinformatic Analysis
See the Supplemental Experimental Procedures.
Establishment of CRISPR MCT4 Cell Lines
Subconfluent Py2T cells were transfected with 2 μg of MCT4 CRISPR/Cas9 KO plasmid and 2 μg of MCT4 HDR plasmid (Santa Cruz Biotechnology; sc-429828 and sc-429828HDR, respectively). Successfully transfected cells were selected by puromycin treatment (5 μg/ml) and fluorescence-activated cell sorting (FACS) sorted based on their RFP expression. Single clones were derived and validated using PCR primers flanking the sequences targeted by the guide RNAs, subsequent sequencing, and western blot analysis. Prior to in vivo experiments, the RFP and puromycin resistance cassettes were removed using infection with adenovirus-expressing Cre recombinase (Ad-Cre).
Extracellular Metabolic Flux Analysis
For details, see the Supplemental Information.
Statistical Analysis
Data analysis and graph generation was performed using GraphPad Prism 6 (GraphPad Prism Software). All experiments performed with mouse samples were analyzed using Mann-Whitney U test. Tumor growth curves are displayed as mean ± SEM. For immunofluorescence analysis, each data point represents one field of view and the mean is displayed. N, number of mice per group. Statistical significance of in vitro experiments was calculated using Student’s t test or ANOVA test, as indicated in the figure legends. Data are displayed as mean ± SD. N, number of independent experiments. n.s., non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Author Contributions
Conceptualization, L.P., R.B., J.H., and G.C.; Methodology, L.P., R.B., and G.C.; Validation, L.P., R.B., E.F., S.D., and R.W.G.; Investigation, L.P., R.B., E.F., and S.D.; Resources, L.P., R.B., E.F., S.D., R.W.G., C.H., and G.C.; Data Curation, R.W.G.; Writing – Original Draft, L.P., R.B., R.W.G., and G.C.; Writing – Review & Editing, L.P., R.B., E.F., S.D., R.W.G., J.H., C.H., and G.C.; Visualization, L.P., R.B., E.F., S.D., and R.W.G.; Supervision, G.C.; Project Administration, G.C.; Funding Acquisition, R.B. and G.C.
Acknowledgments
We thank P. Lorentz (Department of Biomedicine [DBM], University of Basel) for excellent support with microscopy; H. Antoniadis, P. Schmidt, and I. Galm (DBM) for technical support; as well as T. Barthlott, C. Berkemeier, and C. Mayer (DBM) for flow cytometry and R. Ivanek (DBM) for bioinformatics analysis. We highly appreciate the collaboration with S. Dubuis and N. Zamboni (Institute for Systems Biology, ETH Zürich) on metabolomic analysis. We are grateful to D. Gruber, A. Banfi, and O. Pertz (DBM) and M. Hall (Biocenter, University of Basel) for providing reagents. This work was supported by the Swiss Cancer League (5KLS-2846-08-2011) and a MD-PhD fellowship to R.B. by the Swiss National Science Foundation.
Published: April 28, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.04.028.
Accession Numbers
The accession number for the microarray data reported in this paper have been deposited to the NCBI GEO and is available under accession number GEO: GSE78698.
Supplemental Information
References
- Allen E., Miéville P., Warren C.M., Saghafinia S., Li L., Peng M.W., Hanahan D. Metabolic symbiosis enables adaptive resistance to anti-angiogenic therapy that is dependent on mTOR signaling. Cell Rep. 2016;15:1144–1160. doi: 10.1016/j.celrep.2016.04.029. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi N., Hinz S., Brekken R.A., Schwarz M.A., Schwarz R.E. Nintedanib, a triple angiokinase inhibitor, enhances cytotoxic therapy response in pancreatic cancer. Cancer Lett. 2015;358:59–66. doi: 10.1016/j.canlet.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G., Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill R., Fagiani E., Zumsteg A., Antoniadis H., Johansson D., Haefliger S., Albrecht I., Hilberg F., Christofori G. Nintedanib is a highly effective therapeutic for neuroendocrine carcinoma of the pancreas (PNET) in the Rip1Tag2 transgenic mouse model. Clin. Cancer Res. 2015;21:4856–4867. doi: 10.1158/1078-0432.CCR-14-3036. [DOI] [PubMed] [Google Scholar]
- Carmeliet P., Jain R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanovas O., Hicklin D.J., Bergers G., Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Chung A.S., Wu X., Zhuang G., Ngu H., Kasman I., Zhang J., Vernes J.M., Jiang Z., Meng Y.G., Peale F.V. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat. Med. 2013;19:1114–1123. doi: 10.1038/nm.3291. [DOI] [PubMed] [Google Scholar]
- Clem B., Telang S., Clem A., Yalcin A., Meier J., Simmons A., Rasku M.A., Arumugam S., Dean W.L., Eaton J. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008;7:110–120. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- Compagni A., Wilgenbus P., Impagnatiello M.A., Cotten M., Christofori G. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res. 2000;60:7163–7169. [PubMed] [Google Scholar]
- Crawford Y., Ferrara N. VEGF inhibition: insights from preclinical and clinical studies. Cell Tissue Res. 2009;335:261–269. doi: 10.1007/s00441-008-0675-8. [DOI] [PubMed] [Google Scholar]
- Curtarello M., Zulato E., Nardo G., Valtorta S., Guzzo G., Rossi E., Esposito G., Msaki A., Pastò A., Rasola A. VEGF-targeted therapy stably modulates the glycolytic phenotype of tumor cells. Cancer Res. 2015 doi: 10.1158/0008-5472.CAN-13-2037. [DOI] [PubMed] [Google Scholar]
- Doyen J., Trastour C., Ettore F., Peyrottes I., Toussant N., Gal J., Ilc K., Roux D., Parks S.K., Ferrero J.M., Pouysségur J. Expression of the hypoxia-inducible monocarboxylate transporter MCT4 is increased in triple negative breast cancer and correlates independently with clinical outcome. Biochem. Biophys. Res. Commun. 2014;451:54–61. doi: 10.1016/j.bbrc.2014.07.050. [DOI] [PubMed] [Google Scholar]
- Ebos J.M., Kerbel R.S. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat. Rev. Clin. Oncol. 2011;8:210–221. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N. Role of myeloid cells in vascular endothelial growth factor-independent tumor angiogenesis. Curr. Opin. Hematol. 2010;17:219–224. doi: 10.1097/MOH.0b013e3283386660. [DOI] [PubMed] [Google Scholar]
- Ferrara N., Kerbel R.S. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- Ferrara N., Hillan K.J., Gerber H.P., Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115–122. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harris A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Hellström M., Kalén M., Lindahl P., Abramsson A., Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- Hilberg F., Roth G.J., Krssak M., Kautschitsch S., Sommergruber W., Tontsch-Grunt U., Garin-Chesa P., Bader G., Zoephel A., Quant J. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774–4782. doi: 10.1158/0008-5472.CAN-07-6307. [DOI] [PubMed] [Google Scholar]
- Ho J., de Moura M.B., Lin Y., Vincent G., Thorne S., Duncan L.M., Hui-Min L., Kirkwood J.M., Becker D., Van Houten B., Moschos S.J. Importance of glycolysis and oxidative phosphorylation in advanced melanoma. Mol. Cancer. 2012;11:76. doi: 10.1186/1476-4598-11-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz H., Fehrenbacher L., Novotny W., Cartwright T., Hainsworth J., Heim W., Berlin J., Baron A., Griffing S., Holmgren E. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- Jiménez-Valerio G., Martínez-Lozano M., Bassani N., Vidal A., Ochoa-de-Olza M., Suárez C., García-del-Muro X., Carles J., Viñals F., Graupera M. Resistance to antiangiogenic therapies by metabolic symbiosis in renal cell carcinoma PDX models and patients. Cell Rep. 2016;15:1134–1143. doi: 10.1016/j.celrep.2016.04.015. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbel R.S. Issues regarding improving the impact of antiangiogenic drugs for the treatment of breast cancer. Breast. 2009;18(Suppl 3):S41–S47. doi: 10.1016/S0960-9776(09)70271-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar K., Wigfield S., Gee H.E., Devlin C.M., Singleton D., Li J.L., Buffa F., Huffman M., Sinn A.L., Silver J. Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J. Mol. Med. 2013;91:749–758. doi: 10.1007/s00109-013-0996-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutluk Cenik B., Ostapoff K.T., Gerber D.E., Brekken R.A. BIBF 1120 (nintedanib), a triple angiokinase inhibitor, induces hypoxia but not EMT and blocks progression of preclinical models of lung and pancreatic cancer. Mol. Cancer Ther. 2013;12:992–1001. doi: 10.1158/1535-7163.MCT-12-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu V.S., O’Connell J.T., Gonzalez Herrera K.N., Wikman H., Pantel K., Haigis M.C., de Carvalho F.M., Damascena A., Domingos Chinen L.T., Rocha R.M. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014;16:992–1003. doi: 10.1038/ncb3039. 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchiq I., Pouysségur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J. Mol. Med. 2016;94:155–171. doi: 10.1007/s00109-015-1307-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack P.L. Nintedanib: first global approval. Drugs. 2015;75:129–139. doi: 10.1007/s40265-014-0335-0. [DOI] [PubMed] [Google Scholar]
- Michelakis E.D., Sutendra G., Dromparis P., Webster L., Haromy A., Niven E., Maguire C., Gammer T.L., Mackey J.R., Fulton D. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- Miller K., Wang M., Gralow J., Dickler M., Cobleigh M., Perez E.A., Shenkier T., Cella D., Davidson N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007;357:2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- Misteli H., Wolff T., Füglistaler P., Gianni-Barrera R., Gürke L., Heberer M., Banfi A. High-throughput flow cytometry purification of transduced progenitors expressing defined levels of vascular endothelial growth factor induces controlled angiogenesis in vivo. Stem Cells. 2010;28:611–619. doi: 10.1002/stem.291. [DOI] [PubMed] [Google Scholar]
- Morais-Santos F., Granja S., Miranda-Gonçalves V., Moreira A.H., Queirós S., Vilaça J.L., Schmitt F.C., Longatto-Filho A., Paredes J., Baltazar F., Pinheiro C. Targeting lactate transport suppresses in vivo breast tumour growth. Oncotarget. 2015;6:19177–19189. doi: 10.18632/oncotarget.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer R.J., Hutson T.E., Tomczak P., Michaelson M.D., Bukowski R.M., Rixe O., Oudard S., Negrier S., Szczylik C., Kim S.T. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- Pàez-Ribes M., Allen E., Hudock J., Takeda T., Okuyama H., Viñals F., Inoue M., Bergers G., Hanahan D., Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato P.E., Dhup S., Dadhich R.K., Copetti T., Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front. Pharmacol. 2011;2:49. doi: 10.3389/fphar.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintela-Fandino M., Urruticoechea A., Guerra J., Gil M., Gonzalez-Martin A., Marquez R., Hernandez-Agudo E., Rodriguez-Martin C., Gil-Martin M., Bratos R. Phase I clinical trial of nintedanib plus paclitaxel in early HER-2-negative breast cancer (CNIO-BR-01-2010/GEICAM-2010-10 study) Br. J. Cancer. 2014;111:1060–1064. doi: 10.1038/bjc.2014.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond E., Dahan L., Raoul J.L., Bang Y.J., Borbath I., Lombard-Bohas C., Valle J., Metrakos P., Smith D., Vinik A. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011;364:501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- Reck M., Kaiser R., Mellemgaard A., Douillard J.Y., Orlov S., Krzakowski M., von Pawel J., Gottfried M., Bondarenko I., Liao M., LUME-Lung 1 Study Group Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014;15:143–155. doi: 10.1016/S1470-2045(13)70586-2. [DOI] [PubMed] [Google Scholar]
- Rose S. FDA pulls approval for avastin in breast cancer. Cancer Discov. 2011;1:OF1–OF2. doi: 10.1158/2159-8290.CD-ND112311OL-08. [DOI] [PubMed] [Google Scholar]
- Roth G.J., Heckel A., Colbatzky F., Handschuh S., Kley J., Lehmann-Lintz T., Lotz R., Tontsch-Grunt U., Walter R., Hilberg F. Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120) J. Med. Chem. 2009;52:4466–4480. doi: 10.1021/jm900431g. [DOI] [PubMed] [Google Scholar]
- Schoors S., De Bock K., Cantelmo A.R., Georgiadou M., Ghesquière B., Cauwenberghs S., Kuchnio A., Wong B.W., Quaegebeur A., Goveia J. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Sennino B., McDonald D.M. Controlling escape from angiogenesis inhibitors. Nat. Rev. Cancer. 2012;12:699–709. doi: 10.1038/nrc3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M., Ferrara N. Modeling and predicting clinical efficacy for drugs targeting the tumor milieu. Nat. Biotechnol. 2012;30:648–657. doi: 10.1038/nbt.2286. [DOI] [PubMed] [Google Scholar]
- Sonveaux P., Végran F., Schroeder T., Wergin M.C., Verrax J., Rabbani Z.N., De Saedeleer C.J., Kennedy K.M., Diepart C., Jordan B.F. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounni N.E., Cimino J., Blacher S., Primac I., Truong A., Mazzucchelli G., Paye A., Calligaris D., Debois D., De Tullio P. Blocking lipid synthesis overcomes tumor regrowth and metastasis after antiangiogenic therapy withdrawal. Cell Metab. 2014;20:280–294. doi: 10.1016/j.cmet.2014.05.022. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson D., Hanahan D. Translational medicine: Cancer lessons from mice to humans. Nature. 2011;471:316–317. doi: 10.1038/471316a. [DOI] [PubMed] [Google Scholar]
- Waldmeier L., Meyer-Schaller N., Diepenbruck M., Christofori G. Py2T murine breast cancer cells, a versatile model of TGFβ-induced EMT in vitro and in vivo. PLoS ONE. 2012;7:e48651. doi: 10.1371/journal.pone.0048651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick A.N., Drury D.R., Morita T.N. 2-Deoxyglucose; a metabolic block for glucose. Proc. Soc. Exp. Biol. Med. 1955;89:579–582. doi: 10.3181/00379727-89-21881. [DOI] [PubMed] [Google Scholar]
- Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R.C., Spiegelman B.M. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.