

Abstract
The first mutation in a gene associated with a neuronal migration disorder was identified in patients with Kallmann Syndrome, characterized by hypogonadotropic hypogonadism and anosmia. This pathophysiological association results from a defect in the development of the GnRH and the olfactory system. A recent genetic screening of Kallmann Syndrome patients revealed a novel mutation in CCDC141. Little is known about CCDC141, which encodes a coiled-coil domain containing protein. Here, we show that Ccdc141 is expressed in GnRH neurons and olfactory fibers and that knockdown of Ccdc141 reduces GnRH neuronal migration. Our findings in human patients and mouse models predict that CCDC141 takes part in embryonic migration of GnRH neurons enabling them to form a hypothalamic neuronal network to initiate pulsatile GnRH secretion and reproductive function.
Defective neuronal migration contributes to neurological disorders that lead to developmental delay and seizures. The first mutation in a gene associated with a neuronal migration disorder was identified in patients with Kallmann Syndrome (KS). KS is characterized by a combination of anosmia and hypogonadotropic hypogonadism. This pathophysiological association is due to defects during the shared development of GnRH neurons and the olfactory system (1, 2). GnRH neurons originate in the nasal placode, and migrate along olfactory axons into the central nervous system (CNS). Upon entering the CNS, olfactory axons infiltrate the olfactory bulb, whereas GnRH neurons migrate caudally on a subset of nonsensory olfactory axons toward the hypothalamus. Once within the hypothalamus, GnRH neurons form a functional neural circuit that secretes pulses of GnRH into the portal capillary system. Disruption of these developmental processes results in KS (3).
After the discovery of KAL1 in KS (4, 5), several mutations have been identified in neuronal migration disorders including polymicrogyria (6–8), lissencephaly (7, 9–15), schizocephaly (16), periventricular heterotopia (17), microcephaly (8, 9, 16), and Goldberg-Shprintzen syndrome (18). Because neuronal migration requires the dynamic remodeling of the cell's cytoskeleton (19–24), it is not surprising that many of these mutations are in genes encoding cytoskeletal proteins and their binding partners. However, only 1 gene that associated with KS is directly involved in cytoskeletal remodeling (TUBB3). Instead, they have included mutations altering extracellular matrix/cell adhesion proteins (KAL1, HS6ST1), neurogenesis/neuronal differentiation (the fibroblast growth factor [FGF] signaling pathway genes FGF8, FGFR1, FGF17, IL17RD, DUSP6, SPRY4, and FLRT3), chemoattractants/cell survival (SEMA3), PROK2 signaling pathway genes (PROK2 and PROKR2), DNA factors (CHD7, HESX1, and FEZF1), and olfactory ensheathing cells (OECs) (SOX10) (3).
Several KS cases are mediated through oligogenic inheritance (25). Recently, we reported that a mutation in the transcription factor FEZF1 was found in KS patients (26). In one of these families, a mutation in the cytoskeletal associated protein CCDC141 was also found. The FEZF1 mutation showed partial function in vitro, suggesting that the mutated CCDC141 is partially responsible for the overall phenotype (hypogonadotropic hypogonadism and anosmia). Little is known about CCDC141, which encodes a coiled-coil domain containing protein (27). However, CCDC141, in association with disrupted in schizophrenia-1 (Disc1) and myosin II, has been implicated in cortical interneuron migration in mice. Here, we show that Ccdc141 is expressed in GnRH neurons and olfactory fibers during embryonic development and that knockdown of Ccdc141 reduces GnRH neuronal migration. Taken together, these findings in human patients and mouse models predict that cytoskeleton-associated genes may be discovered as secondary mutations in KS contributing to altered neuronal migration.
Materials and Methods
Ethics Committee of the Cukurova University, Faculty of Medicine approved this study, and written informed consent was obtained for each participant. All animal procedures were approved by National Institute of Neurological Disorders and Stroke animal care and use committee and performed in accordance with National Institutes of Health (NIH) guidelines.
Subjects' hormone levels and olfactory function
Plasma hormone levels were analyzed by commercial kits based on solid-phase, 2-site sequential, or competitive chemiluminescent immunometric assay or electrochemi-luminescence immunoassay (Beckman Coulter). Olfactory function was evaluated using the 40-item UPSIT smell identification test (PAR). UPSIT is a validated microencapsulated odor “scratch and sniff” test that correlates with other olfactory tests including odor detection thresholds (28) and thus used to objectively evaluate KS patients. To control for cross-cultural variation of smell identification and thereby to prevent false negatives, a culturally appropriate 20-item-smell test, which was made in the clinic was also administrated. Results of this test were found to be consistent with the UPSIT test ensuring that the patients lacked the ability to detect an odor.
Identification of genes
Genes known to be associated with KS including KAL1, FGFR1, FGF8, NELF, PROK2, PROKR2, CHD7, WDR11, and HS6ST1 were screened by sequencing on an ABI PRISM 3130 autosequencer. A genome-wide single nucleotide polymorphism analysis used 250K NspI SNP microarrays (Affymetrix) and the data were analyzed using AutoSNPa software (http://autozygosity.org). For exome sequencing, briefly, samples were prepared as an Illumina sequencing library. Sequencing libraries were enriched for the desired target using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using Illumina HiSeq 2000 Sequencer (Macrogen). The reads were mapped against University of California Santa Cruz hg19.
Mice
NIH Swiss mice were used for generating nasal explants (see below) and for initial CCDC141 antibody characterization. To delineate the expression of CCDC141 in nasal regions, 2 additional mouse lines were used, GnRH-GFP mice (to identify GnRH cells) (28) and BLBP-Cre mice (to identify OECs) (29) crossed with Rosa-YFP reporter lines (Jackson ImmunoResearch).
Tissue collection
All mice were housed 2–4 per cage in a conventional vivarium at the NIH. Mice were time mated and euthanized in a CO2 chamber followed by cervical dislocation. Embryos (E12.5–E15.5) were removed, immersed in 4% formaldehyde/PBS (1–2 h), cryoprotected overnight in 30% sucrose/PBS, embedded in Tissue Tek OCT, and stored at −80°C until sectioned. Serial sections (14 μm) were cut on a Leica CM 3050S cryostat (Leica Biosystems) and maintained at −80°C until processing.
Immunocytochemistry
Primary antibodies
Rabbit polyclonal (Rb) anti-GnRH (SW-1, 1:3000) (30), mouse monoclonal anti-GnRH (FID3C5, 1:4000; gift from Dr Karande) (31), Rb antiperipherin (peripheral intermediate filament marker; 1:2000; Chemicon), Rb anti-coiled-coil domain-containing protein 141 (CCDC141, 5 μg/mL; Sigma-Aldrich), mouse monoclonal antitubulin III (Tuj1, 1:700; Sigma-Aldrich), monoclonal biotinylated anti-HuC-D (1:100; Invitrogen), and chicken anti-green fluorescent protein (GFP, 1:1000; Abcam).
Explants and embryos were stained as described previously (32). In brief, explants were fixed (4% formaldehyde/PBS) for 30 minutes. After fixation or sectioning, explants and slides, respectively, were washed in PBS, blocked in normal horse serum (10%) with 0.3% Triton X-100 (1 h), then rinsed in PBS before incubating in primary antibody 1–2 nights (4°). After incubation, standard protocols for chromagen staining were used: biotinylated secondary antibodies were either donkey antirabbit-bt (Vector Laboratories) or donkey antimouse-bt (Millipore Bioscience Research Reagents), visualized by ABC/chromagen methods (33). For fluorescent staining, secondary antibodies were avidin-conjugated Cy3 (1 h, 1:1000; Jackson ImmunoResearch) or directly conjugated secondaries (Alexa Fluor 488 or 555; 1 h, 1:1000 donkey antirabbit [Ccdc141], donkey antimouse [GnRH], or donkey antichicken [GFP]; Invitrogen).
Controls
The specificity of the CCDC141 antibody was verified by preincubation of 10 μg/mL (CCDC141 peptide; Sigma) with diluted antibody (see Figure 2 below). In addition, omission of the primary antibody (single stain) resulted in no detectable signal, and omission of the second primary (double stain) resulted in no double-labeled structures (data not shown). Pictures were taken as previously described (20, 32, 34). Fluorescent images were taken on a spinning disk confocal system CSU10 (Yokogawa) mounted on an Eclipse TE200 microscope (Nikon) using an EMCCD ImageM digital camera (Hamamatsu) with I-Vision software (Biovision). Pictures were taken on a Nikon Eclipse E800 with a Retiga SRV camera using QCapture software (in vivo), or a Nikon Eclipse E800 with a Retiga Exi camera using QCapture software (QImaging) (in vitro). Fluorescent images are digitized using a greyscale from 0 to 4095. The images were then processed via. NIH ImageJ software (Wayne Rasband; http://rsbweb.nih.gov/ij/) to pseudocolor.
Figure 2.

Ccdc141 is expressed in migrating GnRH neurons in the developing mouse. A1, Schematic of an E11.5 parasagittal mouse head; *, developing VNO; OE, olfactory epithelium, arrow points to nasal forebrain junction; OB, olfactory bulb. Peripherin-positive olfactory axons (in brown) are shown extending to the olfactory bulb. GnRH neurons (shown in black) migrate on these axon bundles until they cross the nasal forebrain junction (black arrow) and enter the brain. A2, E11.5 embryo stained for peripherin (brown) and GnRH (black), with high-magnification image of boxed area on right. Arrowhead indicates GnRH neurons streaming out from the VNO (*). B1–B4, Fluorescent staining for Ccdc141 (red), GnRH (green), and nuclear staining with DAPI (blue) at E11.5. B1, Ccdc141 staining is expressed in GnRH neurons and in mesenchymal cells. Robust staining is evident in the axonal tracts leaving the VNO where GnRH neurons are located (arrows). B2–B4, Higher magnification showing GnRH/Ccdc141 neurons in the VNO. C1, Schematic of a coronal section of an E14.5 embryo nose; VNO (*), respiratory epithelium (RE), OE, nasal forebrain junction (arrow), and OB. C2, Magnified coronal sections from an E14.5 embryo stained for GnRH (black) and peripherin (brown), showing GnRH neurons at the nasal/forebrain junction (upper box in schematic) and leaving the VNO (*, bottom box in schematic). D, Fluorescent staining of Ccdc141 red at E14.5 showing staining in VNO (*) and olfactory axons (OX). E, Serial section stained with antibody + blocking peptide (10 μg/mL). After primary antibody/primary antibody + blocking peptide, D and E were stained together. No specific staining was detected after blocking peptide was added to the primary antibody. F1–F4, Fluorescent staining for Ccdc141 (red), GnRH (green), and nuclear staining with DAPI (blue) at E14.5. By E14.5, Ccdc141 levels appeared to decrease in mesenchyme cells (F1) yet remained clearly present in sensory axons and GnRH neurons (F2–F4). Scale bars: 100 μm (A), 50 μm (B1 and F1), and 10 μm (B2–B4 and F2–F4).
Primary nasal explants
Explants were generated from the nasal pits of E11.5 NIH Swiss mouse embryos as previously described (33). Briefly, nasal pits were dissected under aseptic conditions in Gey's balanced salt solution (Life Technologies, Inc) supplemented with glucose (Sigma Chemical Co). Explants were adhered onto coverslips by a plasma (Cocalico Biologicals)/thrombin (Sigma) clot and maintained at 37°C in a defined serum-free medium (SFM) in a humidified atmosphere with 5% CO2. On culture day 3, SFM was replaced by fresh SFM containing fluorodeoxyuridine (80μM; Sigma), to inhibit proliferation of dividing olfactory neurons and nonneuronal explant tissue. Both bilateral and unilateral explants were generated (35).
Polymerase chain reaction
Poly(A) amplified cDNA libraries were previously created from individual GnRH cells in nasal explants (35–38). The phenotype and the quality of each single-cell cDNA was confirmed by PCR for GnRH, L19, and β-tubulin (36). The same method was used to generate cDNA libraries from whole explants (35). Primers were designed in the 3′-untranslated region of CCDC141, within 350 bp before the poly adenylation site. All designed primers were screened using BLAST to ensure specificity. For each reaction, 30.5-μL H2O, 5-μL 10× PCR buffer, 4-μL 25mM MgCl2, 5-μL deoxynucleotide mix (Life Technologies; 25 μL of each 100mM dNTP, 900-μL H2O), 2-μL 6.25μM forward primer, 2-μL 6.25μM reverse primer, and 0.5-μL AmpliTaq Gold (Life Technologies) were added to 1-μL template cDNA. For CCDC141 on single cells, a nested PCR was used, ie, 1 μL of the product was run a second time with a second set of primers. Amplified products were run on a 1.5% agarose gel. Specific bands of the predicted size were observed in control total brain, whereas no bands were seen in water (negative control). The sequences of the primers used were: CAAAGGCTCTGGACGGTAAC and TCTTCCCTTCATCAAAAATCTGT (first round), and CTCTGGACGGTAACCTCTGG and TTGATTTTACCCTAGCATGACAAA (second round). GnRH primers: ACTGGTCCTATGGGTTGCGCCCTG and CGGGGCCAGTGGACAGTACATTCG, which bind in exons 2 and 3; separated by approximately 1-kb intronic region. CCDC141 product: 255 and 207 bp for first and second rounds, respectively. Gnrh1 product: 132 bp (absence of 1-kb genomic band indicated no genomic contamination) (data not shown). Positive controls were whole embryo and adult brain cDNAs, prepared as previously described (39). Negative control; sample with no DNA template.
GnRH neuronal migration assays
CCDC141 deficiency was assessed in mouse nasal explants. Two different assays were used after knockdown using small interfering RNA (siRNA) (see below); live imaging to determine GnRH neuronal migration rates and post hoc analysis of total change in the location of GnRH neurons and olfactory axons.
Live imaging
In situ GnRH cell analysis was performed on 11 explants from 2 different culturing dates (CCDC141 siRNA n = 5; control siRNA n = 6). At 4 days in vitro (div), explants were placed into a chamber (Warner Instruments) on top of the microscope stage. Temperature was maintained at 37 ± 2°C and the CO2 concentration at 5% (Live cell; Pathology Devices, Inc). Experiments were performed on a TE200 microscope (Nikon USA) equipped with differential interference contrast optics, a motorized stage (Ludl, Electronic Products Ltd), and an interline charge-coupled device camera (Retiga, Qimaging), piloted by imaging software (IPLab Spectrum; Scanalytics, Inc) on a Macintosh computer. Cells were identified by morphology and then confirmed by post hoc immunostaining for GnRH. Recording of cell positions were monitored every min for up to 1 hour. Images were taken with an ELWD ×20 objective (Nikon). Both linear distance and total distance were measured as previously described (20, 34). Total distance reflects the entire route that the cell moved, whereas linear distance equals the change between the first position and final position. Distance traveled was calculated from the x and y coordinates of the cell center plotted in ImageJ for each frame (total) or first and last frame (linear). Live imagining data was compared by Mann-Whitney U test (20, 34) and considered significantly different if P < .05.
Post hoc imaging
For this analysis, explants from 2–3 different culture dates were fixed at 5 div and stained for GnRH and/or peripherin to assess overall GnRH neuronal migration and olfactory axon outgrowth (36). For control vs Ccdc141 siRNA-treated explants, the distance that GnRH cells migrated (n = 4 explants/group), and the 20 olfactory axon terminals farthest from the center of each explant (n = 3 explants/group) were measured and compared as previously reported (20, 34). Brightfield images in figure 6 below were corrected for uneven illumination with a high-pass filter. Distances of the GnRH neuronal population from the center of the explant were expressed as cumulative frequency distributions for comparison. The 2 treatment groups, in 2 different siRNA treatments, were then compared with the two-way ANOVA to test for differences in overall distance traveled and for interactions of the treatment at different levels within the frequency distribution (which could reflect an effect on a subpopulation of GnRH neurons). Olfactory axon length between the 2 groups was analyzed using an unpaired t test. Statistical analysis was performed in GraphPad Prism.
siRNA electroporation
GnRH neurons emerge from the nasal tissue of unilateral nasal explants at 2 days in vitro (20, 34), at which time they are exposed to the surface of the culture medium and can be effectively transfected. Thus, at this time point, 10 μL of 2% vol/vol control (general-purpose Control siRNA-A sc-37007; Santa Cruz Biotechnology, Inc) or CCDC141 siRNA (sc-108804; Santa Cruz Biotechnology, Inc or Ref. 27) was added to unilateral explants in 10-μL Hanks' balanced salt solution (Thermo Fisher Scientific) at a final concentration of 1% vol/vol siRNA solution. A Nepa21 electroporator was used: 2 poring pulses of 125 V and 5-ms duration with 10% decay separated by 50 ms, followed by 10 transfer pulses (5 positive and 5 negative polarity) of 50 V and 50-ms duration with 40% decay separated by 50 ms. Explants were then washed in SFM and incubated for 2–3 days before live imaging or 3 days before fixation for knockdown assessment and migration analysis (20, 34). In knockdown assessment, GnRH cytoplasmic immunofluorescence was used as a region of interest mask and average CCDC141 immunofluorescence intensity per cell was measured after immunostaining as described above for comparisons between transfected knockdown and control groups. Images were further analyzed using NIH ImageJ software (Wayne Rasband, http://rsbweb.nih.gov/ij/).
Results
The phenotype of the family carrying the identified CCDC141 mutation has been summarized (26). Briefly, a 19-year-old male (who presented first at age 14) had absent pubertal development. His 24-year-old sister suffered from primary amenorrhea and absent breast development. Both patients were anosmic but otherwise healthy. Neither exhibited mental disorders, cleft lip, cleft palate, or other features sometimes associated with KS (40). Neither parent nor other siblings experienced problems in pubertal development or fertility, and all were normosmic. Brain magnetic resonance imaging (MRI) showed aplasia of olfactory bulbs bilaterally in the proband (Figure 1), whereas cortical thickness appeared normal. Hormonal levels of FSH, LH, and estradiol or testosterone were below the normal range (26). Autozygosity mapping identified a nonsense mutation (p.R724X) in the CCDC141 gene (HGNC:26821), which is located in the homozygous region on chromosome 2. Both affected individuals were homozygous for a C to T change at cDNA nucleotide 2170 (NM_173648.3:c.C2170T). This mutation leads to the substitution of arginine at residue 724 for a Stop codon (NP_775919.3:p.R724X). Parents and unaffected siblings were all heterozygous (Figure 1). The transcripts harboring the nonsense mutation in CCDC141 are predicted to undergo nonsense mediated mRNA decay.
Figure 1.

Structural alterations in the affected family vs healthy control. A and B, Magnetic resonance imaging of the brain in coronal sections. Subjects were also described in Ref. 26. A, Healthy adult individual, arrow indicating normal olfactory bulb. B, Aplastic olfactory bulbs in the proband. C, Genotype sequencing of CCDC141 gene shows CCDC141 mutation (p.R724X) (black box indicates mutation site). The top line shows the homozygous wild-type genotype (healthy control individual), the middle line shows the homozygous mutant genotype (proband), and the bottom line shows the heterozygous genotype (mother).
To determine how the loss of CCDC141 contributed to the patient's phenotype, a mouse model was employed. Ccdc141 expression was first characterized in vivo, and then perturbed in explants containing migrating GnRH neurons. In mice, GnRH neurons are first detected in the nasal pit (E10.5–E11.5), arising in the area of the developing vomeronasal organ (VNO) and migrate along olfactory axons to the nasal forebrain junction before reaching their final destination in the brain (Figure 2, A and C). Most GnRH neurons are born between E10.5 and E12.5, and have reached the brain by approximately E16.5. Ccdc141 embryonic staining has not been described earlier than E16.5. To determine whether Ccdc141 was present in migrating GnRH neurons, analysis of Ccdc141 staining was performed in E11.5, E12.5, and E14.5–E15.5 GnRH-GFP mouse embryos. Ccdc141 was detected throughout the nose, and was clearly expressed in GnRH neurons at all 3 developmental points (Figure 2, B and D). Preincubation of CCDC141 peptide with diluted antibody resulted in no specific staining (Figure 2). Staining for the neuronal markers Hu and Tuj1 confirmed Ccdc141 was present in migrating neurons and olfactory/vomeronasal sensory axons in the nasal area (Figure 3A). Ccdc141 did not overlap with BLBP Cre/rosaYFP-positive OECs that surround olfactory fibers and GnRH neurons as they grow/migrate to the olfactory bulb (Figure 3B). Most the GnRH neurons in nasal regions appeared positive for Ccdc141, with expression decreasing as cells entered the forebrain. Because both olfactory axons and GnRH cells were Ccdc141 positive, wrapping of the olfactory axons around the GnRH cells as they migrate in nasal regions makes counting of double labeled cells extremely difficult. One is mainly limited to GnRH cell soma and thus an underestimate can occur if the marker, in this case Ccdc141, is regionally localized to the leading process. However, to address this issue 3 embryos (E12.5 and E15.5) were double stained and GnRH cell soma counted for coexpression of Cdc141 in the nasal region and brain. In nasal regions, 75% of the GnRH cell soma expressed Ccdc141 (n = 82/110). In contrast, no double-labeled GnRH cell soma were detected in the brain (n = 0/20). Robust Ccdc141 staining was also detected in the developing cortex (Figure 3C) as well as cells and sensory axons of the peripheral nervous system such as the trigeminal and dorsal root ganglia (Figure 3, D and E). Because GnRH neurons and olfactory fibers were positive for Ccdc141 between E11.5 and E15.5, disruption of Ccdc141 could certainly impact GnRH/olfactory system development. Thus, the function of Ccdc141 on development of these systems was examined using nasal explants.
Figure 3.

Olfactory neurons and fibers express Ccdc141. A1–A4, Fluorescent staining for Ccdc141 (red) neural markers TUJ1/Hu (green), and nuclear staining with DAPI (blue) in an E14.5 section, highlights expression of Ccdc141 in olfactory sensory axons (arrows) in nasal region, leaving the olfactory epithelium (OE), at the nasal forebrain junction (NFJ) and entering the olfactory bulb (OB); higher magnification of boxed region is shown in A2–A4. B1–B4, In contrast to olfactory axons, Ccdc141 expression (red) was not detected in BLBP-Cre/rosaYFP-positive OECs (green). Ccdc141 (red) was also detected in areas of the developing central (C) and peripheral nervous systems (D and E) and overlapped with early neuronal markers (TUJ1/Hu, green). C1–C3, At E14.5, Ccdc141 was expressed in multiple layers of the developing cortex and in the outer layer (higher magnification) (C2–C3). D and E, At E12.5–E14.5, Ccdc141 expression was detected in multiple sensory ganglia, such as the dorsal root ganglia shown here (cell soma, asterisk; arrow, fibers). Scale bars, 50 μm (A1, B1, C1, and F) and 100 μm (D).
First, we verified that Ccdc141 was expressed in nasal explants in a manner similar to that seen in vivo. PCR confirmed the presence of Ccdc141 transcript in cDNAs made from whole explants (Figure 4A, lanes 1 and 2) and in single GnRH neurons isolated from 4-div explants (Figure 4A, lanes 3–9; 4/7 single cells). Double fluorescence immunostaining confirmed expression of Ccdc141 in most migrating GnRH neurons and olfactory axons (Figure 4B). Because Ccdc141 expression in our model system mimicked that found in vivo, experiments were performed to test Ccdc141 function. Explants were electroporated with control or Ccdc141-targeting siRNA at 2 div, when GnRH neurons emerge from nasal pits. By 4 div, this knockdown reduced Ccdc141 immunofluorescence in GnRH cells by 60% compared with controls (Figure 4, C–E). Defects in migration rates were assessed by live imaging at 4 div (Figure 5) and changes in neuronal advance measured after 5 div (Figure 6). In live imaging studies, GnRH neurons treated with Ccdc141-targeting siRNA showed a reduction of over 25% in both linear and total migration rates (P < .05, Mann-Whitney test). Analysis of explants fixed at 5 div and stained for GnRH to assess overall GnRH cell migration showed that GnRH neurons in Ccdc141 knockdown explants did not migrate as far from the main tissue mass as controls (Figure 6, A–C). No interaction of the treatment was detected along the frequency distribution (two-way ANOVA interaction P > .05), suggesting GnRH neurons were uniformly affected. A second, independent siRNA sequence (27), yielded similar results (Figure 6F). Because Ccdc141 is expressed in other cell types as well as GnRH neurons, migration defects could be indirect. In particular, olfactory axons serve as a substrate for GnRH neuronal migration in nasal regions (33) and migratory defects may occur secondary to changes in olfactory axon growth even though the olfactory neurons themselves do not migrate in this preparation. Thus, a new group of electroporated explants were fixed at 5 div and stained for peripherin. Olfactory axon growth was robust in control and Ccdc141 knockdown explants (Figure 6, D and E). Analysis of axon tips farthest from the explant center showed no statistical difference between groups (control siRNA 1614 ± 56 μm, Ccdc141 siRNA 1706 ± 45 μm, n = 60/group, P = .2, t test). GnRH neurons were closely apposed to olfactory axons in both groups, indicating that adhesion was not impaired by knockdown of Ccdc141 (Figure 6, D and E, inset).
Figure 4.
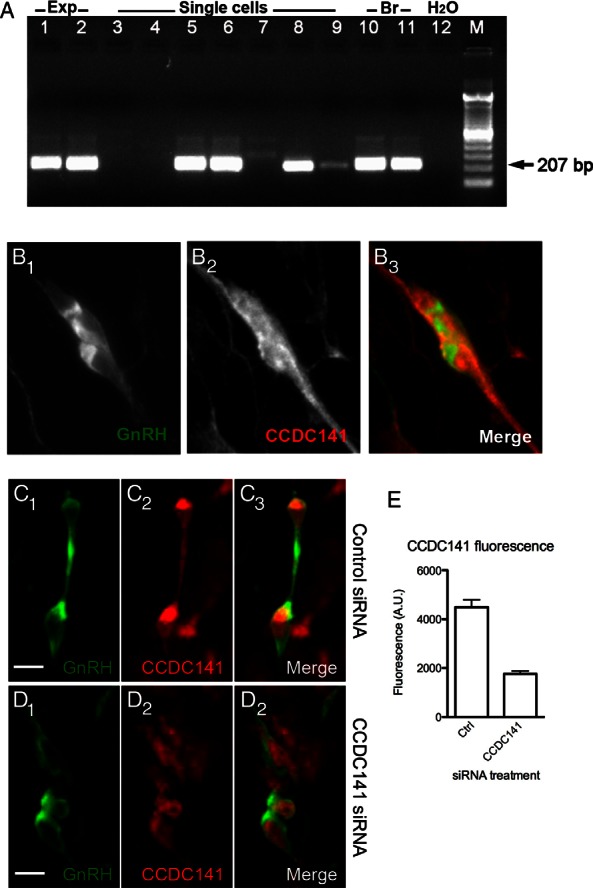
Ccdc141 expression and knockdown in GnRH neurons. A, PCR product of approximately 207 bp indicated presence of Ccdc141 in cDNA made from transcript in 6- and 3-div whole nasal explants (Exp, lanes 1 and 2), and in 4 of 7 cDNA made from individual GnRH cells isolated at 4.5 div (single cells, lanes 3–9). Control brain (Br) cDNAs from embryonic (lane 10) and adult brain (lane 11), and negative control water (lane 12); M, DNA marker. B, Confocal fluorescence immunostaining showing GnRH expression (B1), Ccdc141 expression (B2), and merged image confirming coexpression (B3). C–E, GnRH (C1 and D1) and Ccdc141 (C2 and D2) coexpression (merge, C3 and D3) in explants treated with control (C) and Ccdc141-targeting siRNA (D). E, Measurements of background-subtracted fluorescence indicated levels of cytoplasmic Ccdc141 in GnRH neurons decreased after treatment with Ccdc141-targeting siRNA compared with control siRNA (P < .001, n ≥ 85 GnRH neurons, 2 explants/group). Mann-Whitney U test used for this comparison. Scale bars, 10 μm.
Figure 5.

CCDC141 knockdown reduces GnRH neuronal migration. A and B, Frequency distributions of the linear (A) and total (B) migration rates fit with lognormal curves showing a shift in the distribution in the CCDC141 siRNA-treated cultures vs the controls (P < .05, Mann-Whitney U test, n = 73 control, and n = 40 CCDC141 siRNA-treated cells; error bars, SEM). C and D, Examples of measurement of linear migration (C) and total migration (D) of GnRH neurons over a 40-minute imaging session. C, Linear migration rates represent the displacement of the neuron (black arrow) over the imaging period. D, Total migration rates represent the average of the frame-to-frame movement rates (represented by the simplified time series and trajectory trace). Scale bars, 10 μm.
Figure 6.

CCDC141 knockdown reduces GnRH neuronal migration but not olfactory outgrowth. A and B, Immunopositive GnRH neurons in control (A) or Ccdc141-siRNA-treated explants (B). Distance GnRH neurons (arrowheads, examples) migrated from center of nasal pit (asterisk) outward was measured 3 days after electroporation. Scale bars, 100 μm (A and B). C and F, Cumulative frequency of 2 independent siRNA experiments showing distributions of distances GnRH neurons migrated. In both experiments, a shift to the left after Ccdc141 knockdown was detected (P < .001, two-way ANOVA, n = 4 explants/condition). D and E, Olfactory axon growth was unaffected by electroporation with Ccdc141 siRNA. Images of explants electroporated with control (D) or Ccdc141 siRNA (E) and stained for peripherin (marker of olfactory axons, brown fibers) and GnRH (blue cell bodies). Farthest GnRH neurons visible in the field of view are marked with arrows/arrowheads. Scale bars, 100 μm (D and E). Higher magnification of GnRH neurons (blue) closely apposed to olfactory axons (brown) from explants in A and B, respectively (arrows), are shown in boxes. Scale bars, 10 μm (insets in D and E).
Discussion
An increasing number of KS cases are mediated through oligogenic inheritance, and in fact, oligogenicity might account for most cases (25). In this report, we describe a mutation in CCDC141 in KS patients that also have a partial loss of function mutation in FEZF1 (26). In a cohort of 20 probands with KS, a biallelic mutation in CCDC141 was found. CCDC141 encodes a coiled-coil domain containing protein whose function is uncertain. Using a mouse model the expression and function of CCDC141 during development of the GnRH/olfactory systems was examined. Ccdc141 was found in migrating GnRH neurons and olfactory axons. Knockdown of CCDC141 did not perturb olfactory axon outgrowth but resulted in a decrease in GnRH cell movement and overall migration out of the nasal pit.
In mice, GnRH neurons migrate from the nasal placode to the forebrain between E11.5 and E16.5 (3). During this same period, olfactory axons extend, and OECs migrate, to the developing olfactory bulbs. GnRH neurons migrate on these olfactory axon tracks, associated with OECs. Between E11.5 and E15.5, Ccdc141 was robustly expressed in migrating GnRH neurons, cells in the developing VNO, and olfactory axons, structures which, when perturbed, can contribute to the KS phenotype. During this development time window, Ccdc141 expression was also detected in sensory neurons, cell bodies and axons, in the dorsal root and trigeminal ganglia and within cortical structures within the CNS. Ccdc141 was not expressed in OECs. Between E11.5 and E15.5, Ccdc141 expression decreased in nasal mesenchymal cells, whereas expression in olfactory axons and GnRH cells remained robust. However, as GnRH cells entered the forebrain, Ccdc141 expression appeared to decrease, though general non-GnRH cellular Ccdc141 levels were apparent within the forebrain. Thus, the expression of Ccdc141 in GnRH neurons correlated with migration in nasal regions.
Nasal explants have been successfully used to study the migration of GnRH neurons (20, 21, 34). In this model system large numbers of GnRH cells are maintained and migrate out of the nasal pit in association with extending olfactory axons and OECs. Using nasal explants, we showed that disruption of Ccdc141 impaired GnRH neuronal migration, whereas no changes in olfactory axon outgrowth were detected. Given the intact olfactory axon pathway, migration defects could in principle be caused by alterations to intracellular motility mechanisms, disrupted adhesion to axons, or both. Although neither hypothesis can be excluded at this point, the most parsimonious explanation is that Ccdc141, through its interactions with myosin II, directly affects cellular motility. A calcium signaling pathway to myosin II through calcium/calmodulin-dependent protein kinase kinase, 5′ adenosine monophosphate-activated protein kinase, and Rho A/Rho kinase drives cell motility in GnRH neurons (20). In addition, no loss of adhesion to axons was observed as a consequence of Ccdc141 knockdown (Figure 6). Taken together, these results suggest that defects in motility, rather than adhesion, underlie the migration defects. Impaired migration of GnRH neurons is consistent with the low levels of reproductive hormones measured in the KS patient. The MRI of the proband in the family showed aplasia of the olfactory bulbs, also a common phenotype in patients with KS. The fact that no change was detected in olfactory axon growth in our knockdown experiments supports Ccdc141 disruption of olfactory bulb development rather than olfactory axon ingrowth. Anosmia associated with KS is due to failure of the olfactory/terminal nerve fibers to establish proper contact with the forebrain. This lack of connectivity can result from defects in olfactory neurons or defective formation of olfactory bulb (3). However, we cannot exclude that the combination of slowed GnRH neuronal migration resulting from Cdcc141 disruption and slowed ingrowth of olfactory axons resulting from a partial functioning FezF1 (26) together contributed to aplasia of the olfactory bulbs seen in the KS patients.
Very little is known about the function of CCDC141. To our knowledge, the only other study specifically examining Ccdc141 function also identified a role in cortical neuronal migration (27). Analysis of the MRIs in the present study found no difference in cortical thickness in patients carrying the CCDC141 mutation. Notably, Fezf1 and Fezf2 are both important for neocortical development, with redundant functions such that loss of a single Fez family member is not sufficient to impair early neurogenesis. Fezf2 may compensate for changes in Ccdc141, enabling normal formation of the cortex. However, nuclear translocation, which GnRH neurons exhibit (34), is a hallmark of neuronal migration and regulation of the centrosome in the leading process is important for nuclear translocation during directed migration of neurons (41). Fukuda et al (27) showed that Ccdc141 regulated myosin II and proposed that via interactions with centrosome-associated protein DISC1, CCDC141 may link myosin II activity with centrosome dynamics in the leading process during cortical neuron migration. DISC1 has also been localized to the rear of migrating cortical interneurons (42), where actinomyosin contraction is thought to push the nucleus forward into the leading process (22). Supporting this link, olfactory neuronal precursors derived from patients with schizophrenia and bipolar disorder showed altered DISC1 subcellular distribution and impaired cell migration (43). Live imaging experiments of GnRH neurons demonstrated that actin contraction in both the leading process and the cell rear drive movement (20) through interactions with the microtubule cytoskeleton at the actin cortex (21). In vivo and in vitro, Ccdc141 expression appeared uniformly distributed throughout the GnRH cell soma. Thus, localization of Ccdc141 in GnRH neurons to both the rear of the cell and in the leading process supports the possibility that Ccdc141 may regulate cytoskeletal dynamics in both of these compartments, consistent with decreased migration of GnRH neurons after knockdown of Ccdc141.
In our perturbation experiments, GnRH neurons remained closely apposed to olfactory axons, indicating that adhesion was not altered by knockdown of Ccdc141. Although Ccdc141 was also robustly expressed in olfactory fibers, knockdown in vitro did not reduce axon outgrowth. This may be due to differences in the function of cytoskeletal dynamics in migrating neurons (GnRH) compared with axon extension (olfactory neurons). Manipulations that inhibit migration often have no effect on, or even enhance, axon outgrowth (20). These data suggest that impaired GnRH neuronal migration per se due to the CCDC141 mutation (ie, independent of olfactory neuron axon targeting) can account in part for the failed journey of GnRH neurons. Functions of the known genes associated with KS phenotype generally relate to membrane bound molecules or extracellular factors, which serve as adhesion or guidance molecules (40, 44). CCDC141 on the other hand is associated with cytoskeletal mechanics of neuronal migration.
In conclusion, the CCDC141 mutation reported here is a new example of genetic aberrations associated with KS. These studies strongly suggest that, in humans and mice, CCDC141 takes part in embryonic migration of GnRH neurons enabling them to form a hypothalamic neuronal network to initiate pulsatile GnRH secretion and reproductive function.
Acknowledgments
We thank Dr D. Gutmann for generation of the BLBP-Ccre mice.
This work was supported by the Scientific and Technological Research Council of Turkey Project 109S455, by the Cukurova University Scientific Research Projects, and by the International Centre for Genetic Engineering and Biotechnology Grant CRP/TUR10-01. B.I.H., P.J.C., J.D.R.T., and S.W. were supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke Grant NS002824-24/25. B.I.H. also received funding through the National Institute of General Medical Sciences Postdoctoral Research Associate Program.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CNS
- central nervous system
- Disc1
- disrupted in schizophrenia-1
- div
- days in vitro
- FGF
- fibroblast growth factor
- KS
- Kallmann Syndrome
- MRI
- magnetic resonance imaging
- NIH
- National Institutes of Health
- OEC
- olfactory ensheathing cell
- Rb
- rabbit polyclonal
- SFM
- serum-free medium
- siRNA
- small interfering RNA
- VNO
- vomeronasal organ.
References
- 1. Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161–164. [DOI] [PubMed] [Google Scholar]
- 2. Wray S, Grant P, Gainer H. Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proc Natl Acad Sci USA. 1989;86:8132–8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forni PE, Wray S. GnRH, anosmia and hypogonadotropic hypogonadism–where are we? Front Neuroendocrinol. 2015;36:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Franco B, Guioli S, Pragliola A, et al. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529–536. [DOI] [PubMed] [Google Scholar]
- 5. Ballabio A, Camerino G. The gene for X-linked Kallmann syndrome: a human neuronal migration defect. Curr Opin Genet Dev. 1992;2:417–421. [DOI] [PubMed] [Google Scholar]
- 6. Guerrini R, Mei D, Cordelli DM, Pucatti D, Franzoni E, Parrini E. Symmetric polymicrogyria and pachygyria associated with TUBB2B gene mutations. Eur J Hum Genet. 2012;20:995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jansen AC, Oostra A, Desprechins B, et al. TUBA1A mutations: from isolated lissencephaly to familial polymicrogyria. Neurology. 2011;76:988–992. [DOI] [PubMed] [Google Scholar]
- 8. Poirier K, Saillour Y, Fourniol F, et al. Expanding the spectrum of TUBA1A-related cortical dysgenesis to polymicrogyria. Eur J Hum Genet. 2013;21:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bahi-Buisson N, Poirier K, Boddaert N, et al. Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations. J Med Genet. 2008;45:647–653. [DOI] [PubMed] [Google Scholar]
- 10. des Portes V, Pinard JM, Billuart P, et al. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. [DOI] [PubMed] [Google Scholar]
- 11. Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH. Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993;270:2838–2842. [DOI] [PubMed] [Google Scholar]
- 12. Gleeson JG, Allen KM, Fox JW, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. [DOI] [PubMed] [Google Scholar]
- 13. Jamuar SS, Lam AT, Kircher M, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371:733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reiner O, Carrozzo R, Shen Y, et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein β-subunit-like repeats. Nature. 1993;364:717–721. [DOI] [PubMed] [Google Scholar]
- 15. Sossey-Alaoui K, Hartung AJ, Guerrini R, et al. Human doublecortin (DCX) and the homologous gene in mouse encode a putative Ca2+-dependent signaling protein which is mutated in human X-linked neuronal migration defects. Hum Mol Genet. 1998;7:1327–1332. [DOI] [PubMed] [Google Scholar]
- 16. Romaniello R, Tonelli A, Arrigoni F, et al. A novel mutation in the β-tubulin gene TUBB2B associated with complex malformation of cortical development and deficits in axonal guidance. Dev Med Child Neurol. 2012;54:765–769. [DOI] [PubMed] [Google Scholar]
- 17. Fox JW, Lamperti ED, Ekiolu YZ, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–1325. [DOI] [PubMed] [Google Scholar]
- 18. Drévillon L, Megarbane A, Demeer B, et al. KBP-cytoskeleton interactions underlie developmental anomalies in Goldberg-Shprintzen syndrome. Hum Mol Genet. 2013;22:2387–2399. [DOI] [PubMed] [Google Scholar]
- 19. Falnikar A, Tole S, Baas PW. Kinesin-5, a mitotic microtubule-associated motor protein, modulates neuronal migration. Mol Biol Cell. 2011;22:1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hutchins BI, Klenke U, Wray S. Calcium release-dependent actin flow in the leading process mediates axophilic migration. J Neurosci. 2013;33:11361–11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hutchins BI, Wray S. Capture of microtubule plus-ends at the actin cortex promotes axophilic neuronal migration by enhancing microtubule tension in the leading process. Front Cell Neurosci. 2014;8:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martini FJ, Valdeolmillos M. Actomyosin contraction at the cell rear drives nuclear translocation in migrating cortical interneurons. J Neurosci. 2010;30:8660–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schaar BT, McConnell SK. Cytoskeletal coordination during neuronal migration. Proc Natl Acad Sci USA. 2005;102:13652–13657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Solecki DJ, Trivedi N, Govek EE, Kerekes RA, Gleason SS, Hatten ME. Myosin II motors and F-actin dynamics drive the coordinated movement of the centrosome and soma during CNS glial-guided neuronal migration. Neuron. 2009;63:63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pitteloud N, Durrani S, Raivio T, Sykiotis GP. Complex genetics in idiopathic hypogonadotropic hypogonadism. Front Horm Res. 2010;39:142–153. [DOI] [PubMed] [Google Scholar]
- 26. Kotan LD, Hutchins BI, Ozkan Y, et al. Mutations in FEZF1 cause Kallmann syndrome. Am J Hum Genet. 2014;95:326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fukuda T, Sugita S, Inatome R, Yanagi S. CAMDI, a novel disrupted in schizophrenia 1 (DISC1)-binding protein, is required for radial migration. J Biol Chem. 2010;285:40554–40561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spergel DJ, Krüth U, Hanley DF, Sprengel R, Seeburg PH. GABA- and glutamate-activated channels in green fluorescent protein-tagged gonadotropin-releasing hormone neurons in transgenic mice. J Neurosci. 1999;19:2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hegedus B, Dasgupta B, Shin JE, et al. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell. 2007;1:443–457. [DOI] [PubMed] [Google Scholar]
- 30. Wray S, Gähwiler BH, Gainer H. Slice cultures of LHRH neurons in the presence and absence of brainstem and pituitary. Peptides. 1988;9:1151–1175. [DOI] [PubMed] [Google Scholar]
- 31. Gangatirkar P, Gangadharan S, Narendranath A, Nagpal S, Salunke DM, Karande AA. Monoclonal antibodies to gonadotropin-releasing hormone (GnRH) inhibit binding of the hormone to its receptor. Hybrid Hybridomics. 2002;21:281–286. [DOI] [PubMed] [Google Scholar]
- 32. Forni PE, Taylor-Burds C, Melvin VS, Williams T, Wray S. Neural crest and ectodermal cells intermix in the nasal placode to give rise to GnRH-1 neurons, sensory neurons, and olfactory ensheathing cells. J Neurosci. 2011;31:6915–6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fueshko S, Wray S. LHRH cells migrate on peripherin fibers in embryonic olfactory explant cultures: an in vitro model for neurophilic neuronal migration. Dev Biol. 1994;166:331–348. [DOI] [PubMed] [Google Scholar]
- 34. Casoni F, Hutchins BI, Donohue D, Fornaro M, Condie BG, Wray S. SDF and GABA interact to regulate axophilic migration of GnRH neurons. J Cell Sci. 2012;125:5015–5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kramer PR, Wray S. Novel gene expressed in nasal region influences outgrowth of olfactory axons and migration of luteinizing hormone-releasing hormone (LHRH) neurons. Genes Dev. 2000;14:1824–1834. [PMC free article] [PubMed] [Google Scholar]
- 36. Giacobini P, Kopin AS, Beart PM, Mercer LD, Fasolo A, Wray S. Cholecystokinin modulates migration of gonadotropin-releasing hormone-1 neurons. J Neurosci. 2004;24:4737–4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sharifi N, Reuss AE, Wray S. Prenatal LHRH neurons in nasal explant cultures express estrogen receptor β transcript. Endocrinology 2002;143:2503–2507. [DOI] [PubMed] [Google Scholar]
- 38. Toba Y, Tiong JD, Ma Q, Wray S. CXCR4/SDF-1 system modulates development of GnRH-1 neurons and the olfactory system. Dev Neurobiol. 2008;68:487–503. [DOI] [PubMed] [Google Scholar]
- 39. Tiong J, Locastro T, Wray S. Gonadotropin-releasing hormone-1 (GnRH-1) is involved in tooth maturation and biomineralization. Dev Dyn. 2007;236:2980–2992. [DOI] [PubMed] [Google Scholar]
- 40. Topaloglu AK, Kotan LD. Molecular causes of hypogonadotropic hypogonadism. Curr Opin Obstet Gynecol. 2010;22:264–270. [DOI] [PubMed] [Google Scholar]
- 41. Marín O, Valiente M, Ge X, Tsai LH. Guiding neuronal cell migrations. Cold Spring Harb Perspect Biol. 2010;2:a001834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steinecke A, Gampe C, Valkova C, Kaether C, Bolz J. Disrupted-in-Schizophrenia 1 (DISC1) is necessary for the correct migration of cortical interneurons. J Neurosci. 2012;32:738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Muñoz-Estrada J, Benítez-King G, Berlanga C, Meza I. Altered subcellular distribution of the 75-kDa DISC1 isoform, cAMP accumulation, and decreased neuronal migration in schizophrenia and bipolar disorder: implications for neurodevelopment. CNS Neurosci Ther. 2015;21:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wray S. From nose to brain: development of gonadotrophin-releasing hormone-1 neurones. J Neuroendocrinol. 2010;22:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]