

Abstract
The human cytosolic sulfotransferases (SULTs) regulate hundreds, perhaps thousands, of small molecule metabolites and xenobiotics via transfer of a sulfuryl moiety (−SO3) from PAPS (3′-phosphoadenosine 5′-phosphosulfate) to the hydroxyls and primary amines of the recipients. In liver, where it is abundant, SULT1A1 engages in modifying metabolites and neutralizing toxins. The specificity of 1A1 is the broadest of any SULT, and understanding its selectivity is fundamental to understanding its biology. Here, for the first time, we show that SULT1A1 substrates separate naturally into two classes: those whose affinities are either enhanced ~20-fold (positive synergy) or unaffected (neutral synergy) by the presence of a saturating nucleotide. kcat for the positive-synergy substrates is shown to be ~100-fold greater than that of neutral-synergy compounds; consequently, the catalytic efficiency (kcat/Km) is approximately 3 orders of magnitude greater for the positive-synergy species. All-atom dynamics modeling suggests a molecular mechanism for these observations in which the binding of only positive-synergy compounds causes two phenylalanine residues (F81 and 84) to reposition and “sandwich” the phenolic moiety of the substrates, thus enhancing substrate affinity and positioning the nucleophilic oxygen for attack. Molecular dynamics movies reveal that the neutral-synergy compounds “wander” about the active site, infrequently achieving a reactive position. In-depth analysis of select point mutants strongly supports the model and provides an intimate view of the interdependent catalytic functions of subsections of the active site.
Graphical abstract
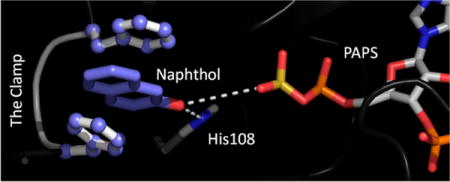
The cytosolic sufotransferases (SULTs) regulate the activities of small-molecule metabolites via transfer of the sulfuryl-moiety (−SO3) from a universal nucleotide donor (PAPS, 3′-phosphoadenosine 5′-phosphosulfate) to the hydroxyls and primary amines of the acceptors. While general principles, such as sulfonation prevents signaling compounds from binding to their receptors and shortens (often dramatically) their terminal half-lives by enhancing their solubility, are useful starting points for understanding the biological effects of sulfonation, the manner in which the activity of a given metabolite is modified is often more complex. Consider thyroid hormone, a ubiquitous regulator of energy metabolism1,2 —here, sulfotranferases selectively sulfonate the most active form of the hormone (T3) and, in so doing, prevent it from binding its receptor and cause it to become a 200-fold more efficient substrate for deiodinases, which irreversibly inactivate the hormone; or hydroxycholesterols, which regulate cholesterol homeostasis by binding the liver X receptor—in this case, sulfonation transitions the signaling molecule from an effective agonist to antagonist with a yet higher affinity;3,4 or finally, dopamine, whose rapid sulfotransferase mediated inactivation in brain may be the root cause of the shaking tremors experienced by the majority of late-stage Parkinson’s patients.5,6 There are numerous similar examples,7–10 many of which have been linked to human disease.11–14
SULT1A1, the focus of this paper, is the most abundant SULT in liver, where it engages in the extensive sulfonation of metabolites and xenobiotics, including numerous drugs and toxins. Only recently have we begun to understand just how remarkably complex and adaptive this enzyme can be.15–18 For example, its specificity changes as a function of the occupancy of its two PAPS-binding sites. When only a single subunit is occupied, the nucleotide stabilizes an active-site conformation that sterically restricts access to the acceptor-binding pocket and strongly biases selectivity toward smaller, roughly planar acceptors (e.g., steroid-like structures).17,18 Nucleotide affinity for the second binding site is far lower (81-fold) than that for the first, and binding of the second nucleotide “lifts” the steric restrictions, allowing the enzyme to field a far greater range of substrate geometries. Further, the enzyme turns over 8-fold faster when it is doubly occupied. The in vivo concentrations of PAPS in human tissues suggest that SULT1A1 is singly nucleotide occupied in tissues that have little need for the detoxifying functions of the enzyme, and doubly occupied in those (e.g., liver) where the need is greatest.17 Thus, remarkably, the substrate specificity of the enzyme appears to be tissue-dependent. In addition to its PAPS-dependent behavior, the enzyme harbors two functionally independent allosteric binding sites, each of which binds a different complex class of compounds.16,19,20 Thus, the enzyme seems well equipped to adjust its catalytic behavior to its environment.
This study explores the substrate specificity of SULT1A1. The work reveals, for the first time, that small planar acceptors separate into two classes: those whose affinities increase ~20-fold when PAPS is bound (positive-synergy compounds) and those whose affinities are not affected (neutral-synergy compounds). Positive-synergy compounds turn over ~100-fold faster than neutral compounds, and the combined effects on affinity and turnover give them an enormous (~3 orders of magnitude) catalytic advantage over their neutral-synergy counterparts. All-atom dynamics modeling suggests that only positive-synergy substrates cause the active-site residues F81 and 84 to reposition, sandwiching the reactive phenolic moiety of the substrate. The resulting structure resembles a “molecular clamp” that appears to enhance substrate affinity and increase the rate of turnover by “fixing” the substrate’s nucleophilic hydroxyl in an active-site position that is highly conducive to reaction. The catalytic behaviors of select point mutants strongly support the model.
MATERIALS AND METHODS
Materials
The materials and sources used in this study are as follows. Acetaminophen (Acet), apomorphine (AP), dithiothreitol (DTT), dehydroepiandrosterone (DHEA), ethylenediaminetetraacetic acid (EDTA), 17-β-estradiol (E2), fulvestrant (Ful), L-glutathione (reduced), 1-hydroxypyrene (1-HP), imidazole, isopropyl thio-β-D-galactopyranoside (IPTG), Lysogeny broth (LB), lysozyme, β-mercaptoethanol, 1-naphthol (1-Nap), 2-naphthol (2-Nap), pepstatin A, resveratrol (Res), and sodium phosphate were the highest grade available from Sigma. PAPS and [35S]PAPS were synthesized in house21 and were >98% pure. Ampicillin, HEPES, KOH, MgCl2, NaCl, KCl, LiCl, and phenylmethanesulfonyl fluoride (PMSF) were purchased from Fisher Scientific. Glutathione- and nickel-chelating resins and PreScission protease were obtained from GE Healthcare. Competent Escherichia coli [BL21(DE3)] was purchased from Novagen. The QuikChange mutagenesis kit was purchased from Agilent Technologies, and mutagenic primers were purchased from Fisher Custom Oligos. An Amicon Ultra Centrifugal Filter [molecular weight cutoff (MWC) of 10000] was purchased from EMD Millipore. PEI-F anion exchange TLC sheets were purchased from Merck, KGaA.
Computer and Software
The simulations were performed on a Parallel Quantum Solutions QS32-2670C-XS8 computer. PQS Molecular Builder was purchased from Parallel Quantum Solutions. A GOLD license was obtained from the Cambridge Crystallographic Data Center. The source code for GROMACS 4.5 was downloaded from http://www.GROMACS.org under the GROMCAS General Public License (GPL). AMBER and Ambertools 10.0 were obtained from the University of California, San Francisco.
Methods
Molecular Dynamics Simulations
A ligand-free model of SULT1A1 was constructed from the SULT1A1·PAP [Protein Data Bank (PDB) entry 4GRA]18 binary structure, and missing atoms were inserted by homology modeling with SWISS-MODEL.22 PAPS was constructed from PAP using PQS Molecular Builder. The models were protonated at pH 7 and energy minimized in GROMACS.23 The initial placement of acceptors was done by docking with GOLD.24 Molecular dynamics simulations were performed using GROMACS23 with the Amber force field.25 Briefly, energy parameter files for acceptors and PAPS were generated using AmberTools.26 The initial structures were energy minimized and then equilibrated by allowing the system to evolve in 100 ps steps at a simulated temperature of 310 K until the protein root-mean-square deviation was stable. Once equilibrated, simulations were run for 1.0 ns. All measurements and energy calculations were made using fully equilibrated systems.
Protein Purification
Wild-type and mutant SULT1A1 proteins were expressed in E. coli [BL21(DE3)] from a coding region that was codon-optimized for E. coli expression. Proteins were expressed with a PreScission-protease-cleavable, N-terminal His/GST/MBP tag from a pGEX-6P vector.21 SULT expression and purification were performed as described previously.18 The cells were grown in LB with ampicillin (100 μg mL−1) at 37 °C to an OD600 of 0.6. Induction was initiated with IPTG (0.30 mM), and cells were grown overnight at 18 °C. The cells were pelleted and resuspended in cold (4 °C) lysis buffer [lysozyme (0.10 mg/mL), PMSF (290 μM), pepstatin A (1.5 μM), β-mercaptoethanol (5.0 mM), KCl (0.40 M), and NaPO4 (50 mM) (pH 7.2)], sonicated, and centrifuged (35000g for 1.0 h at 4 °C) to remove debris. The supernatant was loaded onto a Chelating Sepharose Fast Flow column charged with Ni2+. The enzyme was then eluted with imidazole (10 mM, pH 7.2) onto a Glutathione Sepharose column followed by elution with glutathione (10 mM, pH 7.2). The fusion protein was digested with Precision Protease and dialyzed against HEPES/K+ (50 mM, pH 7.5), DTT (1.5 mM), and KCl (100 mM) at 4 °C overnight. The protein was then passed through a second GST column to remove the His/GST/MBP tag and PreScission protease. The enzyme was concentrated using an Amicon Ultra Centrifugal Filter (MWC of 10000). The protein concentration was determined by UV absorbance (ɛ280 = 53.8 mM−1 cm−117). The protein purity was >95%, as determined by Coomassie staining of sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels. The enzyme was flash-frozen and stored at −80 °C.
Mutagenesis
All SULT1A1 mutants were generated by polymerase chain reaction mutagenesis using the QuikChange mutagenesis kit, and mutations were confirmed by sequencing of the entire coding region.
Equilibrium Binding Fluorescence Titrations
Acceptor binding to SULT1A1 was monitored by changes in the intrinsic fluorescence of the enzyme (λex = 290 nm; λem = 340 nm) with a Carry Eclipse spectrofluorometer. Ligands were successively added to a solution containing SULT1A1 (10 nM, dimer), MgCl2 (5.0 mM), and NaPO4 (25 mM) at pH 7.2 and 25 ± 2 °C. Dilution at the titration end points was <2.0% in all cases. Acceptors were added from stocks containing 50% ethanol or dimethyl sulfoxide (DMSO). Controls confirmed that addition of ethanol or DMSO does not cause detectable fluorescence changes. Titrations were performed in duplicate. The data were averaged and least-squares fit to a model that assumes a single binding site per monomer. In all cases, ligand concentrations were varied from approximately 0.10Kd to 20Kd. Titrations that measure acceptor binding to binary complexes contained PAP (10 μM), at 33Kd E.
Initial-Rate Parameters in Table 1
Table 1.
SULT1A1 Substrate Synergies and Catalytic Efficiencies
| acceptor |
Kd (μM)
|
synergy
|
kcat (s−1) | Km (μM) | efficiency
|
|
|---|---|---|---|---|---|---|
| E | E·PAP | Kd E/Kd E·PAP | kcat/Km (μM−1 s−1) | |||
| E2a | 1.0 | 0.9 | 0.90 | 1.2 | 1.0 | 1.2 |
| DHEAb | 0.68 | 0.74 | 0.91 | 0.75 | 0.77 | 0.97 |
| Resc | 1.1 | 0.85 | 1.4 | 1.5 | 1.6 | 0.93 |
| Acetd | 0.50 | 0.018 | 28 | 100 | 0.022 | 4500 |
| 1-Nape | 1.3 | 0.07 | 18 | 100 | 0.077 | 1300 |
| 2-Napf | 1.5 | 0.06 | 25 | 110 | 0.063 | 1700 |
| APg | 0.88 | 0.047 | 20 | 58 | 0.051 | 1100 |
| 1-HPh | 5.5 | 0.019 | 290 | 120 | 0.018 | 3300 |
E2, estradiol.
DHEA, dehydroepiandrosterone.
Res, resveratrol.
Acet, acetaminophen.
1-Nap, 1-naphthol.
2-Nap, 2-naphthol.
AP, apomorphine.
1-HP, 1-hydroxypyrene.
Rates were determined by monitoring the conversion of [35S]PAPS to [35S]acceptor for all acceptors except 1-HP. In all cases, consumption of the concentration-limiting substrate was <5% of product formed at the reaction end point. Reaction conditions were as follows: SULT1A1 (5.0 nM, dimer), acceptor (10 concentrations that varied from 0.10Kd E·PAP to 30Kd E·PAP in equal increments in double-reciprocal space), MgCl2 (5.0 mM), and NaPO4 (50 mM) at pH 7.2 and 25 ± 2 °C. Reactions were initiated by addition of [35S]PAPS (10 μM, specific activity of 4.3 Ci mmol−1) and stopped by addition of NaOH (0.10 M, final concentration) at time intervals adjusted for each substrate. Each time point measurement was determined in triplicate, and averaged values were used in subsequent analysis. Quenched solutions were neutralized with HCl, boiled for 1.0 min, and centrifuged, and supernatants were spotted onto PEI-F anion exchange TLC sheets. Radiolabeled reactants were separated using a 0.90 M LiCl mobile phase8 and quantitated using STORM imaging.27 Velocities at each acceptor concentration were obtained by least-squares fitting of four-point progress. 1-HP sulfation was monitored by fluorescence (λex = 320 nm; λem = 380 nm).17 Because SULT1A1 is partially substrate-inhibited, Km and Vmax were obtained by least-squares fitting of the velocities to the equation for partial substrate inhibition v = (Vmax[S] + Vinh[S]2/Ki)/(Km + [S] + [S]2/K2),27,28 where Km and Vmax are Michaelis–Menten constants, Ki is the substrate inhibition constant, and Vinh is the velocity at infinite concentration of substrate.
Trapping Experiments: The Order of Substrate Binding. Trapping Neutral-Synergy Substrates
A pulse solution containing SULT1A1 [30 μM (monomer), 25Kd E] and [3H]E2 (1.0 μM, 131 Ci mmol−1) in buffer [MgCl2 (5.0 mM) and NaPO4 (25 mM) at pH 7.2 and 25 ± 2 °C] was rapidly diluted [200-fold (v/v)] by hand into a stirred chase solution containing PAPS (0.50 mM, 1500Kd E) and unlabeled E2 (10 μM, 15Kd E) in the identical buffer. Once the solution had been mixed, the enzyme rapidly forms the ternary complex (<50 ms27) and [3H]E2 is diluted 2000-fold. The reactions were run for 3.0 s, which is long enough relative to kcat (1.0 s−1) to capture the majority of product that might form from [3H]E2 before its departure from the enzyme, and short enough to minimize the formation of the labeled product from [3H]E2 that has entered the bulk solution. Simulations using rate constants from previous work18,27 predict that under these conditions 97% of the [3H]E2 bound to the enzyme at time zero will be “trapped” in the ternary substrate complex (E·PAPS·[3H]E2); 69% of the complex is predicted to form product if the mechanism is random and 2.3% if it is ordered.
Reactions were quenched by addition of NaOH (0.10 M, final). [3H]E2 and [3H]E2S were separated by extracting the quenched solution with chloroform [1:5 (v/v)], vortexing, centrifuging (5.0 min at 1000g), and extracting a second time with chloroform. Aqueous-phase [3H]E2S was quantitated by scintillation counting using a PerkinElmer W450624 spectrometer. The reactions were performed in triplicate and revealed that 67 ± 2.0% of the [3H]E2 formed product. Control reactions in which the labeled acceptor was added to the chase provide an upper limit for labeled product formation in the case of the ordered mechanism. The control reactions, also run in triplicate, converted 4.0 ± 1.7% of [3H]E2 to product.
Trapping Positive-Synergy Substrates
1-HP was used to assess whether the reaction mechanism of positive-synergy substrates is random or ordered. SULT1A1 [240 μM (monomer), 44Kd E] and 1-HP (100 μM, 18Kd E) were preincubated in buffer [MgCl2 (5.0 mM) and NaPO4 (25 mM) at pH 7.2 and 25 ± 2 °C], and the solution was then diluted [1000-fold (v/v)] by hand into a well-stirred solution containing PAPS (10 μM, 28Kd E, 560Kd E·pNP, and 8120Kd E·1-HP) and pNP (200 μM, 17Kd E and 334Kd E·PAPS) in the identical buffer. 1-HP sulfonation was monitored via the fluorescence change of 1-HPS (λex = 320 nm; λem = 375 nm).17 A control reaction that was identical except that it was initiated by addition of SULT1A1 was used obtain an upper limit for 1-HPS formation in the case of an ordered mechanism. To accurately determine the 1-HPS concentration, a standard [1-HPS] versus fluorescence curve was generated under conditions identical to those used in the trapping experiments (200 μM pNP, 10 μM PAPS, 5.0 mM MgCl2, and 25 mM NaPO4 at pH 7.2 and 25 ± 2 °C). Simulations predict that under these conditions 96% of the 1-HP bound to the enzyme at t0 will be “trapped” by the addition of the nucleotide in the substrate ternary complex (E·PAPS·1-HP) in ~25 ms. Three seconds is the minimum time following initiation of reaction that a measurement can be made. At 3.0 s, simulations predict that 96% of the ternary complex will produce product if the mechanism is random and 9% if it is ordered. Experiments, performed in triplicate, showed 95 ± 4% of the bound 1-HP formed 1-HPS after 3.0 s; controls gave 5 ± 2% 1-HPS formation.
Initial-Rate Kinetics Using E2
Classical initial-rate studies of the wild type and I89A were performed. Initial rates were determined by least-squares fitting of duplicate, four-point progress curves at each of the 16 conditions defined by a 4 × 4 matrix of substrate concentrations. PAPS and E2 were varied from 0.20Km to 5.0Km in equal increments in double-reciprocal space. Reaction conditions were as follows: SULT1A1 (5.0 nM, dimer), PAPS (0.060–1.5 μM), [3H]E2 (0.30–7.5 μM, specific activity of 15 Ci mmol−1), MgCl2 (5.0 mM), and NaPO4 (50 mM) at pH 7.2 and 25 ± 2 °C. Reactions were initiated by addition of SULT1A1 and quenched by addition of NaOH (0.20 M, final). Quenched solutions (30 μL) were neutralized by addition of 170 μL of NaPO4 (50 mM, pH 7.2). Nonsulfated acceptors were removed by two successive extractions with 1.0 mL of chloroform. The sulfated acceptor was quantified by scintillation counting the aqueous phase using a PerkinElmer W450624 scintillation spectrometer. In all cases, E2 was the concentration-limiting substrate and its consumption was ≤5% of that consumed at the reaction end point. The initial-rate data were globally fit to a random bi-bi model using the SEQUENO algorithm of Cleland.29
Initial-Rate Kinetics Using 2-Naphthol
The rate of 2-naphthol sulfonation was determined by monitoring the conversion of [35H]PAPS to [35H]naphthol. The [35H]PAPS was synthesized and purified (>98%) in house.21 Initial rates were determined by least-squares fitting of duplicate, four-point progress curves under each of the 16 conditions defined by a 4 × 4 matrix of substrate concentrations. PAPS and naphthol were varied from 0.20Km to 5.0Km in equal increments in double-reciprocal space. In all cases, consumption of the concentration-limiting substrate was <5% of that product at the reaction end point. Reaction conditions were as follows: wild-type or I89A SULT1A1 (5.0 nM, dimer), 2-naphthol (0.010–0.25 μM), MgCl2 (5.0 mM), and NaPO4 (50 mM) at pH 7.2 and 25 ± 2 °C. Reactions were initiated by addition of [35H]PAPS (0.020–0.50 μM, specific activity of 4.3 Ci mmol−1) and stopped by addition of NaOH (0.10 M, final). Quenched solutions were neutralized with HCl, boiled for 1.0 min, and centrifuged, and supernatants were spotted onto PEI-F anion exchange TLC sheets. Radiolabeled reactants were separated using a 0.9 M LiCl mobile phase8 and quantitated using STORM imaging.27 Initial-rate parameters were obtained by globally fitting data to a random bi-bi model using the SEQUENO29 algorithm of Cleland.
Assays of the Non-Active Lower-Lip Mutants (F81A, F84A, and E85S)
The initial rate of E2 sulfonation was determined at a saturating concentrations of PAPS (4.0 μM, ~15Km WT) and [H3]E2 (25 μM, ~20Km WT, specific activity of 15 Ci mmol−1). The conditions were identical to those described in the E2 assay section above except that [SULT1A1]dimer = 100 nM. The lower limit of detection under these conditions is ~0.5 active-site equivalents of product. No product was detected at 60 min; hence, the mutant activity is ≤0.012% of wild-type activity. Acceptor binding to each mutant was tested using E2 and Nap at concentrations as high as their solubility would allow (25 and 100 μM, respectively); binding was not detected.
RESULTS AND DISCUSSION
SULT1A1 Substrate Synergy
Table 1 lists dissociation and initial rate parameters for a series of SULT1A1 acceptors. Dissociation constants were determined by fitting equilibrium titrations that monitor binding via ligand-induced changes in SULT1A1 intrinsic fluorescence (see Materials and Methods). A representative titration is presented in Figure 1. Initial-rate parameters were obtained as described in Materials and Methods. The synergy between the nucleotide and a given acceptor is given in Table 1 as the ratio of acceptor affinities for the E and E·PAP forms of SULT1A1 (i.e., Kd E/Kd E·PAP) and is a measure of their interaction energy. Positive-synergy acceptors are those for which the affinity is enhanced when a nucleotide is bound; neutral-synergy acceptors exhibit synergies near one (i.e., near-zero interaction energies). In every case, positive synergy correlates with a dramatic increase in kcat, which, together with the effects on binding, results in efficiency enhancements as high as ~104.
Figure 1.
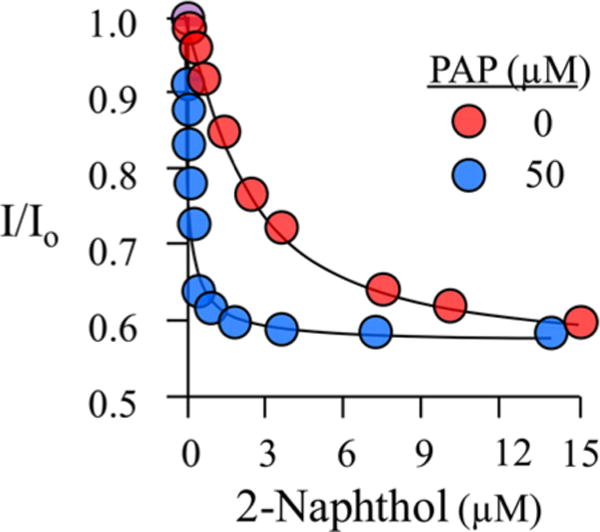
Equilibrium binding of 2-naphthol to the E and E·PAP forms of SULT1A1. Binding was monitored via changes in the intrinsic fluorescence of SULT1A1 (λex = 290 nm; λem = 340 nm). The fluorescence intensity is given relative to that observed in the absence of 2-Nap (I/I0). The solution consisted of SULT2A1 [20 nM (dimer)], PAP [0 (red) or 50 μM (blue)], MgCl2 (5.0 mM), and NaPO4 (50 mM) at pH 7.2 and 25 ± 2 °C. Each point represents the average of three independent determinations, and the lines through the points are the behaviors predicted by a best-fit, single-site binding model.
This analysis is without precedent in the SULT literature and reveals that specific substrates are recognized by SULT1A1 in ways that give them enormous catalytic advantage. Understanding the molecular basis of this enhanced reactivity is the focus of the remainder of this paper.
A Note on the SULT1A1 Mechanism
The SULT literature is at odds on the issue of the order of substrate binding to 1A1. Early initial-rate studies favored a random mechanism;30,31 that is, either donor or acceptor can bind in the absence of its “partner”, which can then add to form a productive ternary complex (i.e., one that can yield product). Later, interpretations of isotope-exchange-at-equilibrium (IEE) experiments favored an ordered mechanism in which PAPS adds first and PAP departs first in the product release phase.32 It is well established that IEE experiments often cannot distinguish ordered from random mechanisms that include a dead-end complex,33 and the acceptor used in the IEE studies (i.e., 1-napthol) forms a tight dead-end complex with PAP (see Table 1). A variety of recent lines of evidence favor a random mechanism for SULT1A1,16,17,27,28 and an in-depth study has shown that the mechanism of a close relative, SULT2A1, is random.27
Substrate trapping experiments21,27,34 were used to resolve the order-of-binding issue and in so doing determine whether the E·acceptor complexes listed in Table 1 are catalytically competent. These experiments, detailed in Materials and Methods, involve rapid mixing of a small volume of a pulse solution, in which the acceptor is nearly entirely enzyme-bound, with a large volume of a chase solution, which contains donor (PAPS) at a highly saturating concentration and a competing acceptor, also at a high concentration. The goal is to have the ternary complex form rapidly, following dilution, relative to dissociation of the pulse acceptor from the binary complex, and to set the dilution and competitive acceptor concentration such that product formation from the pulse acceptor that dissociates is negligible. Under these conditions, any pulse acceptor product must derive from acceptor that was productively bound in the pulse.
Experiments were performed with neutral (E2) and positive-synergy (1-HP) substrates, and the experimental designs used radiolabeled and nonradiolabeled pulse-chase strategies. The experiments were performed in triplicate with the result that 67 ± 2.0% of the neutral-synergy and 95 ± 4% of the positive-synergy acceptors bound in the pulse were converted to product. Hence, acceptors bind productively, and the mechanism is random.
A Molecular Model of Positive Synergy
In an attempt to define the molecular basis of positive synergy, molecular dynamics models of SULT1A1 ternary complexes with positive-synergy (acetaminophen) and neutral-synergy (estradiol) acceptors were constructed and compared (see Materials and Methods). The X-ray structures of the E·PAP and E·PAP·E2 backbones (PDB entries 4GRA and 2D06) are nearly identical, suggesting that the E·PAP structure is representative of the neutral-synergy conformation. The X-ray and GROMACS-generated E·PAP structures are also virtually identical; consequently, the GROMACS structure was used in subsequent simulations as a mock neutral-synergy structure.
To reveal differences in the ways in which SULT1A1 responds to Acet and E2, the ligands were docked (using GOLD24) into an equilibrated E·PAP complex in which only side chains are allowed to reposition, and the resulting rigid-backbone structures were used as input for Gromacs8,23 simulations in which all atoms were subsequently allowed to relax. The rigid and relaxed structures of the E·PAP·E2 complex are nearly identical to one another and to their corresponding X-ray structure. In contrast, the structure of an active-site loop changed substantially as the E·PAP structure “relaxed” around Acet (see Figure 2). To avoid being misled by possible idiosyncrasies associated with the acceptor pair of Acet and E2, the same docking exercise was repeated with a second positive-and neutral-synergy substrate pair (2-Nap and DHEA). The results were virtually identical to those obtained using the pair of Acet and E2. The 2-Nap structure is discussed below; the DHEA structure, while not discussed, is provided in the Supporting Information.
Figure 2.
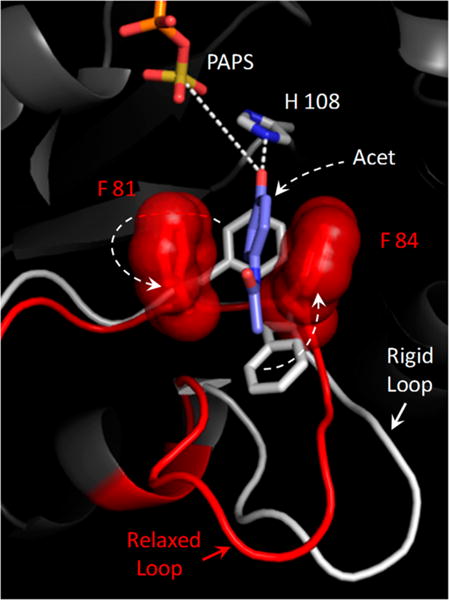
Structural basis of positive synergy. The active-site structural changes predicted to occur when a positive-synergy substrate (acetaminophen, Acet) binds to SULT1A1 are shown. The white segment indicates the disposition of the active-site “lower lip” when Acet is docked into a neutral-synergy conformation (see the text) in which the backbone is held fixed. The red segment indicates how the lower lip switches into what appears to be a positive-synergy conformation when the backbone becomes mobile and the system can “relax” around Acet. This conformational change shifts F81 and F84 into positions that sandwich the benzyl ring of the substrate. Consistent with experimental findings, the molecular “clamp” that forms seems likely to both enhance the affinity of the positive-synergy substrates and to increase the rate of turnover by positioning the nucleophilic hydroxyl for both activation (by hydrogen bonding to H108) and attack at the sulfuryl group. Simulations performed with additional positive- and neutral-synergy substrates suggest that only positive-synergy substrates induce the rearrangement of F81 and F84.
Active-Site Changes
The active-site structural changes predicted to occur as SULT1A1 “relaxes” around Acet are depicted in Figure 2. The rigid protein is colored white; the relaxed protein is colored red. While slight structural changes occur in several non-active-site regions, the most significant active-site change is the rearrangement of a small loop (F81–S91) that interacts with the substrate. We refer to this loop at the lower lip because it is beneath the active-site cap and forms a piece of the “mouth” of the active site (see Lip–Cap Interactions). A prominent feature of the lower-lip rearrangement is the repositioning of F81 and F84. As SULT1A1 responds to Acet, the benzyl side chains of these residues “flip” into positions that sandwich the substrate ring, forming a molecular “clamp” that seems likely to substantially increase the affinity for the substrate and to activate catalysis by fixing the nucleophilic hydroxyl at a position where it can both be activated by the conserved general base H10835 and attack the sulfuryl group (i.e., ~3.0 Å to the H108 ɛ-nitrogen and ~5.5 Å to the sulfur atom of the sulfuryl group).
The view that F81 and 84 are engaged in enhancing the binding and turnover of positive-synergy substrates is supported by calculations of the interaction energies and mobility [i.e., root-mean-square fluctuations (rmsfs)] of these residues in positive- and neutral-synergy complexes (i.e., E·PAPS·Acet and E·PAPS·E2, respectively). Here, interaction energy refers to the time-averaged energy of interaction of the R groups of F81 and F84 with their environments. The interaction energies and rmsf calculations assess the extent to which F81 and F84 are stabilized and “stiffened” as they move between neutral- and positive-synergy environments. The F81 and F84 parameters were calculated and compared to those of all other residues in the protein. Less than two percent of residues are stabilized, and <1% are stiffened as much or more than F81 or F84.
General support for the model is given by the excellent agreement of calculated and experimental acceptor binding free energies: calculations predict PAPS binding will increase Acet and naphthol affinities 35- and 37-fold, respectively, without affecting the binding of E2 or DHEA; experimentation reveals 28- and 18-fold increases, respectively, and no detectable effect on E2 or DHEA binding.
Wandering Nucleophile
Positioning of reactive moieties is a fundamental means of achieving catalysts.36,37 To delve further into the possible role of catalytic positioning in enhancing the turnover of positive-synergy substrates, movies of Gromacs simulations (see the Supporting Information) were created to visualize how acceptors might move about the cavity in the E·PAPS·Acet and E·PAPS·E2 complexes. Throughout the 1.0 ns simulations, Acet remains essentially stationary and well-positioned for catalysis; E2, however, wanders extensively, rarely achieving a catalytic position. The wandering of E2 is depicted in Figure 3, which overlays its positions from several frames of the movie. Simulation predicts that E2 will spend only ~2% of its time in the active site in a catalytic position. Thus, kcat should decrease ~50-fold in moving from Acet to E2, which, given the sophistication of the calculation, is reasonably similar to the 92-fold effect determined experimentally. Here again, results with 2-Nap and DHEA were similar to those obtained with Acet and E2.
Figure 3.
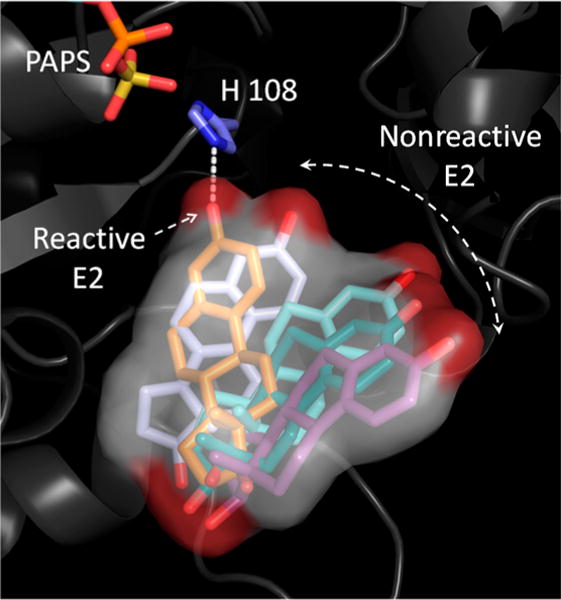
Wandering nucleophile. Snapshots from a GROMACS simulation reveal how E2 (a neutral-synergy substrate) wanders in and out of catalytic position when bound in the E·PAPS·E2 complex. E2 is estimated to spend approximately 2% of its time in a catalytically active position. Positive-synergy substrates are predicted to spend ~100% of the time in an active position, and this difference may explain the roughly 100-fold differences in kcat associated with positive- and neutral-synergy substrates. Similar behaviors were observed with two positive-synergy and two neutral-synergy acceptors: Acet and 2-Nap, respectively, and E2 and DHEA, respectively. Movies that present the predicted motions of these acceptors in the SULT1A1 active-site cavities are available as Supporting Information.
Testing the Model
Strategy
The prediction that the binding of select substrates organizes the lower lip into a high-efficiency catalytic conformation was tested by evaluating whether mutations that destabilize the high-efficiency structure result in a loss of synergy. If the low- and high-efficiency conformations of the lower lip interconvert independently from the remainder of the catalytic machinery, the destabilizing mutations may cause only the loss of positive-synergy traits. For example, if 2-Nap has identical affinity for the E form of the wild type and a mutant enzyme but did not have enhanced affinity for the E·PAP form of the mutant, only the positive-synergy component of binding is lost; the nonsynergistic binding interactions remain intact. Similarly, if a mutation decreased the rate of turnover of 2-Nap to neutral-synergy levels but had no effect on E2 turnover, the catalytic machinery remains intact. If the individual mutations cause both effects, the positive-synergy binding and turnover functions are likely colocalized to the same 10-residue stretch of amino acids, the lower lip.
Lip–Cap Interactions
SULT1A1, like many of its siblings, harbors a 30-reside active-site cap (shaded blue, Figure 4) that closes in response to nucleotide binding.17,18,27,38 As it closes, the cap forms a “pore” at the entrance to the acceptor-binding pocket that sterically screens large acceptors but does not affect binding of acceptors that are small enough to pass through it.18 The acceptor-binding region of the closed cap directly contacts the lower lip and appears to stabilize the positive-synergy conformation. As the nucleotide departs and the cap “peels” away from its base,18 the lip–cap contacts will disengage and likely render the lip incapable of forming a stable high-efficiency conformation.
Figure 4.
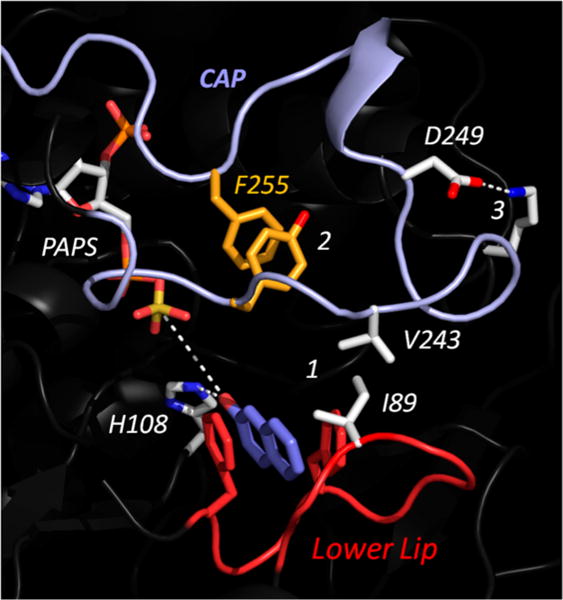
Linkages that stabilize cap closure and the positive-synergy conformation of the lower lip. This structure, which is of the E·PAPS· 2-Nap complex, was predicted using GROMACS. 2-Nap is a positive-synergy acceptor, and the enzyme has assumed the same positive-synergy conformation seen in Figure 1. The R group interactions labeled 1–3 appear to stabilize the closed state of the cap; they are broken as the cap opens. All of the linkages are located in the acceptor-half active-site cap. The cap is colored light blue, and the lower lip is colored red. Each linkage was disrupted via mutagenesis, and the mutants were tested for cap opening, nucleotide binding, and positive synergy.
An in-depth study of the cap of SULT2A138 revealed that it subdivides into two “halves”, a nucleotide half and an acceptor half, and led to the ability to mutagenically uncouple nucleotide binding from closure of the acceptor segment. Analogous SULT1A1 mutants would allow one to test how opening just the segment of the cap that touches the lower lip effects the positive-synergy interactions. This approach tests the model’s predictions without mutating the lip or the residues that it contacts; they are left to engage freely.
Cap Mutations
Using prior SULT2A1 studies as a template,38 the three cap linkages selected for mutagenesis are labeled 1–3 in Figure 4. Link 1 is a direct-contact hydrophobic link between the cap (V243) and lower lip (I89). In an attempt to weaken this interaction, V243 was replaced with G. Link 2, an intracap ring stacking interaction, forms an interesting “kink” in the cap that appears to stabilize its closed structure. Link 2 was weakened by replacing the F255 benzyl moiety with a methyl group (i.e., F255A). The third link stabilizes the cap by forming a salt bridge between D249 (a cap residue) and L147, located in an adjacent protein segment. This interaction was weakened by replacing the carboxylate oxygens of D249 with a hydroxyl group (i.e., D249S). All of the mutants expressed well in E. coli and were purified to >95% homogeneity (see Materials and Methods).
Assaying the Mutants
The consequences of the cap mutations were assessed by monitoring their effects on the binding interactions of the nucleotide and acceptor. In the absence of bound ligands, the equilibrium constant for the closed–open isomerization of the SULT1A1 cap favors the open state (Kiso = [cap]closed/[cap]open, > 0.0518,39). In contrast, when a nucleotide (PAP or PAPS) is bound, Kiso favors the closed conformation (Kiso E·Nuc = 2517,18). Consequently, the affinities of acceptors too large to pass through the pore (large acceptors) decrease when a nucleotide is bound by a factor given to a near approximation18,39 by Kiso E·Nuc (i.e., 26-fold). If acceptor-binding determinants other than those associated with cap closure are not affected by the mutation, the affinities of small acceptors will not be altered. Thus, by studying the effects of the nucleotide on the binding of small and large acceptors, the effects of the mutation on cap isomerization and the intrinsic binding properties of the acceptor pocket can be evaluated. E2 and fulvestrant are a homologous small and large acceptor pair, respectively, used previously in SULT1A1 studies.18 Their binding is readily monitored via significant decreases (~25%) in the intrinsic fluorescence of the enzyme. Fulvestrant, an FDA-approved selective estrogen receptor modulator, is a C7 derivative of E2 whose substituent is large enough to prevent it from passing through the acceptor pore of the closed cap.
To assess whether a mutation affects nucleotide protein interactions other than those associated with the cap, the affinity of the nucleotide for the mutant and wild-type enzymes was compared. The absence of a detectable change in affinity is taken as an indication that the “other” interactions remained largely intact.
Assay Results
The effects of the cap mutations on the interactions of large and small acceptors with nucleotide-bound and -free SULT1A1 are compiled in Table 2. Within error, the mutations do not affect the binding of E2 to either E or E·PAP. In contrast, the nucleotide-induced 21-fold decrease in the affinity of fulvestrant seen in the wild-type enzyme does not occur in any of the cap mutants. Fulvestrant binds with equal affinity to E and E·PAP; moreover, its affinities for the E forms of the mutants and wild-type enzyme are identical. Thus, the mutations appear to destabilize the closed form of the cap without affecting the pocket’s other binding functions. Finally, the affinity of PAP is not influenced by the mutations; the nucleotide-binding functions remain fully intact. In summary, the mutations appear to have uncoupled only the nucleotide’s ability to stabilize the pore; all other acceptor- and nucleotide-binding functions remain near those of the wild type.
Table 2.
Ligand Binding to Wild-Type and Mutant SULT1A1
| SULT1A1 |
Kd (μM)a
|
||||||
|---|---|---|---|---|---|---|---|
| E2
|
fulvestrant
|
PAP
|
2-Nap
|
||||
| E | E·PAP | E | E·PAP | E | E | E·PAP | |
| WT | 1.0 (0.1) | 0.9 (0.2) | 1.7 (0.3) | 35 (4) | 0.27 (0.02) | 1.7 (0.3) | 0.077 (0.02) |
| V243G | 0.9 (0.1) | 1.6 (0.2) | 1.3 (0.2) | 1.5 (0.3) | 0.28 (0.01) | 2.2 (0.2) | 2.7 (0.3) |
| F255A | 0.9 (0.2) | 0.9 (0.2) | 1.5 (0.1) | 1.4 (0.2) | 0.27 (0.02) | 2.0 (0.2) | 1.7 (0.3) |
| D249S | 0.9 (0.1) | 1.5 (0.3) | 1.6 (0.2) | 1.2 (0.3) | 0.30 (0.01) | 2.2 (0.2) | 2.1 (0.3) |
| I89A | 1.5 (0.3) | 1.6 (0.2) | 1.1 (0.1) | 22 (3) | 0.31 (0.04) | 1.8 (0.1) | 1.9 (0.2) |
Parentheses enclose standard error estimates.
Coupling of Nucleotide Binding, Pore Formation, and Positive Synergy
To assess whether pore formation and positive synergy are coupled, the effects of nucleotide on the affinity of 2-Nap, a positive-synergy substrate, for each cap mutant were determined and compared to the effect on the wild type (Table 2). In each case, the 22-fold enhancement in the affinity of 2-Nap seen in the wild type vanishes; positive synergy is lost. Thus, closure of the acceptor “flap” is required to stabilize the positive-synergy conformation of the enzyme. This model, which is well supported by the data (Table 2), provides a cogent molecular rationale that explains how binding of a nucleotide transmits a signal across the cap to the lower lip by closing a flap that directly contacts and stabilizes the positive-synergy conformation, a beautiful example of molecular communication between distal subsections of an active site.
A Lower-Lip Mutant
To directly test the dependence of positive synergy on the lower lip, it was mutagenized and the effects of the mutations were determined. Four point mutations, each at a different position in the lip, were constructed (F81A, F84A, E85S, and I89A). All of the mutations expressed well in E. coli and were stable toward purification; however, only one (i.e., I89A) was active. Neither binding nor turnover could be detected in the remaining three (see Materials and Methods). Consistent with the structure shown in Figure 4, replacement of the isopropyl group of I89 with a methyl group is expected to weaken the interaction between the lower lip and cap. The binding studies (Table 2) reveal that, within error, the I89A mutation completely abolishes positive synergy, thus confirming a direct role for the lower lip in establishing positive synergy.
It is interesting that while the I89A mutation prevents positive synergy, it does not prevent cap closure. This is evident in the results of the fulvestrant binding studies, which reveal that nucleotide binding causes a 22-fold decrease in the affinity of fulvestrant for both the wild-type and mutant enzymes (Table 2). Thus, the mutation uncouples the positive-synergy functions of the lower lip from cap closure. The possibility that these functions are separable is plausible given that they are associated with structural changes in separate regions of the protein. This separability is supported by modeling, which suggests that in lieu of interactions with I89, V243 can “tuck” up under the cap to form other stabilizing interactions, and by the experimental finding that the wild-type cap closes when neutral-synergy compounds are bound.40 In summary, closure of the acceptor half of the cap is necessary but not sufficient to elicit positive synergy, and the positive-synergy conformation is not required for cap closure. It is as if positive synergy requires both the I89–V243 interaction and a substrate capable of stabilizing the active conformation, a molecular “linchpin” that acts to “fasten” the system in the active conformation.
Nap Wanders in the I89A Mutant
If the I89A mutation did no more than prevent the lower lip from achieving the positive-synergy conformation, it would, in essence, “lock” the enzyme in a neutral-synergy state and thus prevent it from distinguishing positive- from neutral-synergy acceptors. If so, positive-synergy substrates, like their neutral-synergy counterparts, would be expected to wander in the mutant active-site cavity, causing a decrease in kcat relative to that of the wild-type enzyme. GROMACS simulations of the I89A mutant predict that with both PAPS and Nap bound, the positive-synergy conformation does not form in the mutant, and that Nap wanders, like a neutral-synergy substrate, in the active site (see the movies in the Supporting Information).
The wandering-nucleophile model predicts that the kcat of 2-Nap will be decreased by the I89A mutation. To test this, the initial-rate parameters for Nap were determined using the wild-type and mutant enzymes. As a control to assess whether the sulfuryl-transfer “machinery” of the mutant remains intact, initial-rate parameters for a neutral-synergy acceptor (E2) were also determined. The 4 × 4 data used to obtain the parameters are presented in Supplemental Figure 1. The E2 parameters for the wild-type and mutant enzymes are nearly identical (Table 3), whereas the 2-Nap parameters reveal that the mutation causes a 19-fold decrease in kcat, a 54-fold increase in Km, and a 650-fold decrease catalytic efficiency. These fold changes are in line with those observed upon comparison of the wild-type parameters of positive- and neutral-synergy substrates. In summary, the data suggest that the mutation causes the enzyme to remain in the neutral-synergy conformation and support the wandering-nucleophile mechanism.
Table 3.
Effect of the I89A Mutation on Initial-Rate Parameters
| enzyme | acceptor | Km (μM) | kcat (s−1) | kcat/Km |
|---|---|---|---|---|
| WT | E2a | 1.2 (0.1)c | 1.0 (0.05) | 0.83 (0.02) |
| I89A | 1.2 (0.2) | 0.9 (0.03) | 0.75 (0.09) | |
| WT | 2-Napb | 0.05 (0.01) | 111 (8) | 2220 (500) |
| I89A | 2.7 (0.2) | 5.8 (0.2) | 3.4 (0.5) |
E2, estradiol.
2-Nap, 2-naphthol.
Parentheses enclose standard error estimates.
CONCLUSIONS
A study of the binding and reactivity of SULT1A1 substrates has revealed that they separate into two groups: those whose affinities are enhanced ~20-fold in the presence of saturating nucleotide (positive-synergy substrates) and those whose affinities are unaffected by nucleotide (neutral-synergy substrates). kcat for the positive-synergy group is ~100-fold greater that that for the neutral-synergy compounds; thus, the catalytic efficiencies (kcat/Km) of the positive-synergy compounds are 3–4 orders of magnitude greater than those of their neutral-synergy counterparts.
A molecular model for positive synergy is proposed in which binding of a positive-synergy substrate causes F81 and F84 to “switch” into positions that “sandwich” the phenolic moiety of the substrate and thus stabilize its nucleophilic hydroxyl in a reactive position. Unlike their positive-synergy counterparts, neutral-synergy acceptors are predicted to wander in the active site, rarely achieving a reactive position. The major tenets of this model were tested using site direct mutants with the result that the mutants behave precisely as predicted by the model.
Several additional insights were obtained during the course of the work. Notably, the order of substrate binding has been resolved, and binding is random; it was shown that the acceptor half of the active-site cap of SULT1A1 can be uncoupled from the binding of the nucleotide.
Supplementary Material
Acknowledgments
Funding
National Institutes of Health Grant RO1 GM106158.
ABBREVIATIONS
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- PAPS
3′-phosphoadenosine 5′-phosphosulfate
- PAP
3′,5′-diphosphoadenosine
- WT
wild type
Footnotes
Supporting Information
- Initial-rate studies of wild-type and I89A SULT1A1 turnover with 2-Nap and E2 (PDF)
- Molecular dynamics movies of neutral- and positive-synergy (E2 and DHEA, and Acet and NAP) acceptors bound to the E·PAPS complex of wild-type and I89A SULT1A1 (MOV)
- Molecular dynamics movies of neutral- and positive-synergy (E2 and DHEA, and Acet and NAP) acceptors bound to the E·PAPS complex of wild-type and I89A SULT1A1 (MOV)
- Molecular dynamics movies of neutral- and positive-synergy (E2 and DHEA, and Acet and NAP) acceptors bound to the E·PAPS complex of wild-type and I89A SULT1A1 (MOV)
- Molecular dynamics movies of neutral- and positive-synergy (E2 and DHEA, and Acet and NAP) acceptors bound to the E·PAPS complex of wild-type and I89A SULT1A1 (MOV)
- Molecular dynamics movies of neutral- and positive-synergy (E2 and DHEA, and Acet and NAP) acceptors bound to the E·PAPS complex of wild-type and I89A SULT1A1 (MOV)
Notes
The authors declare no competing financial interest.
References
- 1.Visser TJ. Role of sulfation in thyroid hormone metabolism. Chem-Biol Interact. 1994;92:293–303. doi: 10.1016/0009-2797(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 2.Spaulding SW, Smith TJ, Hinkle PM, Davis FB, Kung MP, Roth JA. Studies on the biological activity of triiodothyronine sulfate. J Clin Endocrinol Metab. 1992;74:1062–1067. doi: 10.1210/jcem.74.5.1533227. [DOI] [PubMed] [Google Scholar]
- 3.Bai Q, Xu L, Kakiyama G, Runge-Morris MA, Hylemon PB, Yin L, Pandak WM, Ren S. Sulfation of 25-hydroxycholesterol by SULT2B1b decreases cellular lipids via the LXR/SREBP-1c signaling pathway in human aortic endothelial cells. Atherosclerosis. 2011;214:350–356. doi: 10.1016/j.atherosclerosis.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cook IT, Duniac-Dmuchowski Z, Kocarek TA, Runge-Morris M, Falany CN. 24-Hydroxycholesterol Sulfation by Human Cytosolic Sulfotransferases: Formation of Monosulfates and Disulfates, Molecular Modeling, Sulfatase Sensitivity and Inhibition of LXR Activation. Drug Metab Dispos. 2009;37:2069–2078. doi: 10.1124/dmd.108.025759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calabresi P, Di Filippo M, Ghiglieri V, Tambasco N, Picconi B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol. 2010;9:1106–1117. doi: 10.1016/S1474-4422(10)70218-0. [DOI] [PubMed] [Google Scholar]
- 6.Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- 7.Meisheri KD, Cipkus LA, Taylor CJ. Mechanism of action of minoxidil sulfate-induced vasodilation: a role for increased K+ permeability. J Pharmacol Exp Ther. 1988;245:751–760. [PubMed] [Google Scholar]
- 8.Cook I, Wang T, Falany CN, Leyh TS. High Accuracy In-Silico Sulfotransferase Models. J Biol Chem. 2013;288:34494–34501. doi: 10.1074/jbc.M113.510974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parker CR. Dehydroepiandrosterone and dehydroepiandrosterone sulfate production in the human adrenal during development and aging. Steroids. 1999;64:640–647. doi: 10.1016/s0039-128x(99)00046-x. [DOI] [PubMed] [Google Scholar]
- 10.Kotov A, Falany JL, Wang J, Falany CN. Regulation of estrogen activity by sulfation in human Ishikawa endometrial adenocarcinoma cells. J Steroid Biochem Mol Biol. 1999;68:137–144. doi: 10.1016/s0960-0760(99)00022-9. [DOI] [PubMed] [Google Scholar]
- 11.Falany JL, Macrina N, Falany CN. Regulation of MCF-7 breast cancer cell growth by beta-estradiol sulfation. Breast Cancer Res Treat. 2002;74:167–176. doi: 10.1023/a:1016147004188. [DOI] [PubMed] [Google Scholar]
- 12.Tranheim Kase E, Andersen B, Nebb HI, Rustan AC, Hege Thoresen G. 22-Hydroxycholesterols regulate lipid metabolism differently than T0901317 in human myotubes. Biochim Biophys Acta, Mol Cell Biol Lipids. 2006;1761:1515–1522. doi: 10.1016/j.bbalip.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Falany JL, Falany CN. Regulation of estrogen sulfotransferase in human endometrial adenocarcinoma cells by progesterone. Endocrinology. 1996;137:1395–1401. doi: 10.1210/endo.137.4.8625916. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Falany CN. Elevated hepatic SULT1E1 activity in mouse models of cystic fibrosis alters the regulation of estrogen responsive proteins. J Cystic Fibrosis. 2007;6:23–30. doi: 10.1016/j.jcf.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Leyh TS, Cook I, Wang T. Structure, dynamics and selectivity in the sulfotransferase family. Drug Metab Rev. 2013;45:423–430. doi: 10.3109/03602532.2013.835625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cook I, Wang T, Falany CN, Leyh TS. The allosteric binding sites of sulfotransferase 1A1. Drug Metab Dispos. 2015;43:418–423. doi: 10.1124/dmd.114.061887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang T, Cook I, Leyh TS. 3′-Phosphoadenosine 5′-phosphosulfate allosterically regulates sulfotransferase turnover. Biochemistry. 2014;53:6893–6900. doi: 10.1021/bi501120p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cook I, Wang T, Almo SC, Kim J, Falany CN, Leyh TS. The gate that governs sulfotransferase selectivity. Biochemistry. 2013;52:415–424. doi: 10.1021/bi301492j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coughtrie MW, Johnston LE. Interactions between dietary chemicals and human sulfotransferases-molecular mechanisms and clinical significance. Drug Metab Dispos. 2001;29:522–528. [PubMed] [Google Scholar]
- 20.Vietri M, De Santi C, Pietrabissa A, Mosca F, Pacifici GM. Inhibition of human liver phenol sulfotransferase by nonsteroidal anti-inflammatory drugs. Eur J Clin Pharmacol. 2000;56:81–87. doi: 10.1007/s002280050725. [DOI] [PubMed] [Google Scholar]
- 21.Sun M, Leyh TS. The human estrogen sulfotransferase: a half-site reactive enzyme. Biochemistry. 2010;49:4779–4785. doi: 10.1021/bi902190r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 23.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 24.Verdonk ML, Berdini V, Hartshorn MJ, Mooij WT, Murray CW, Taylor RD, Watson P. Virtual screening using protein-ligand docking: avoiding artificial enrichment. J Chem Inf Model. 2004;44:793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Cieplak P, Kollman PA. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules. J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 26.Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA. AMBER 14. University of California; San Francisco: 2014. [Google Scholar]
- 27.Wang T, Cook I, Falany CN, Leyh TS. Paradigms of Sulfotransferase Catalysis - The Mechanism of SULT2A1. J Biol Chem. 2014;289:26474–26480. doi: 10.1074/jbc.M114.573501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gamage NU, Tsvetanov S, Duggleby RG, McManus ME, Martin JL. The structure of human SULT1A1 crystallized with estradiol. An insight into active site plasticity and substrate inhibition with multi-ring substrates. J Biol Chem. 2005;280:41482–41486. doi: 10.1074/jbc.M508289200. [DOI] [PubMed] [Google Scholar]
- 29.Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 30.Whittemore RM, Pearce LB, Roth JA. Purification and kinetic characterization of a phenol-sulfating form of phenol sulfotransferase from human brain. Arch Biochem Biophys. 1986;249:464–471. doi: 10.1016/0003-9861(86)90023-8. [DOI] [PubMed] [Google Scholar]
- 31.Duffel MW, Jakoby WB. On the mechanism of aryl sulfotransferase. J Biol Chem. 1981;256:11123–11127. [PubMed] [Google Scholar]
- 32.Tyapochkin E, Cook PF, Chen G. Isotope exchange at equilibrium indicates a steady state ordered kinetic mechanism for human sulfotransferase. Biochemistry. 2008;47:11894–11899. doi: 10.1021/bi801211t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrison JF, Cleland WW. Isotope Exchange Studies of the Mechanism of the Reaction Catalyzed by Adenosine Triphosphate:Creatine Phosphotransferase. J Biol Chem. 1966;241:673–683. [PubMed] [Google Scholar]
- 34.Rose IA, O’Connell EL, Litwin S. Determination of the rate of hexokinase-glucose dissociation by the isotope-trapping method. J Biol Chem. 1974;249:5163–5168. [PubMed] [Google Scholar]
- 35.Pedersen LC, Petrotchenko E, Shevtsov S, Negishi M. Crystal structure of the human estrogen sulfotransferase-PAPS complex: evidence for catalytic role of Ser137 in the sulfuryl transfer reaction. J Biol Chem. 2002;277:17928–17932. doi: 10.1074/jbc.M111651200. [DOI] [PubMed] [Google Scholar]
- 36.Page MI, Jencks WP. Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect. Proc Natl Acad Sci U S A. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirby AJ. Effective Molarities for Intramolecular Reactions. Vol. 17. Academic Press Inc; New York: 1980. [Google Scholar]
- 38.Cook I, Wang T, Almo SC, Kim J, Falany CN, Leyh TS. Testing the Sulfotransferase Molecular Pore Hypothesis. J Biol Chem. 2013;288:8619–8626. doi: 10.1074/jbc.M112.445015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cook I, Wang T, Falany CN, Leyh TS. A Nucleotide-Gated Molecular Pore Selects Sulfotransferase Substrates. Biochemistry. 2012;51:5674–5683. doi: 10.1021/bi300631g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gamage NU, Duggleby RG, Barnett AC, Tresillian M, Latham CF, Liyou NE, McManus ME, Martin JL. Structure of a human carcinogen-converting enzyme, SULT1A1. Structural and kinetic implications of substrate inhibition. J Biol Chem. 2003;278:7655–7662. doi: 10.1074/jbc.M207246200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.